Discovery of Potent Dual EGFR/HER2 Inhibitors Based on Thiophene Scaffold Targeting H1299 Lung Cancer Cell Line

 , ,
, ,  , ,
, ,  ,
,
Abstract
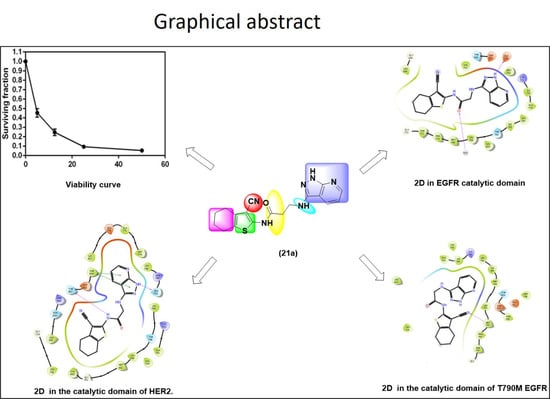
:
1. Introduction
Rationale and Design
2. Results and Discussion
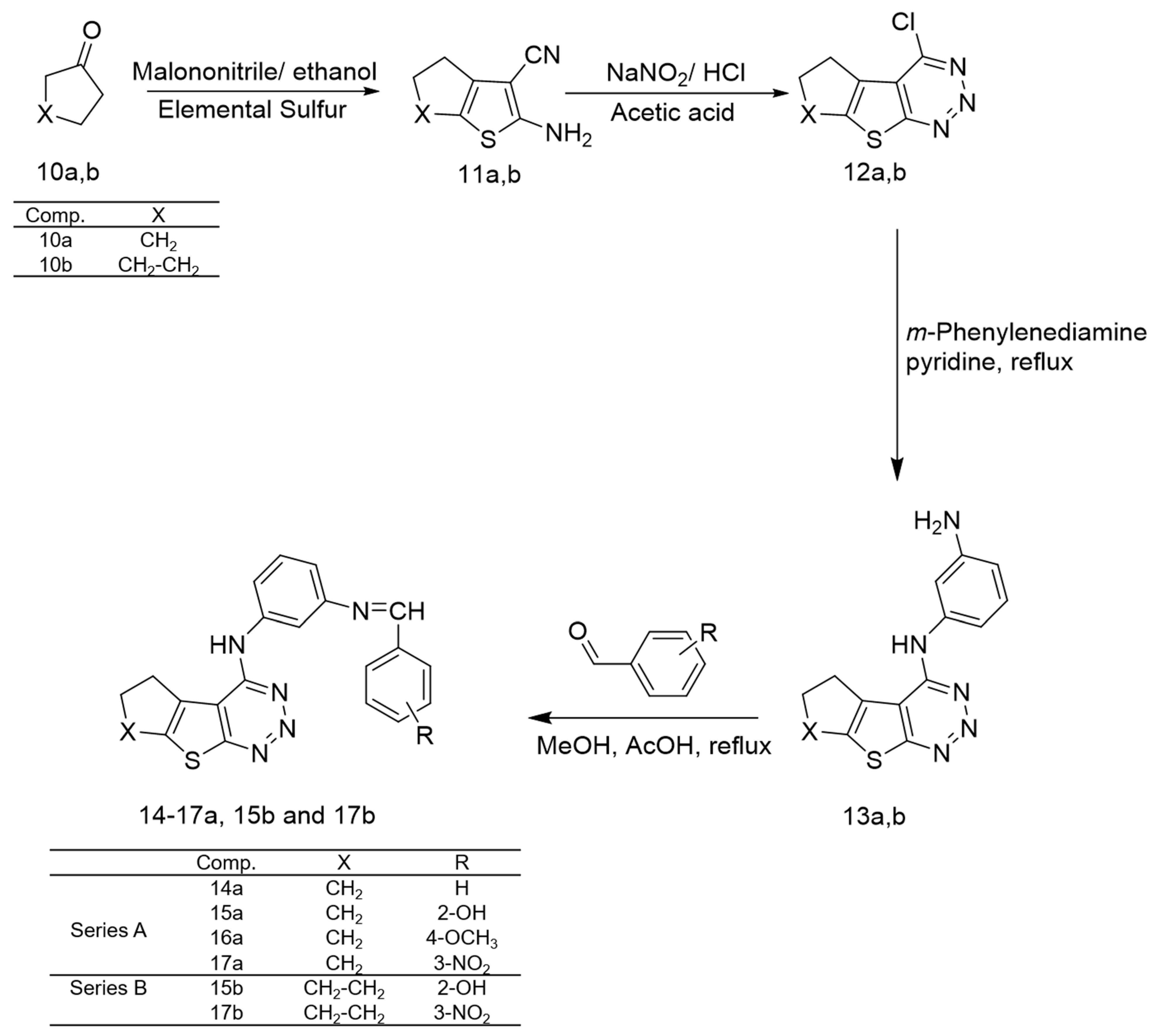
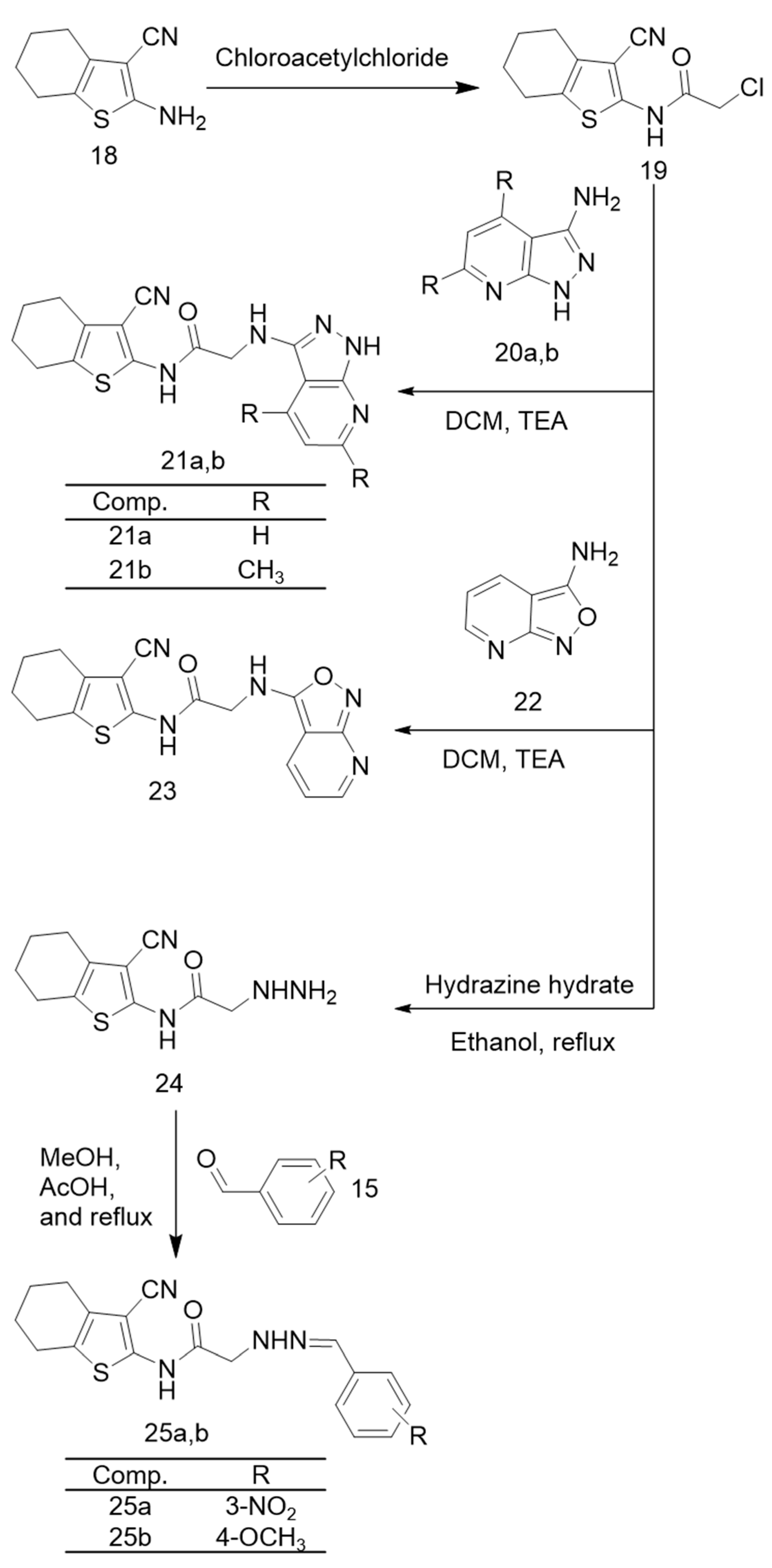
2.1. Chemistry
2.2. Biological Evaluation
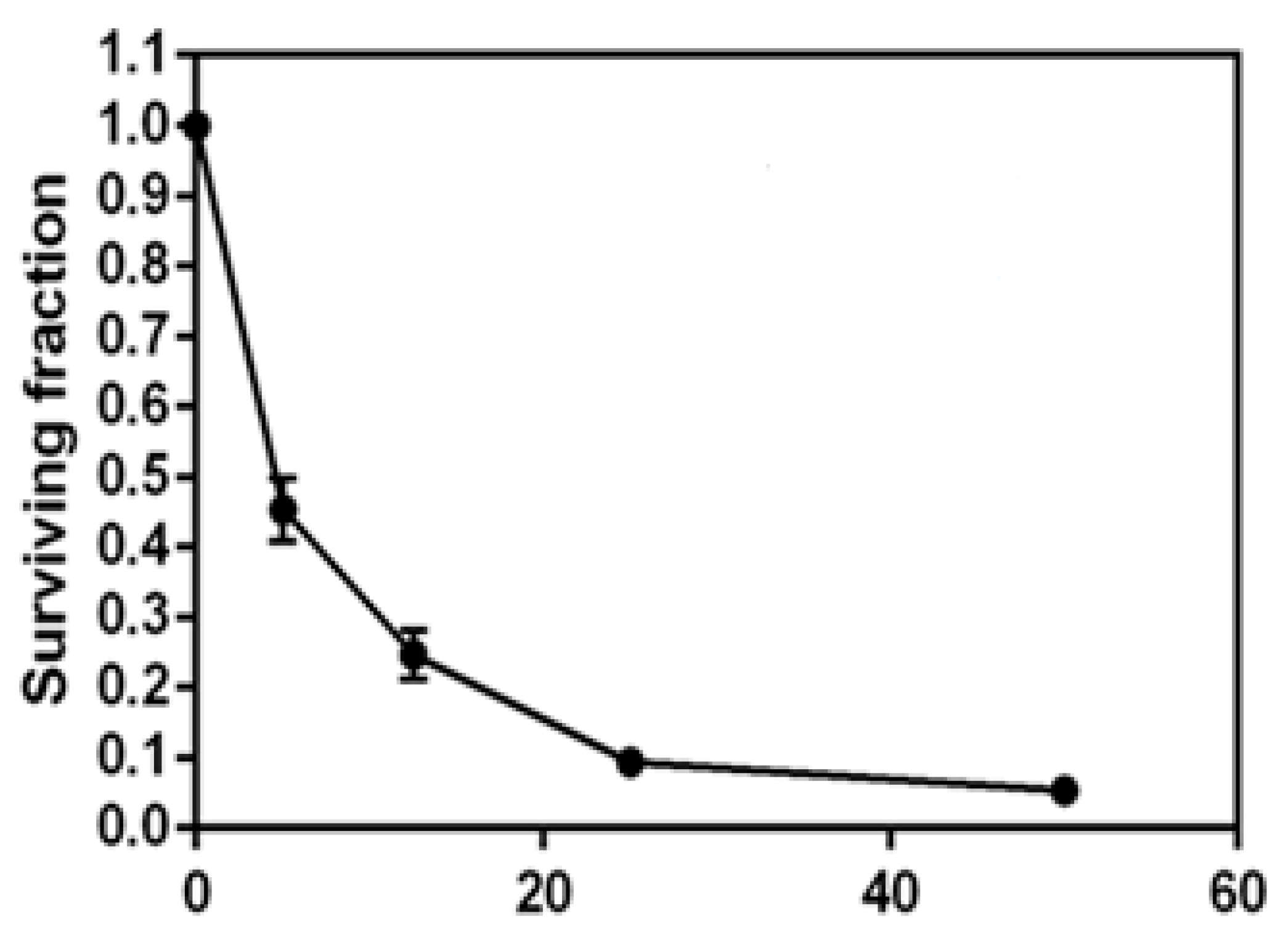
2.2.1. In Vitro Cytotoxic Activity against H1299 Cell Line
2.2.2. EGFR and HER2 Kinase Inhibitory Assay
2.3. Molecular Modeling
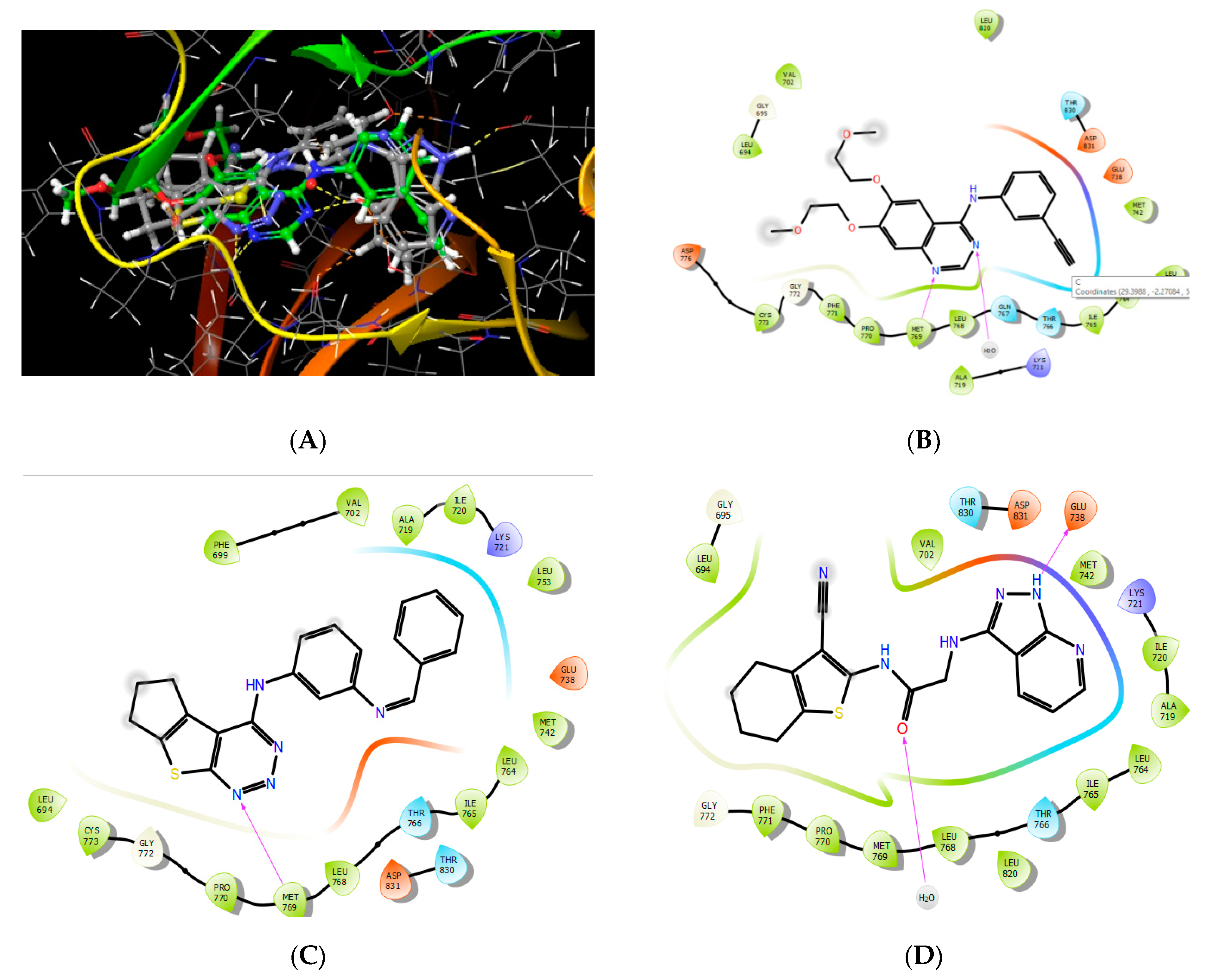
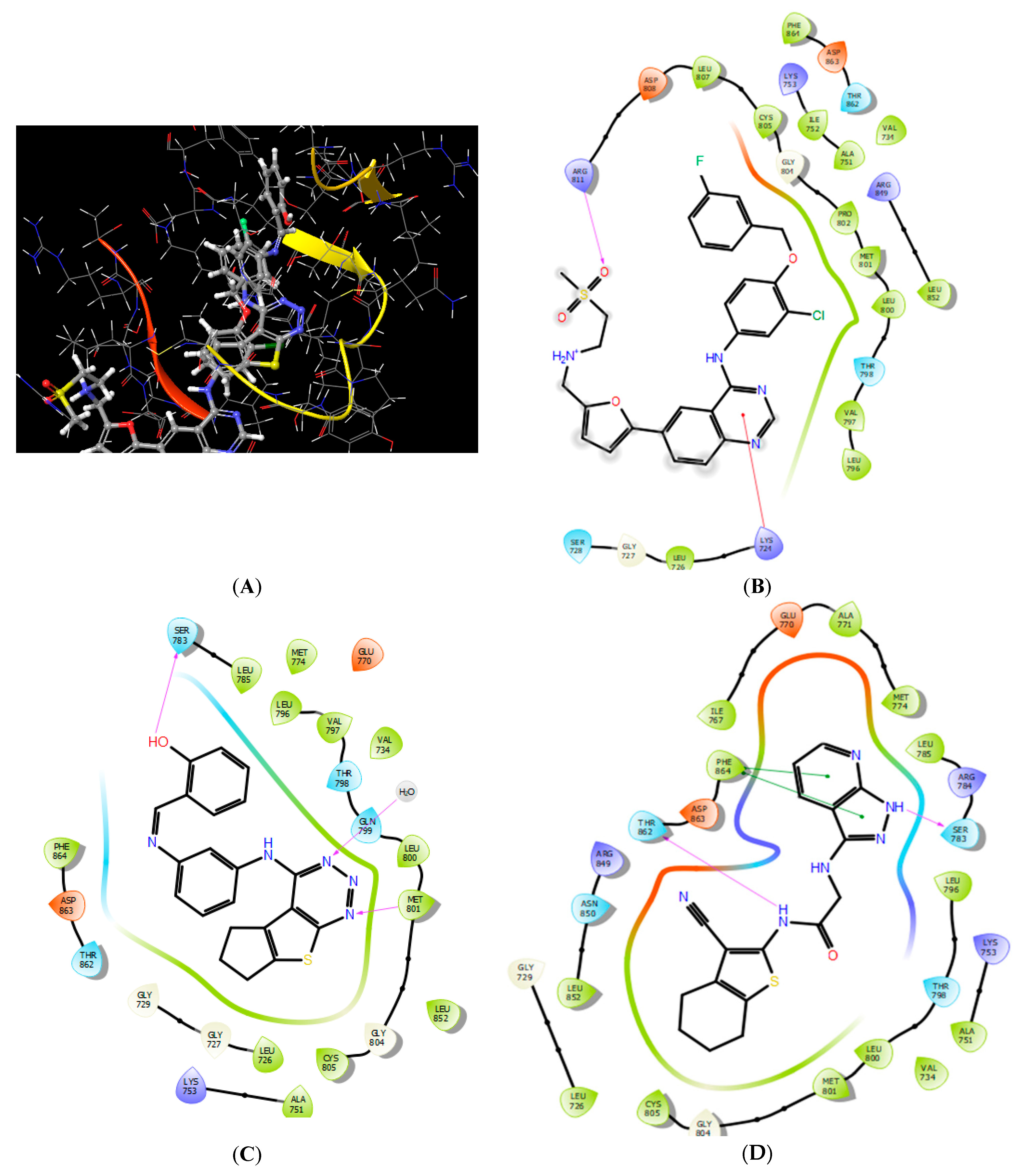
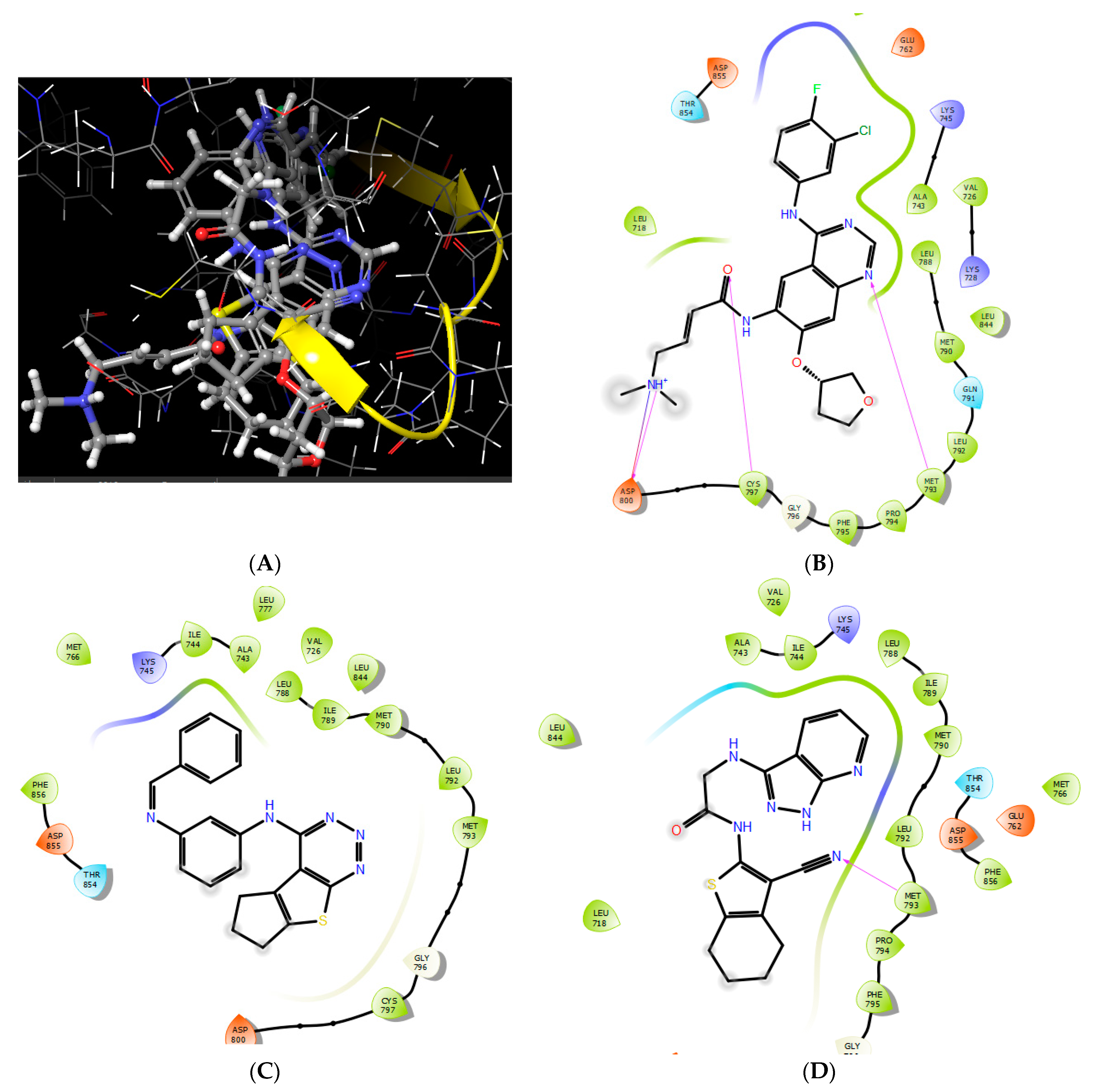
2.3.1. Molecular Docking
2.3.2. Physicochemical Properties and In Silico ADME Prediction
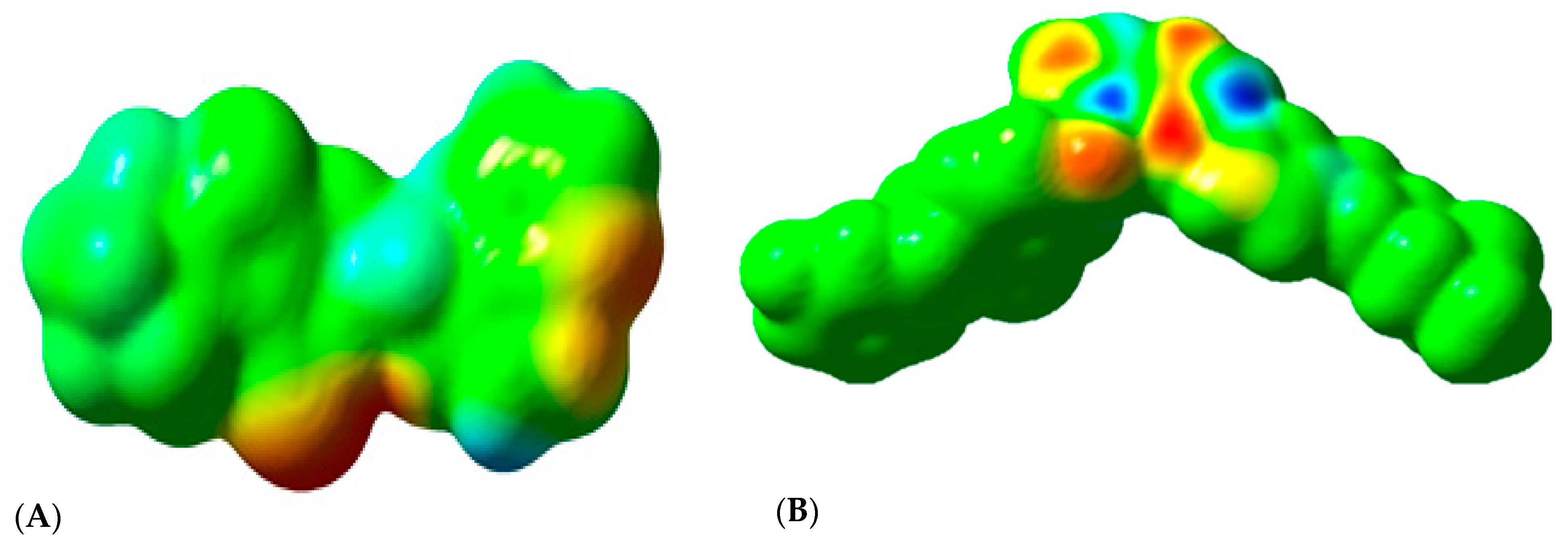
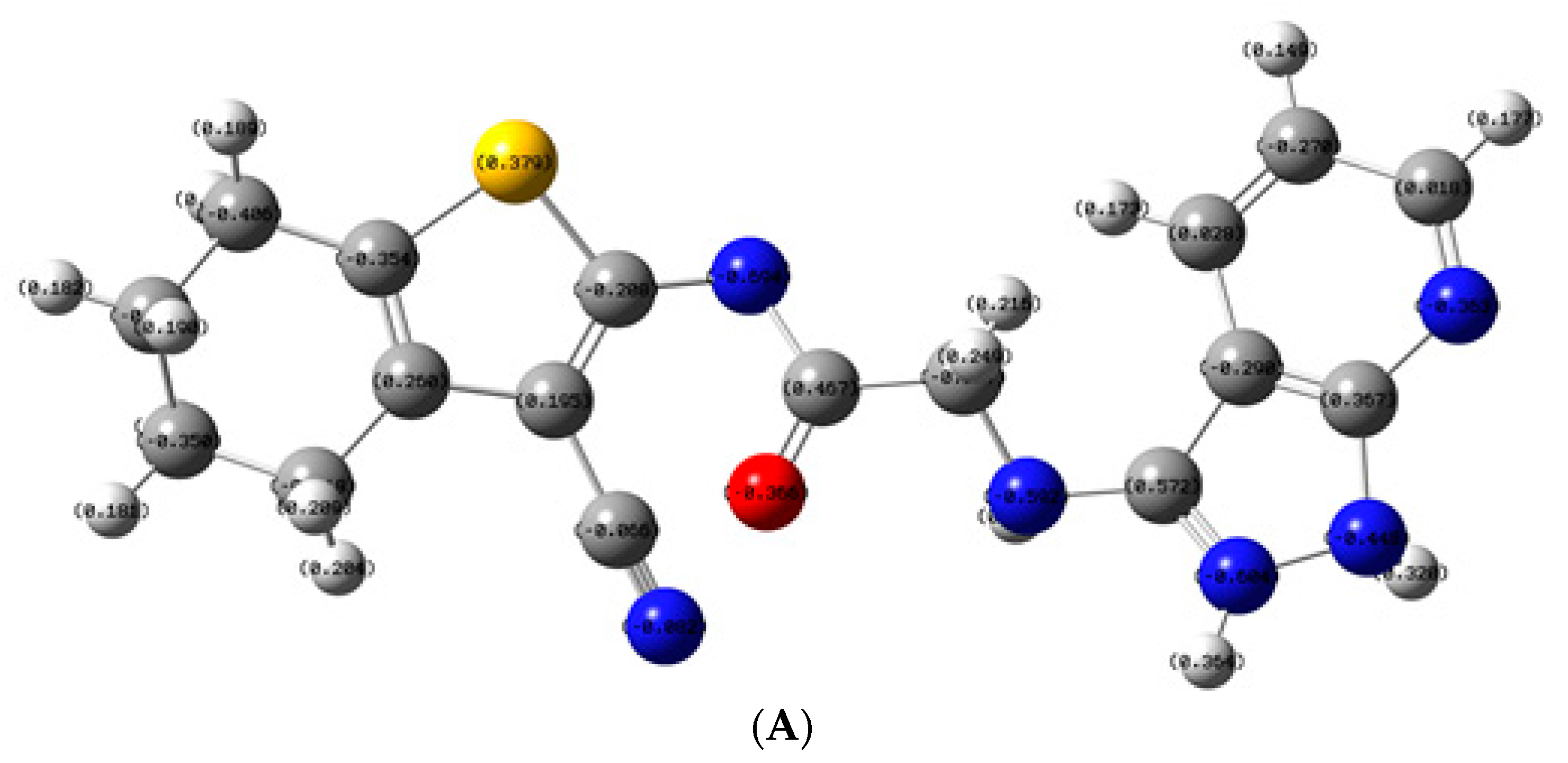
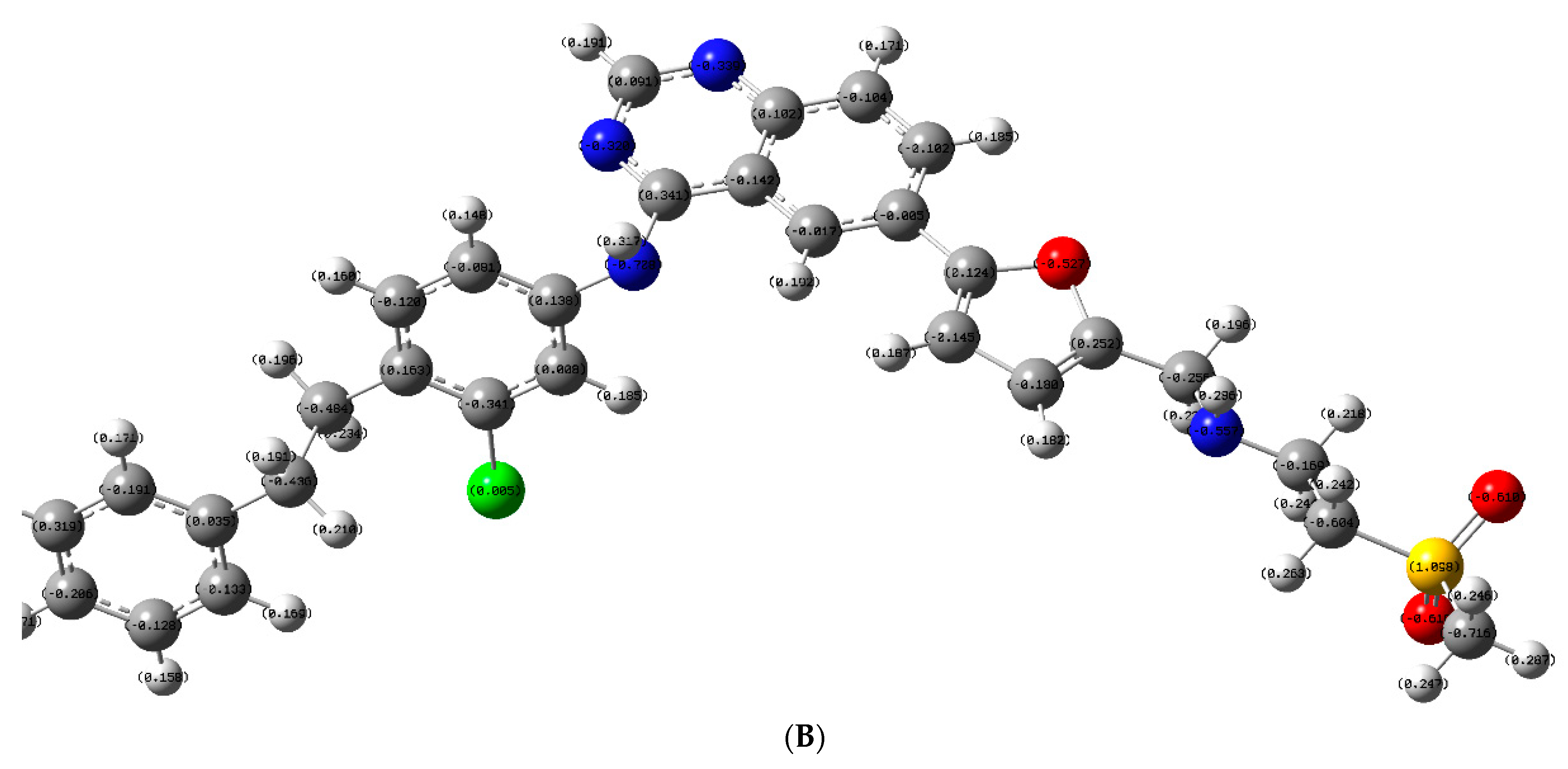
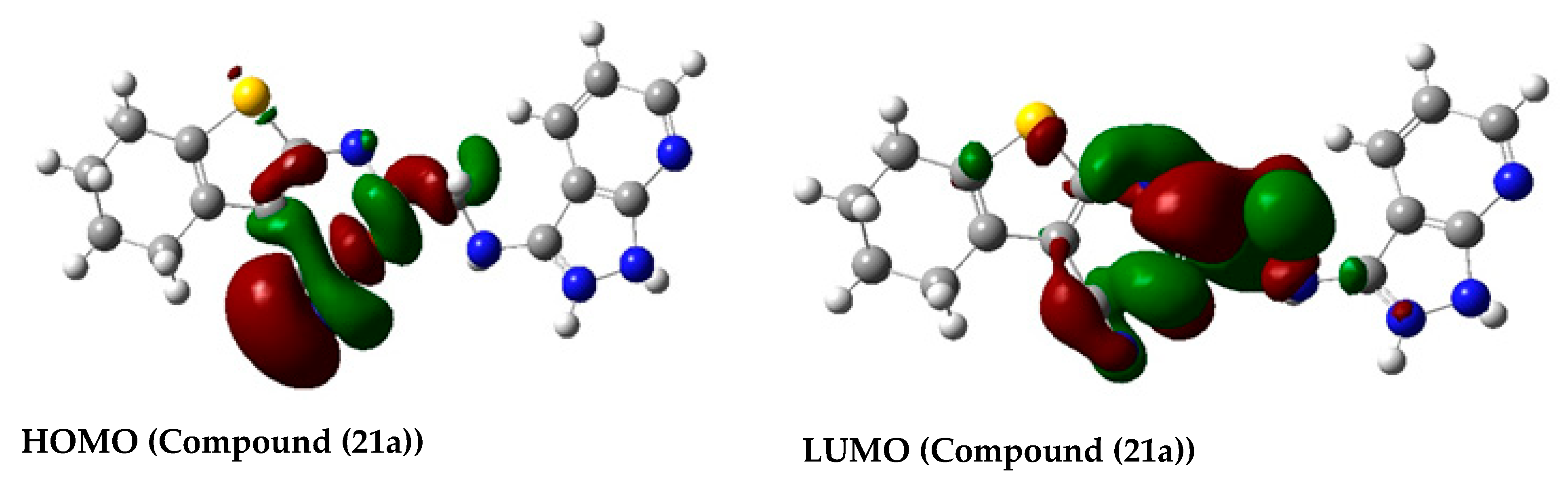
