
Understanding Physicochemical Mechanisms of Sequential Infiltration Synthesis toward Rational Process Design for Uniform Incorporation of Metal Oxides


Abstract
: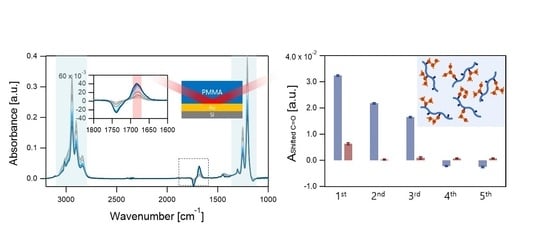
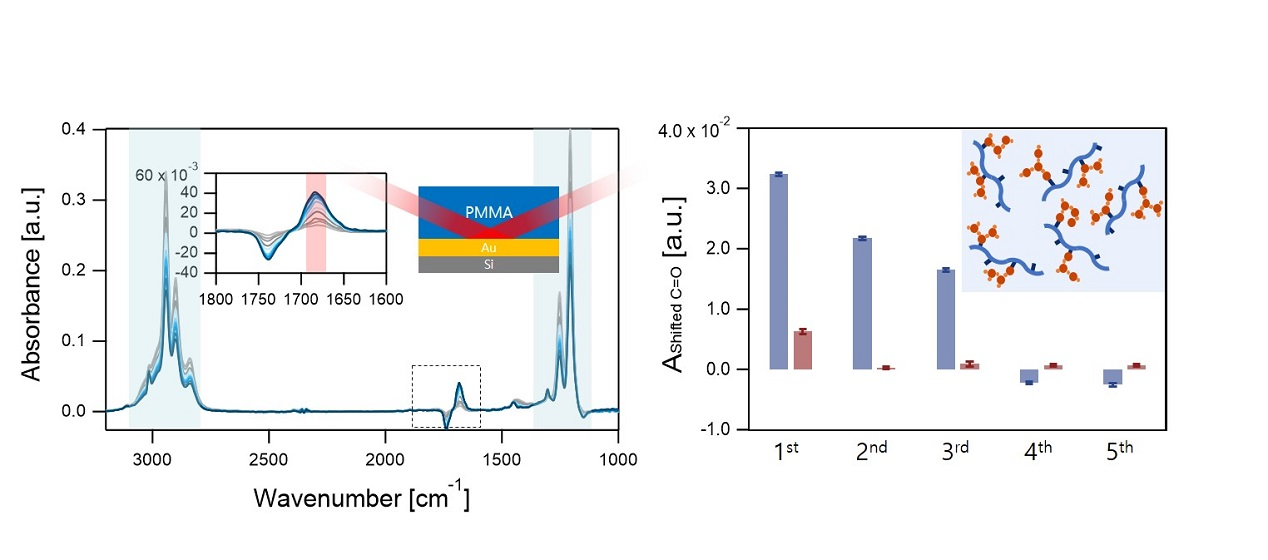{kind=link}
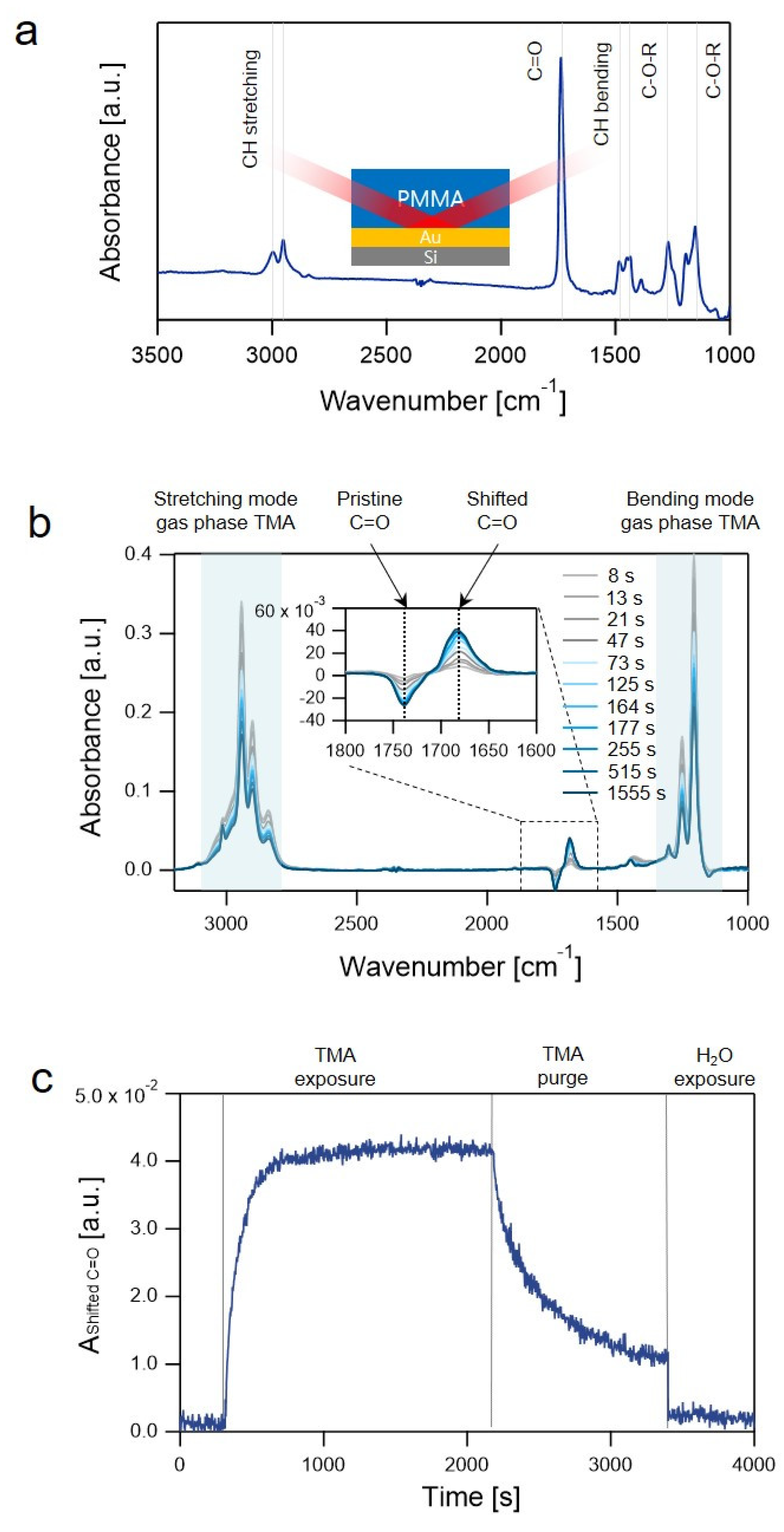{kind=link}
{kind=link}
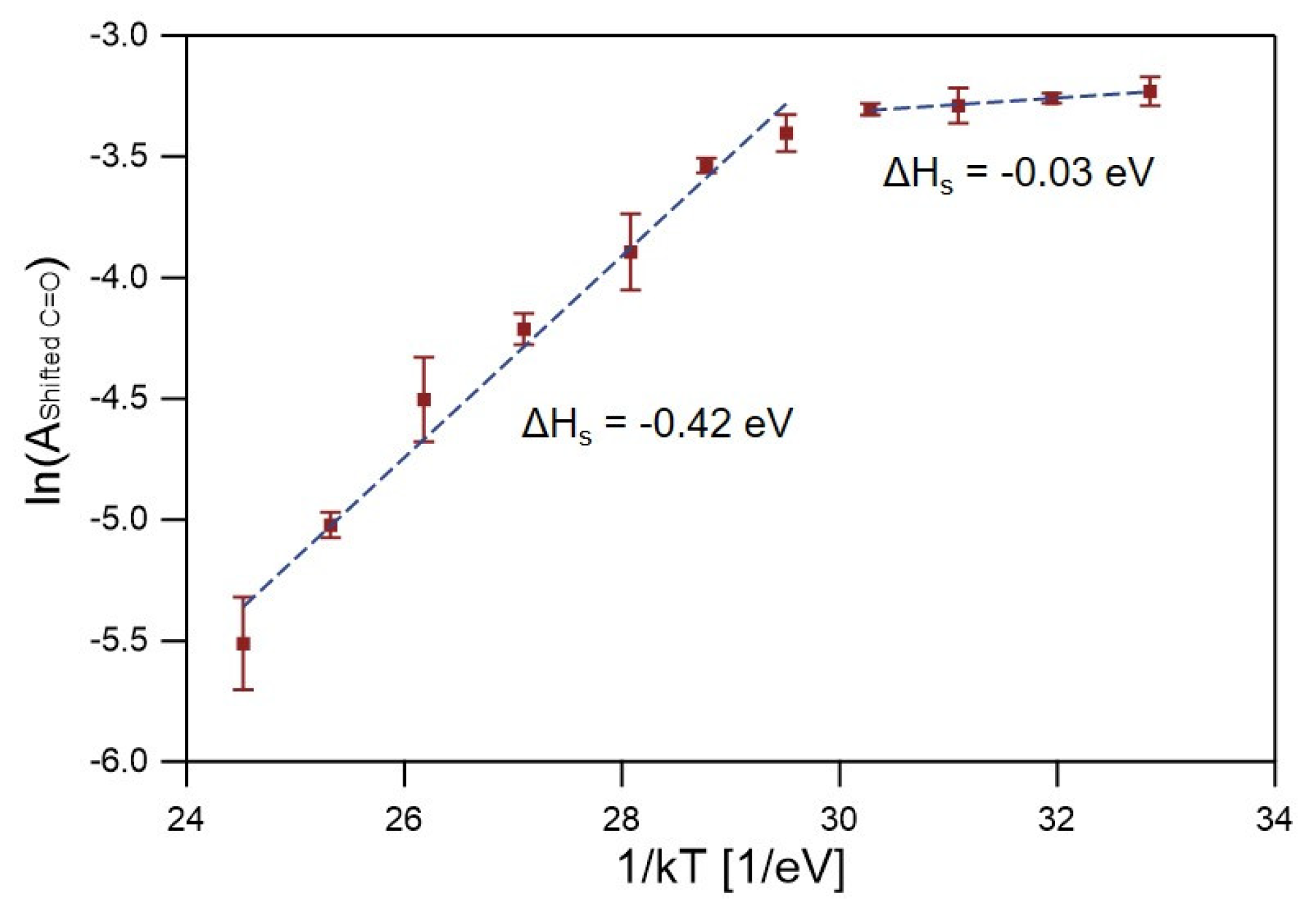
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. SIS
2.3. In Situ FTIR Spectroscopy
2.4. Other Characterization Methods
3. Results
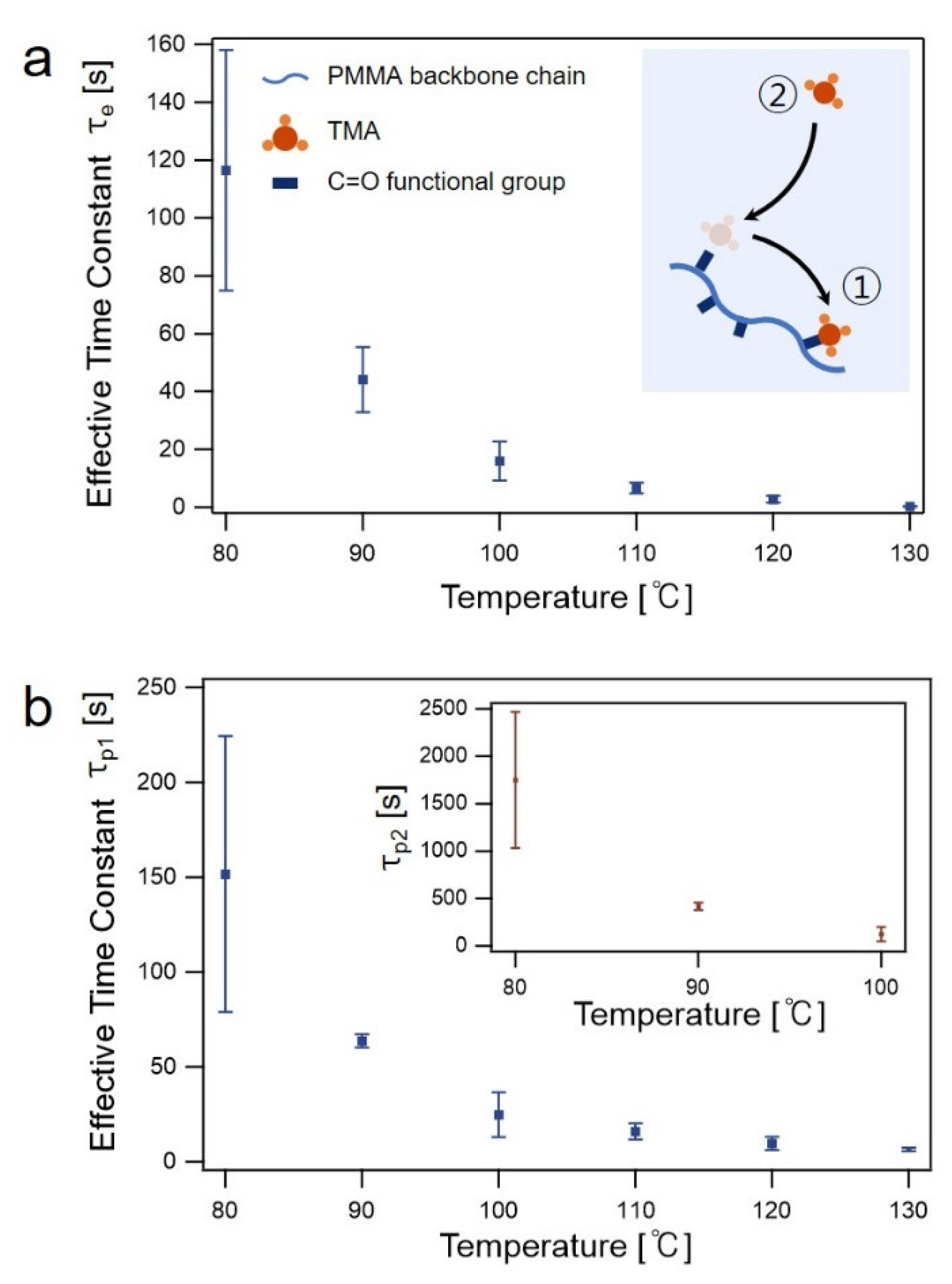
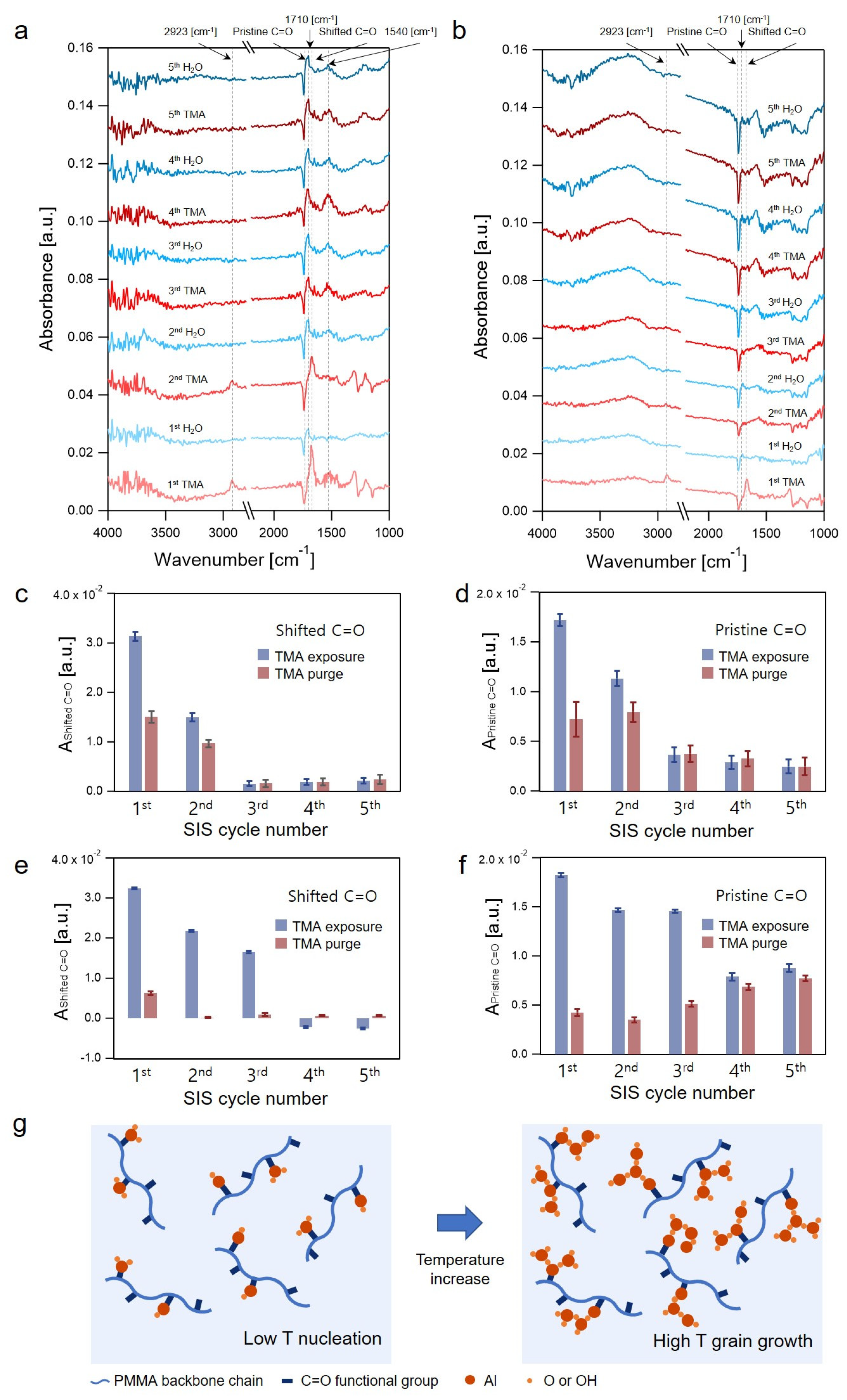
3.1. In Situ FTIR Spectroscopy
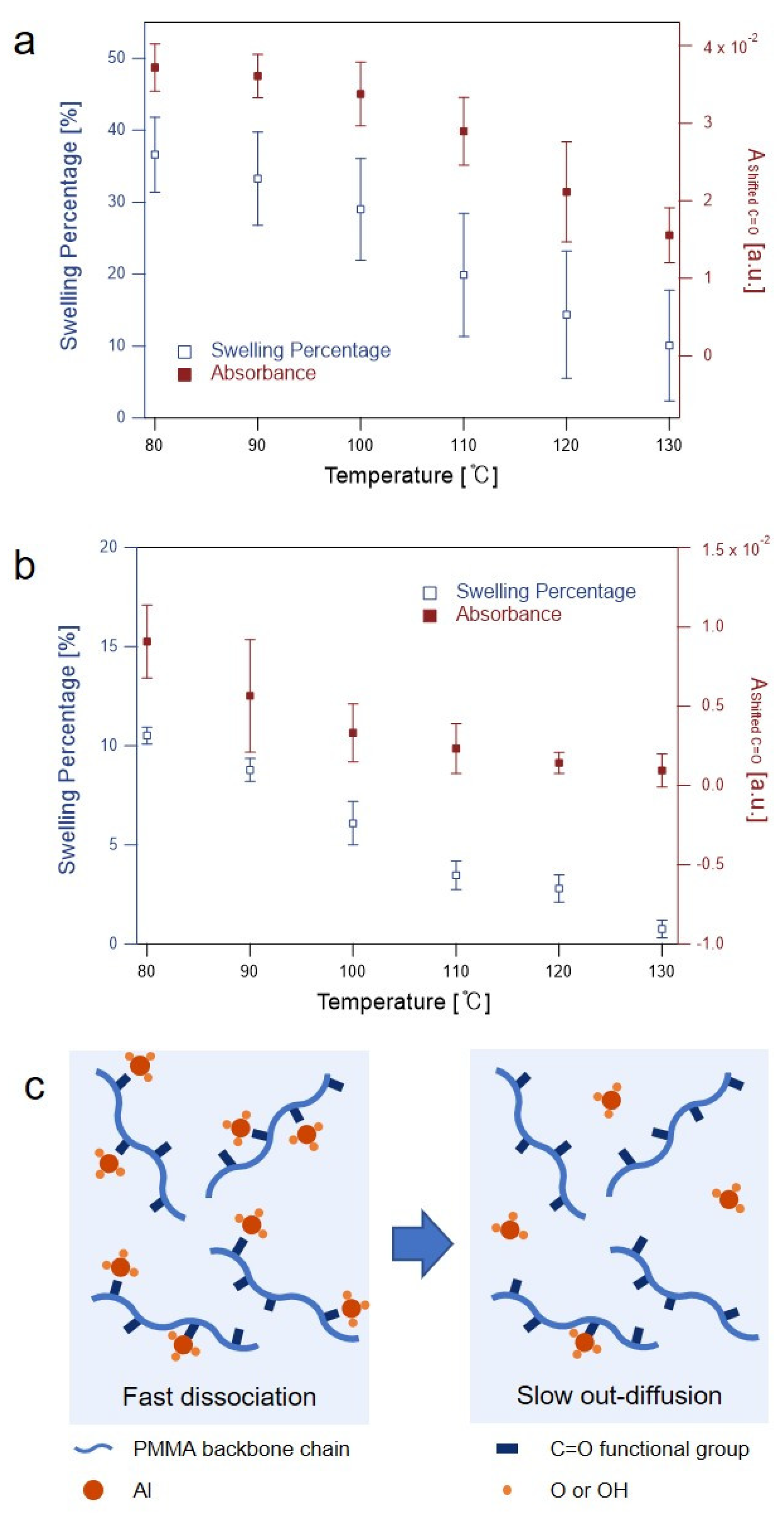
3.2. Precursor Sorption
3.3. Precursor Diffusion
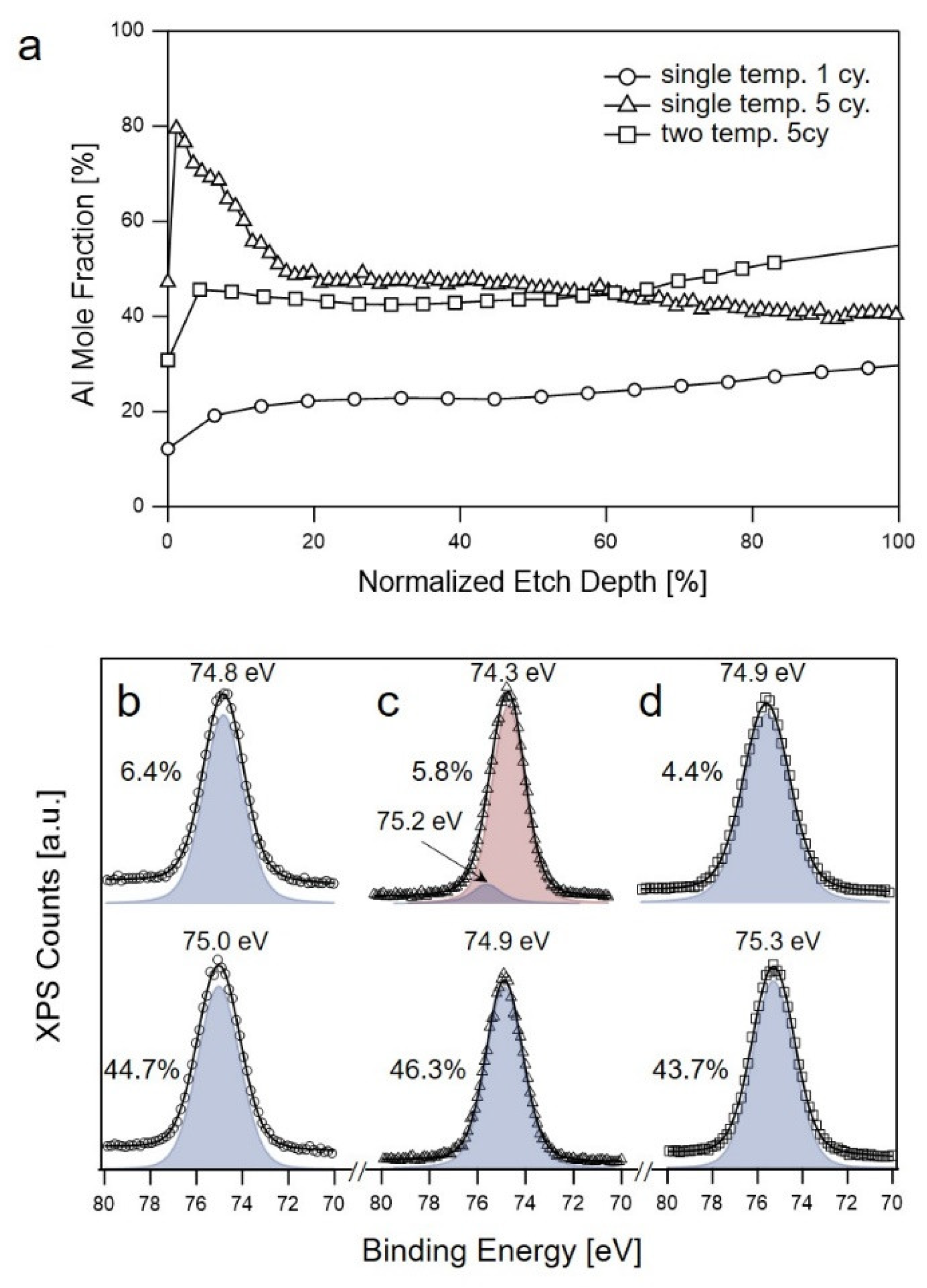
3.4. Uniform Distribution of Aluminum Oxides/Hydroxides
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Waldman, R.Z.; Mandia, D.J.; Yanguas-Gil, A.; Martinson, A.B.F.; Elam, J.W.; Darling, S.B. The chemical physics of sequential infiltration synthesis—A thermodynamic and kinetic perspective. J. Chem. Phys. 2019, 151, 190901. [Google Scholar] [CrossRef] [PubMed]
- Leng, C.Z.; Losego, M.D. Vapor phase infiltration (VPI) for transforming polymers into organic–inorganic hybrid materials: A critical review of current progress and future challenges. Mater. Horiz. 2017, 4, 747–771. [Google Scholar] [CrossRef]
- Cara, E.; Murataj, I.; Milano, G.; De Leo, N.; Boarino, L.; Ferrarese Lupi, F. Recent Advances in Sequential Infiltration Synthesis (SIS) of Block Copolymers (BCPs). Nanomaterials 2021, 11, 994. [Google Scholar] [CrossRef] [PubMed]
