
Discovery and Characterization of a Novel MASTL Inhibitor MKI-2 Targeting MASTL-PP2A in Breast Cancer Cells and Oocytes

 , and
, and 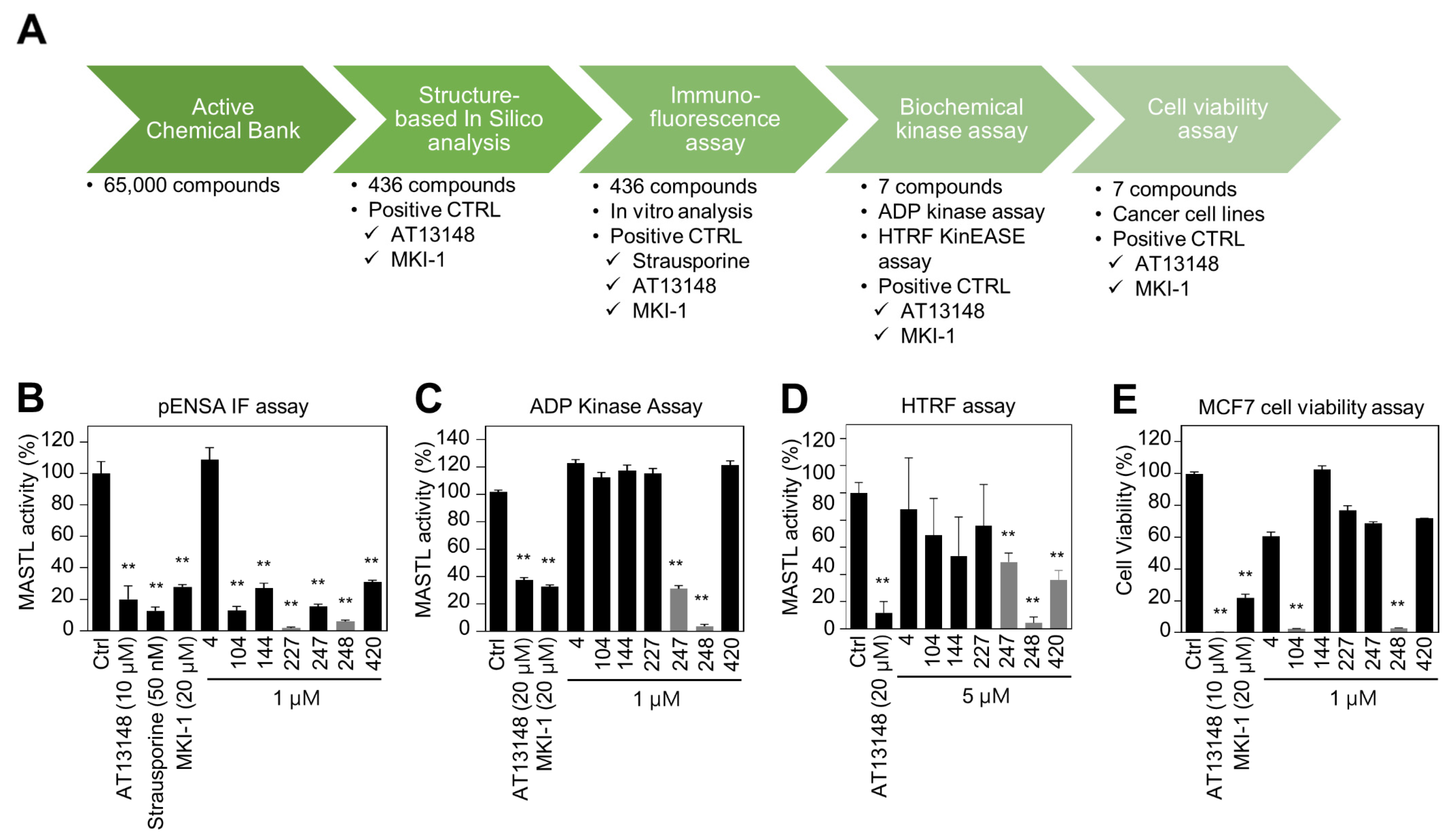{