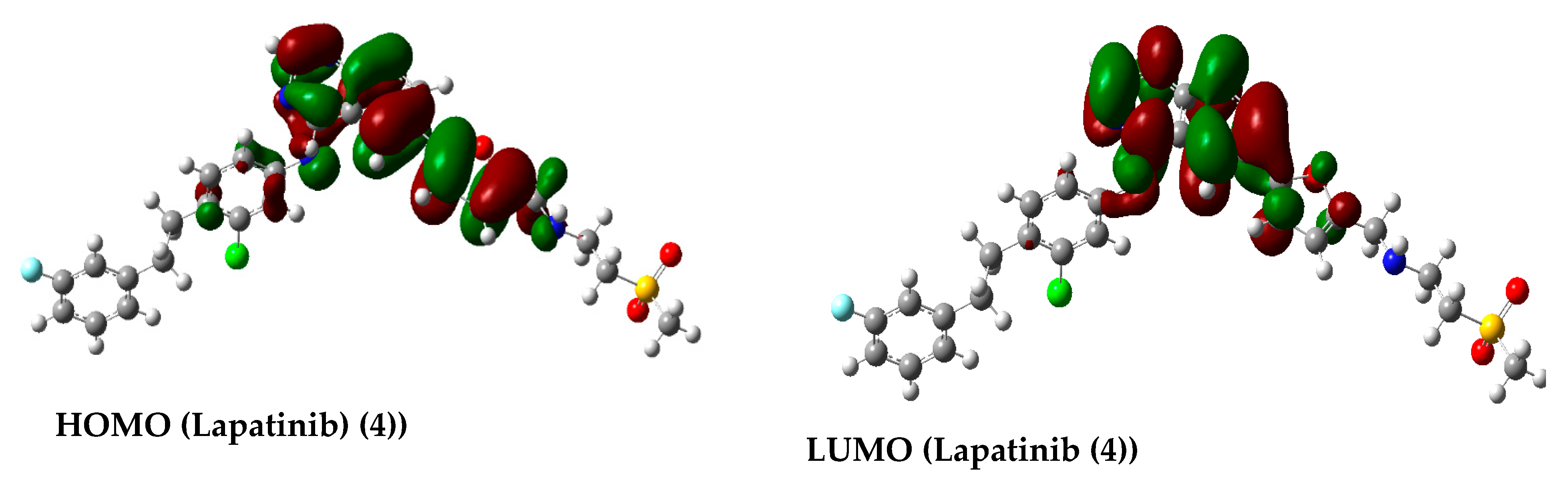
2.3.3. Molecular Orbital Energy Study and Molecular Electrostatic Potential (MESP)
3. Materials and Methods
3.1. Instrument
3.2. Chemicals and Reagents
3.3. Experimental
Chemistry
3.4. Biological Assays
3.4.1. Cytotoxic Activity against H1299
3.4.2. Measurement of Potential EGFR/HER2 Inhibitory Activity (IC50)
3.5. Molecular Modeling Study
3.5.1. Protein Preparation for Docking Study
3.5.2. Ligand Preparation
3.5.3. Molecular Docking
3.5.4. In Silico ADME Prediction
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhao, J.; Xia, Y. Targeting Her2 Alterations in Non–Small-Cell Lung Cancer: A Comprehensive Review. JCO Precis. Oncol. 2020, 4, 411–425. [Google Scholar] [CrossRef]
- Li, X.; Wu, J.; Cao, H.; Ma, R.; Wu, J.; Zhong, Y.; Feng, J. Blockade of DNA Methylation Enhances the Therapeutic Effect of Gefitinib in Non-Small Cell Lung Cancer Cells. Oncol. Rep. 2013, 29, 1975–1982. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.N.; Kim, S.; Yun, M.R.; Kim, H.R.; Lim, S.M.; Kim, M.; Hong, K.; Kim, S.; Kim, H.; Pyo, K.; et al. Er2, a Novel Human Anti-Egfr Monoclonal Antibody Inhibit Tumor Activity in Non-Small Cell Lung Cancer Models. Lung Cancer 2016, 95, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Yassen, A.S.A.; Elshihawy, H.E.A.E.A.; Said, M.M.A.; Abouzid, K.A.M. Molecular Modelling and Synthesis of Quinazoline-Based Compounds as Potential Antiproliferative Agents. Chem. Pharm. Bull. 2014, 62, 454–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charles, S.; Futreal, A.; Eisen, T. Her2-Targeted Therapies in Non–Small Cell Lung Cancer. Clin. Cancer Res. 2006, 12, 4377s–4383s. [Google Scholar]
- Jianming, Z.; Yang, P.L.; Gray, N.S. Targeting Cancer with Small Molecule Kinase Inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar]
- Elmetwally, S.A.; Saied, K.F.; Eissa, I.H.; Elkaeed, E.B. Design, Synthesis and Anticancer Evaluation of Thieno [2, 3-D] Pyrimidine Derivatives as Dual Egfr/Her2 Inhibitors and Apoptosis Inducers. Bioorgan. Chem. 2019, 88, 102944. [Google Scholar] [CrossRef]
- Emeline, L.; Barluenga, S.; Moras, D.; Wurtz, J.; Winssinger, N. Cysteine Mapping in Conformationally Distinct Kinase Nucleotide Binding Sites: Application to the Design of Selective Covalent Inhibitors. J. Med. Chem. 2011, 54, 1347–1355. [Google Scholar]
- Smith, A.J.; Zhang, X.; Leach, A.G.; Houk, K.N. Beyond Picomolar Affinities: Quantitative Aspects of Noncovalent and Covalent Binding of Drugs to Proteins. J. Med. Chem. 2009, 52, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Lanning, B.R.; Whitby, L.R.; Dix, M.M.; Douhan, J.; Gilbert, A.M.; Hett, E.C.; Johnson, T.O.; Joslyn, C.; Kath, J.C.; Niessen, S. A Road Map to Evaluate the Proteome-Wide Selectivity of Covalent Kinase Inhibitors. Nat. Chem. Biol. 2014, 10, 760–767. [Google Scholar] [CrossRef] [Green Version]