- Berman, D.; Shevchenko, E. Design of functional composite and all-inorganic nanostructured materials via infiltration of polymer templates with inorganic precursors. J. Mater. Chem. C 2020, 8, 10604–10627. [Google Scholar] [CrossRef]
- Lee, S.M.; Pippel, E.; Gösele, U.; Dresbach, C.; Qin, Y.; Chandran, C.V.; Bräuniger, T.; Hause, G.; Knez, M. Greatly increased toughness of infiltrated spider silk. Science 2009, 324, 488–492. [Google Scholar] [CrossRef]
- Tiwale, N.; Subramanian, A.; Kisslinger, K.; Lu, M.; Kim, J.; Stein, A.; Nam, C.-Y. Advancing next generation nanolithography with infiltration synthesis of hybrid nanocomposite resists. J. Mater. Chem. C 2019, 7, 8803–8812. [Google Scholar] [CrossRef]
- Tseng, Y.-C.; Peng, Q.; Ocola, L.E.; Czaplewski, D.A.; Elam, J.W.; Darling, S.B. Etch properties of resists modified by sequential infiltration synthesis. J. Vac. Sci. Technol. B 2011, 29, 06FG01. [Google Scholar] [CrossRef]
- Barry, E.; Mane, A.U.; Libera, J.A.; Elam, J.W.; Darling, S.B. Advanced oil sorbents using sequential infiltration synthesis. J. Mater. Chem. 2017, 5, 2929–2935. [Google Scholar] [CrossRef]
- Yu, Y.; Li, Z.; Wang, Y.; Gong, S.; Wang, X. Sequential Infiltration Synthesis of Doped Polymer Films with Tunable Electrical Properties for Efficient Triboelectric Nanogenerator Development. Adv. Mater. 2015, 27, 4938–4944. [Google Scholar] [CrossRef]