- Shuhang, W.; Cang, S.; Liu, D. Third-Generation Inhibitors Targeting Egfr T790 m Mutation in Advanced Non-Small Cell Lung Cancer. J. Hematol. Oncol. 2016, 9, 34. [Google Scholar]
- Yu, H.A.; Tian, S.K.; Drilon, A.E.; Borsu, L.; Riely, G.J.; Arcila, M.E.; Ladanyi, M. Acquired Resistance of Egfr-Mutant Lung Cancer to a T790m-Specific Egfr Inhibitor: Emergence of a Third Mutation (C797s) in the Egfr Tyrosine Kinase Domain. JAMA Oncol. 2015, 1, 982–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuo, L.; Wang, L.; Feng, M.; Yi, Y.; Zhang, W.; Liu, W.; Li, L.; Liu, Z.; Li, Y.; Ma, X. New Acrylamide-Substituted Quinazoline Derivatives with Enhanced Potency for the Treatment of Egfr T790m-Mutant Non-Small-Cell Lung Cancers. Bioorgan. Chem. 2018, 77, 593–599. [Google Scholar]
- Siyuan, Y.; Tang, C.; Wang, B.; Zhang, Y.; Zhou, L.; Xue, L.; Zhang, C. Design, Synthesis and Biological Evaluation of Novel Egfr/Her2 Dual Inhibitors Bearing a Oxazolo [4, 5-G] Quinazolin-2 (1h)-One Scaffold. Eur. J. Med. Chem. 2016, 120, 26–36. [Google Scholar]
- Mohamed, S.; Elshihawy, H. Synthesis, Anticancer Activity and Structure-Activity Relationship of Some Anticancer Agents Based on Cyclopenta (B) Thiophene Scaffold. Pak. J. Pharm. Sci. 2014, 27. [Google Scholar]
- Ranza, E.; Aziz, Y.M.A.; Elgawish, M.S.; Elewa, M.; Elshihawy, H.A.; Said, M.M. Pharmacophore Modeling, 3d-Qsar, Synthesis, and Anti-Lung Cancer Evaluation of Novel Thieno [2, 3-D] [1, 2, 3] Triazines Targeting Egfr. Arch. Pharm. 2020, 353, 1900108. [Google Scholar]
- Adesh, D.; Srivastava, S.K.; Srivastava, S.D. Conventional and Microwave Assisted Synthesis of 2-Oxo-4-Substituted Aryl-Azetidine Derivatives of Benzotriazole: A New Class of Biological Compounds. Bioorgan. Med. Chem. Lett. 2011, 21, 569–573. [Google Scholar]