- Moshonov, M.; Frey, G.L. Directing Hybrid Structures by Combining Self-Assembly of Functional Block Copolymers and Atomic Layer Deposition: A Demonstration on Hybrid Photovoltaics. Langmuir 2015, 31, 12762–12769. [Google Scholar] [CrossRef]
- Obuchovsky, S.; Levin, M.; Levitsky, A.; Frey, G.L. Morphology visualization of P3HT:Fullerene blends by using subsurface atomic layer deposition. Org. Electron. 2017, 49, 234–241. [Google Scholar] [CrossRef]
- Obuchovsky, S.; Frankenstein, H.; Vinokur, J.; Hailey, A.K.; Loo, Y.-L.; Frey, G.L. Mechanism of Metal Oxide Deposition from Atomic Layer Deposition inside Nonreactive Polymer Matrices: Effects of Polymer Crystallinity and Temperature. Chem. Mater. 2016, 28, 2668–2676. [Google Scholar] [CrossRef]
- Berman, D.; Guha, S.; Lee, B.; Elam, J.W.; Darling, S.B.; Shevchenko, E.V. Sequential Infiltration Synthesis for the Design of Low Refractive Index Surface Coatings with Controllable Thickness. ACS Nano 2017, 11, 2521–2530. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Ashraf, A.; Xin, H.; Tong, X.; Sutter, P.; Eisaman, M.D.; Black, C.T. Sub-50-nm self-assembled nanotextures for enhanced broadband antireflection in silicon solar cells. Nat. Commun. 2015, 6, 5963. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Segal-Peretz, T.; Oruc, M.E.; Suh, H.S.; Wu, G.; Nealey, P.F. Fabrication of Nanoporous Alumina Ultrafiltration Membrane with Tunable Pore Size Using Block Copolymer Templates. Adv. Funct. Mater. 2017, 27, 1701756. [Google Scholar] [CrossRef]