- Al-Obaid, A.M.; Abdel-Hamide, S.G.; El-Kashef, H.A.; Alaa, A.M.; El-Azab, A.S.; Al-Khamees, H.A.; El-Subbagh, H.I. Substituted Quinazolines, Part 3. Synthesis, in Vitro Antitumor Activity and Molecular Modeling Study of Certain 2-Thieno-4 (3h)-Quinazolinone Analogs. Eur. J. Med. Chem. 2009, 44, 2379–2391. [Google Scholar] [CrossRef]
- Canan, K.; Ayhan-Kılcıgil, G.; Özbey, S.; Kaynak, F.B.; Kaya, M.; Çoban, T.; Can-Eke, B. Synthesis and Antioxidant Properties of Novel N-Methyl-1, 3, 4-Thiadiazol-2-Amine and 4-Methyl-2h-1, 2, 4-Triazole-3 (4h)-Thione Derivatives of Benzimidazole Class. Bioorgan. Med. Chem. 2008, 16, 4294–4303. [Google Scholar]
- Philip, S.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening. JNCI J. Nat. Cancer Inst. 1990, 28, 1107–1112. [Google Scholar]
- 1 Product Results for Neratinib. Available online: https://www.selleckchem.com/search.html?searchDTO.searchParam=Neratinib&sp=Neratinib (accessed on 20 November 2020).