- McGuinness, E.K.; Zhang, F.; Ma, Y.; Lively, R.P.; Losego, M.D. Vapor Phase Infiltration of Metal Oxides into Nanoporous Polymers for Organic Solvent Separation Membranes. Chem. Mater. 2019, 31, 5509–5518. [Google Scholar] [CrossRef]
- Yang, H.-C.; Waldman, R.Z.; Chen, Z.; Darling, S.B. Atomic layer deposition for membrane interface engineering. Nanoscale 2018, 10, 20505–20513. [Google Scholar] [CrossRef]
- Pleshek, D.; Tran, J.; Li, Y.; Shirani, A.; Shevchenko, E.V.; Berman, D. Swelling-Assisted Sequential Infiltration Synthesis of Nanoporous ZnO Films with Highly Accessible Pores and Their Sensing Potential for Ethanol. ACS Appl. Mater. Interfaces 2021, 13, 35941–35948. [Google Scholar] [CrossRef]
- Chakrabarti, B.; Chan, H.; Alam, K.; Koneru, A.; Gage, T.E.; Ocola, L.E.; Divan, R.; Rosenmann, D.; Khanna, A.; Grisafe, B.; et al. Nanoporous Dielectric Resistive Memories Using Sequential Infiltration Synthesis. ACS Nano 2021, 15, 4155–4164. [Google Scholar] [CrossRef]
- Petit, R.R.; Li, J.; Van de Voorde, B.; Van Vlierberghe, S.; Smet, P.F.; Detavernier, C. Atomic Layer Deposition on Polymer Thin Films: On the Role of Precursor Infiltration and Reactivity. ACS Appl. Mater. Interfaces 2021, 13, 46151–46163. [Google Scholar] [CrossRef]
- Hill, G.T.; Lee, D.T.; Williams, P.S.; Needham, C.D.; Dandley, E.C.; Oldham, C.J.; Parsons, G.N. Insight on the Sequential Vapor Infiltration Mechanisms of Trimethylaluminum with Poly(methyl methacrylate), Poly(vinylpyrrolidone), and Poly(acrylic acid). J. Phys. Chem 2019, 123, 16146–16152. [Google Scholar] [CrossRef]
- Dandley, E.C.; Needham, C.D.; Williams, P.S.; Brozena, A.H.; Oldham, C.J.; Parsons, G.N. Temperature-dependent reaction between trimethylaluminum and poly(methyl methacrylate) during sequential vapor infiltration: Experimental and ab initio analysis. J. Mater. Chem. C 2014, 2, 9416–9424. [Google Scholar] [CrossRef]
- Waldman, R.Z.; Choudhury, D.; Mandia, D.J.; Elam, J.W.; Nealey, P.F.; Martinson, A.B.F.; Darling, S.B. Sequential Infiltration Synthesis of Al2O3 in Polyethersulfone Membranes. JOM 2019, 71, 212–223. [Google Scholar] [CrossRef]
- Cianci, E.; Nazzari, D.; Seguini, G.; Perego, M. Trimethylaluminum Diffusion in PMMA Thin Films during Sequential Infiltration Synthesis: In Situ Dynamic Spectroscopic Ellipsometric Investigation. Adv. Mater. Interfaces 2018, 5, 1801016. [Google Scholar] [CrossRef]
- Caligiore, F.E.; Nazzari, D.; Cianci, E.; Sparnacci, K.; Laus, M.; Perego, M.; Seguini, G. Effect of the Density of Reactive Sites in P(S-r-MMA) Film during Al2O3 Growth by Sequential Infiltration Synthesis. Adv. Mater. Interfaces 2019, 6, 1900503. [Google Scholar] [CrossRef]
- Snelgrove, M.; McFeely, C.; Shiel, K.; Hughes, G.; Yadav, P.; Weiland, C.; Woicik, J.C.; Mani-Gonzalez, P.G.; Lundy, R.; Morris, M.A.; et al. Analysing trimethylaluminum infiltration into polymer brushes using a scalable area selective vapor phase process. Adv. Mater. 2021, 2, 769–781. [Google Scholar] [CrossRef]