- Gewald, K. Heterocyclen Aus Ch-Aciden Nitrilen, Vii. 2-Amino-Thiophene Aus A-Oxo-Mercaptanen Und Methylenaktiven Nitrilen. Chem. Berichte 1965, 98, 3571–3577. [Google Scholar] [CrossRef]
- Lou, Z.; Li, Z.; Chunfen, X.; Yong, Y.; Fanbo, Z.; Kaixun, H. Anticancer Activities of Some Arylcarbamoylalkyltriphenylphosphonium Chlorides. Med. Chem. Res. 2007, 16, 380–391. [Google Scholar]
- Kundariya, D.S.; Bheshdadia, B.M.; Joshi, N.K.; Patel, P.K. Synthesis, Characterization and Pharmacological Evaluation of Some Novel Schiff Bases Containing 1h-Pyrazolo [3, 4-B] Pyridine Moiety. Int. J. ChemTech. Res. 2011, 44, 4385–4392. [Google Scholar]
- Available online: http://www.bpsbioscience.com/biochemical-based-assays (accessed on 21 November 2020).
- Elrayess, R.; Elgawish, M.S.; Elewa, M.; Nafie, M.S.; Elhady, S.S.; Yassen, A.S.A. Synthesis, 3d-Qsar, and Molecular Modeling Studies of Triazole Bearing Compounds as a Promising Scaffold for Cyclooxygenase-2 Inhibition. Pharmaceuticals 2020, 13, 370. [Google Scholar] [CrossRef] [PubMed]
- Ghareb, N.; El-Sayed, N.M.; Abdelhameed, R.; Yamad, K.; Elgawish, M.S. Toward a treatment of diabesity: Rational design, synthesis and biological evaluation of benzene-sulfonamide derivatives as a new class of PTP-1B inhibitors. Bioorgan. Chem. 2019, 86, 322–338. [Google Scholar] [CrossRef] [PubMed]
- Elgawish, M.S.; Kishikawa, N.; Helal, M.A.; Ohyama, K.; Kuroda, N. Molecular modeling and spectroscopic study of quinone–protein adducts: Insight into toxicity, selectivity, and reversibility. Toxicol. Res. 2015, 4, 843–847. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound No. | IC50 against H1299 |
|---|---|
| 13a | 28.79 nM |
| 14a | 25.68 nM |
| 15a | 32.81 nM |
| 16a | 34.91 nM |
| 17a | 54.80 nM |
| 13b | 25 nM |
| 15b | 34.00 nM |
| 17b | 38.13 nM |
| 21a | 12.50 nM |
| 21b | 13.68 nM |
| 23 | 18.41 nM |
| 25a | 18.53 nM |
| 25b | 18.47 nM |
| Gefitinib (2) | 40 µM |
| Compound No. | IC50 against EGFR (nM) | IC50 against HER2 (nM) |
|---|---|---|
| (21a) | 0.47 | 0.14 |
| Gefitinib (2) | 1.9 | -- |
| Imatinib (1) | 0.11 | 0.06 |
| Neratinib (6) | 92 [21] | 59 [21] |
| ADME Prediction Parameters | Compound (15a) | Compound (21a) | Lapatinib (4) |
|---|---|---|---|
| mol MW a | 371.4 | 352.1 | 581.06 |
| Donor-HB b | 1 | 2.25 | 1 |
| Accept-HB c | 5 | 5.7 | 8.25 |
| QPlogPo/w d | 4.3 | 2.4 | 6.3 |
| PSA e | 62.2 | 116.8 | 95.35 |
| QPlogS f | –5.6 | –6.2 | –8.2 |
| QPPCaco g | 1143.7 | 96.8 | 187.5 |
| QPlogBB h | –0.6 | –1.9 | 0.8 |
| QPPMDCK i | 1080 | 59.5 | 376.5 |
| QPlogKhsa j | 0.55 | 0.26 | 1.19 |
| #rotor k | 5 | 5 | 10 |
| % Human Oral Absorption l | 100 | 76.9 | 78.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elrayess, R.; Abdel Aziz, Y.M.; Elgawish, M.S.; Elewa, M.; Yassen, A.S.A.; Elhady, S.S.; Elshihawy, H.A.; Said, M.M. Discovery of Potent Dual EGFR/HER2 Inhibitors Based on Thiophene Scaffold Targeting H1299 Lung Cancer Cell Line. Pharmaceuticals 2021, 14, 9. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14010009
Elrayess R, Abdel Aziz YM, Elgawish MS, Elewa M, Yassen ASA, Elhady SS, Elshihawy HA, Said MM. Discovery of Potent Dual EGFR/HER2 Inhibitors Based on Thiophene Scaffold Targeting H1299 Lung Cancer Cell Line. Pharmaceuticals. 2021; 14(1):9. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14010009
Chicago/Turabian StyleElrayess, Ranza, Yasmine M. Abdel Aziz, Mohamed Saleh Elgawish, Marwa Elewa, Asmaa S. A. Yassen, Sameh S. Elhady, Hosam A. Elshihawy, and Mohamed M. Said. 2021. "Discovery of Potent Dual EGFR/HER2 Inhibitors Based on Thiophene Scaffold Targeting H1299 Lung Cancer Cell Line" Pharmaceuticals 14, no. 1: 9. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14010009