- Hermans, J.J.; Baij, L.; Koenis, M.; Keune, K.; Iedema, P.D.; Woutersen, S. 2D-IR spectroscopy for oil paint conservation: Elucidating the water-sensitive structure of zinc carboxylate clusters in ionomers. Sci. Adv. 2019, 5, eaaw3592. [Google Scholar] [CrossRef]
- He, X.; Waldman, R.Z.; Mandia, D.J.; Jeon, N.; Zaluzec, N.J.; Borkiewicz, O.J.; Ruett, U.; Darling, S.B.; Martinson, A.B.F.; Tiede, D.M. Resolving the Atomic Structure of Sequential Infiltration Synthesis Derived Inorganic Clusters. ACS Nano 2020, 14, 14846–14860. [Google Scholar] [CrossRef]
- Biswas, M.; Libera, J.A.; Darling, S.B.; Elam, J.W. New Insight into the Mechanism of Sequential Infiltration Synthesis from Infrared Spectroscopy. Chem. Mater. 2014, 26, 6135–6141. [Google Scholar] [CrossRef]
- Leng, C.Z.; Losego, M.D. A physiochemical processing kinetics model for the vapor phase infiltration of polymers: Measuring the energetics of precursor-polymer sorption, diffusion, and reaction. Phys. Chem. Chem. Phys. 2018, 20, 21506–21514. [Google Scholar] [CrossRef]
- Weisbord, I.; Shomrat, N.; Azoulay, R.; Kaushansky, A.; Segal-Peretz, T. Understanding and Controlling Polymer–Organometallic Precursor Interactions in Sequential Infiltration Synthesis. Chem. Mater. 2020, 32, 4499–4508. [Google Scholar] [CrossRef]
- Ren, Y.; McGuinness, E.K.; Huang, C.; Joseph, V.R.; Lively, R.P.; Losego, M.D. Reaction–Diffusion Transport Model to Predict Precursor Uptake and Spatial Distribution in Vapor-Phase Infiltration Processes. Chem. Mater. 2021, 33, 5210–5222. [Google Scholar] [CrossRef]
- Ito, S.; Ozaki, Y.; Nakamura, T.; Nakagawa, M. Depth profiles of aluminum component in sequential infiltration synthesis-treated electron beam resist films analyzed by time-of-flight secondary ion mass spectrometry. Jpn. J. Appl. Phys. 2020, 59, SIIC03. [Google Scholar] [CrossRef]
- Kim, J.J.; Suh, H.S.; Zhou, C.; Mane, A.U.; Lee, B.; Kim, S.; Emery, J.D.; Elam, J.W.; Nealey, P.F.; Fenter, P.; et al. Mechanistic understanding of tungsten oxide in-plane nanostructure growth via sequential infiltration synthesis. Nanoscale 2018, 10, 3469–3479. [Google Scholar] [CrossRef]
- Biswas, M.; Libera, J.A.; Darling, S.B.; Elam, J.W. Polycaprolactone: A Promising Addition to the Sequential Infiltration Synthesis Polymer Family Identified through In Situ Infrared Spectroscopy. ACS Appl. Polym. Mater. 2020, 2, 5501–5510. [Google Scholar] [CrossRef]
- Waldman, R.Z.; Jeon, N.; Mandia, D.J.; Heinonen, O.; Darling, S.B.; Martinson, A.B.F. Sequential Infiltration Synthesis of Electronic Materials: Group 13 Oxides via Metal Alkyl Precursors. Chem. Mater. 2019, 31, 5274–5285. [Google Scholar] [CrossRef]
- Elam, J.W.; Biswas, M.; Darling, S.B.; Yanguas-Gil, A.; Emery, J.D.; Martinson, A.B.F.; Nealey, P.F.; Segal-Peretz, T.; Peng, Q.; Winterstein, J.; et al. New Insights into Sequential Infiltration Synthesis. ECS Trans. 2015, 69, 147–157. [Google Scholar] [CrossRef]
- Biswas, M.; Libera, J.A.; Darling, S.B.; Elam, J.W. Kinetics for the Sequential Infiltration Synthesis of Alumina in Poly(methyl methacrylate): An Infrared Spectroscopic Study. J. Phys. Chem 2015, 119, 14585–14592. [Google Scholar] [CrossRef]
- Peng, Q.; Tseng, Y.-C.; Darling, S.B.; Elam, J.W. A Route to Nanoscopic Materials via Sequential Infiltration Synthesis on Block Copolymer Templates. ACS Nano 2011, 5, 4600–4606. [Google Scholar] [CrossRef]
- Berens, A.R.; Hopfenberg, H.B. Diffusion of organic vapors at low concentrations in glassy PVC, polystyrene, and PMMA. J. Membr. Sci. 1982, 10, 283–303. [Google Scholar] [CrossRef]
- Guerra-Nuñez, C.; Döbeli, M.; Michler, J.; Utke, I. Reaction and Growth Mechanisms in Al2O3 deposited via Atomic Layer Deposition: Elucidating the Hydrogen Source. Chem. Mater. 2017, 29, 8690–8703. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ham, J.; Ko, M.; Choi, B.; Kim, H.-U.; Jeon, N. Understanding Physicochemical Mechanisms of Sequential Infiltration Synthesis toward Rational Process Design for Uniform Incorporation of Metal Oxides. Sensors 2022, 22, 6132. https://0-doi-org.brum.beds.ac.uk/10.3390/s22166132
Ham J, Ko M, Choi B, Kim H-U, Jeon N. Understanding Physicochemical Mechanisms of Sequential Infiltration Synthesis toward Rational Process Design for Uniform Incorporation of Metal Oxides. Sensors. 2022; 22(16):6132. https://0-doi-org.brum.beds.ac.uk/10.3390/s22166132
Chicago/Turabian StyleHam, Jiwoong, Minkyung Ko, Boyun Choi, Hyeong-U Kim, and Nari Jeon. 2022. "Understanding Physicochemical Mechanisms of Sequential Infiltration Synthesis toward Rational Process Design for Uniform Incorporation of Metal Oxides" Sensors 22, no. 16: 6132. https://0-doi-org.brum.beds.ac.uk/10.3390/s22166132