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
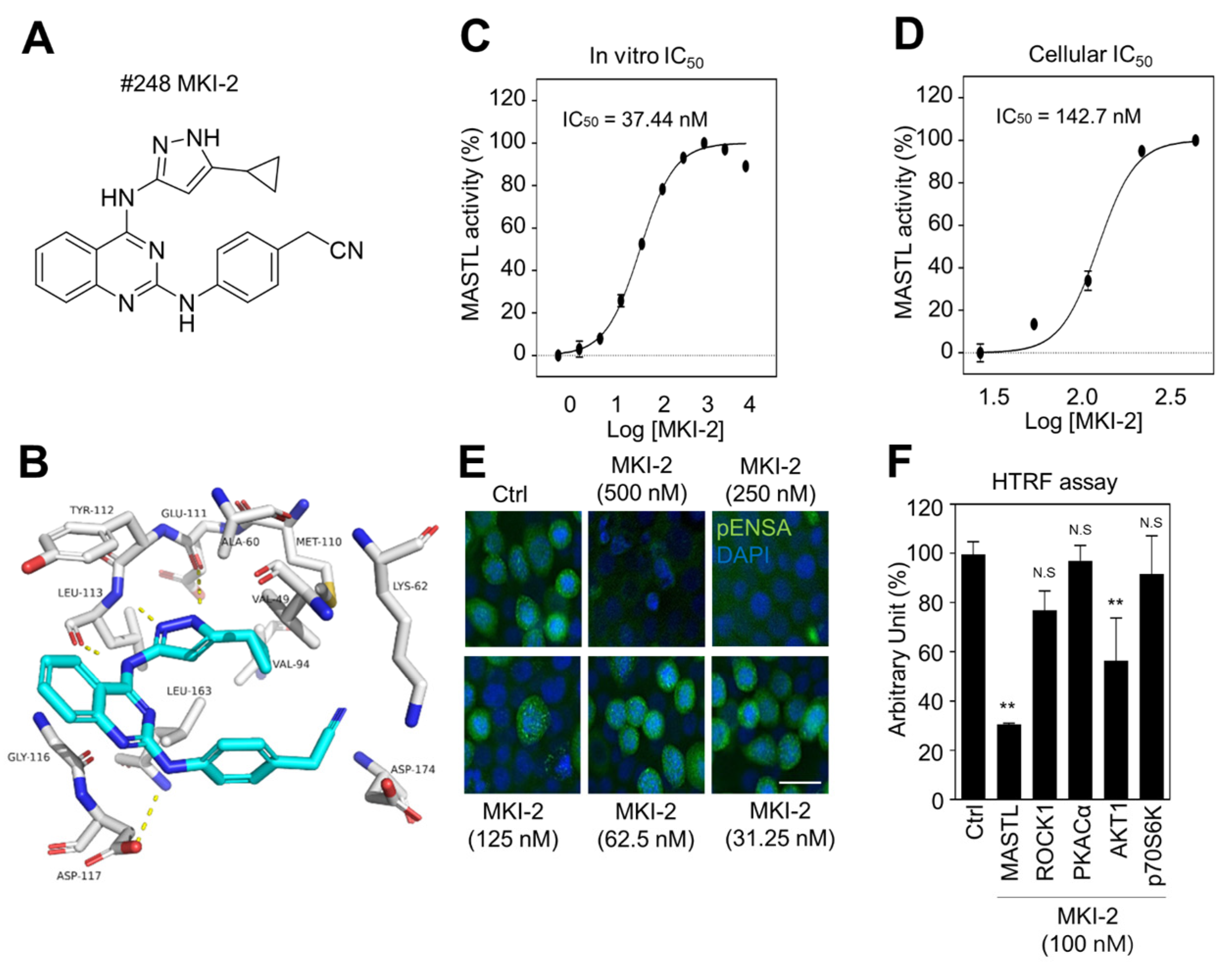
2.1. Identification and Validation of MASTL Inhibitor MKI-2 In Silico and In Vitro
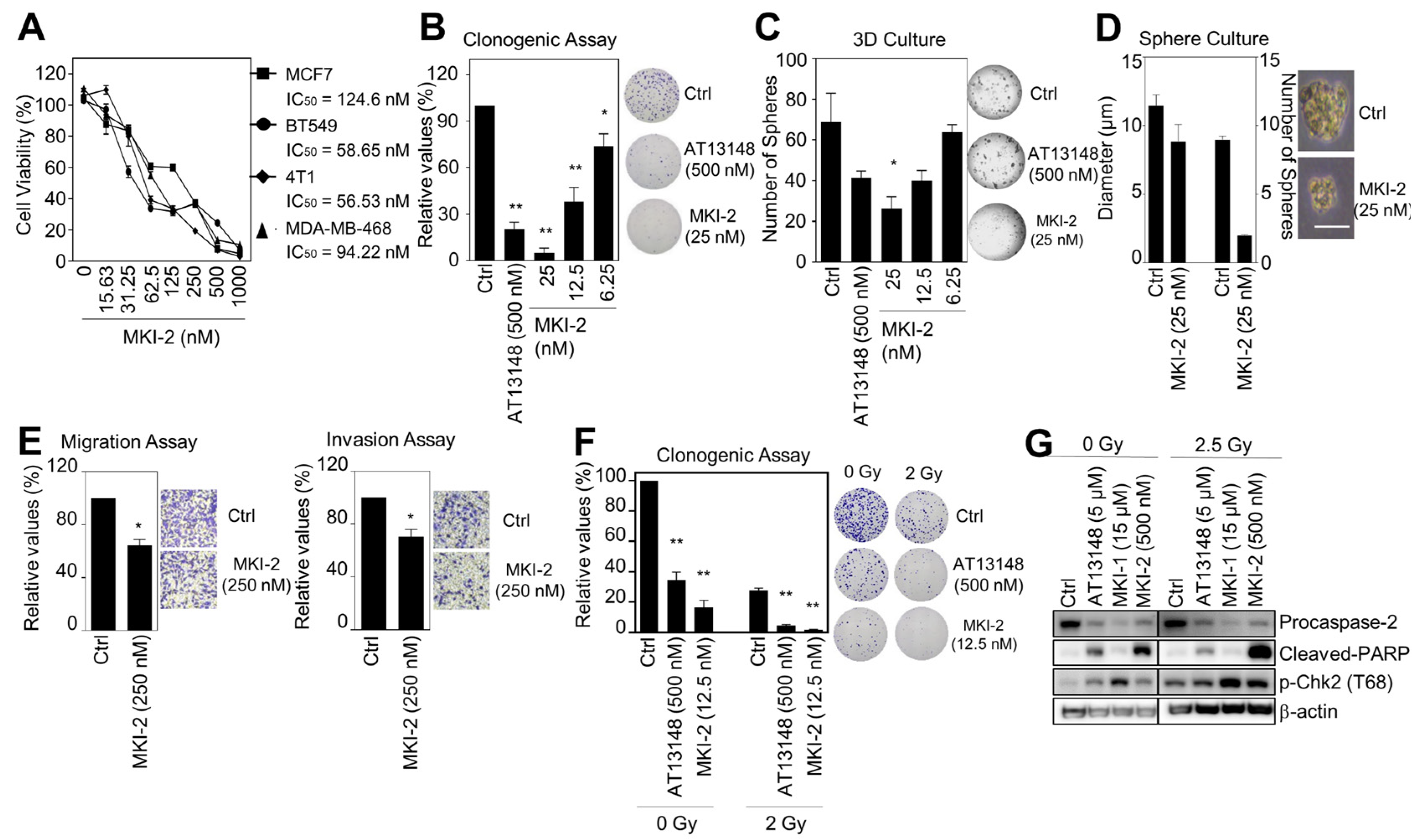
2.2. MKI-2 Induces Mitotic Catastrophe of Breast Cancer Cells via MASTL-PP2A
2.3. MKI-2 Inhibits the Oncogenic Properties and Enhances the Radiosensitivity of Breast Cancer Cells
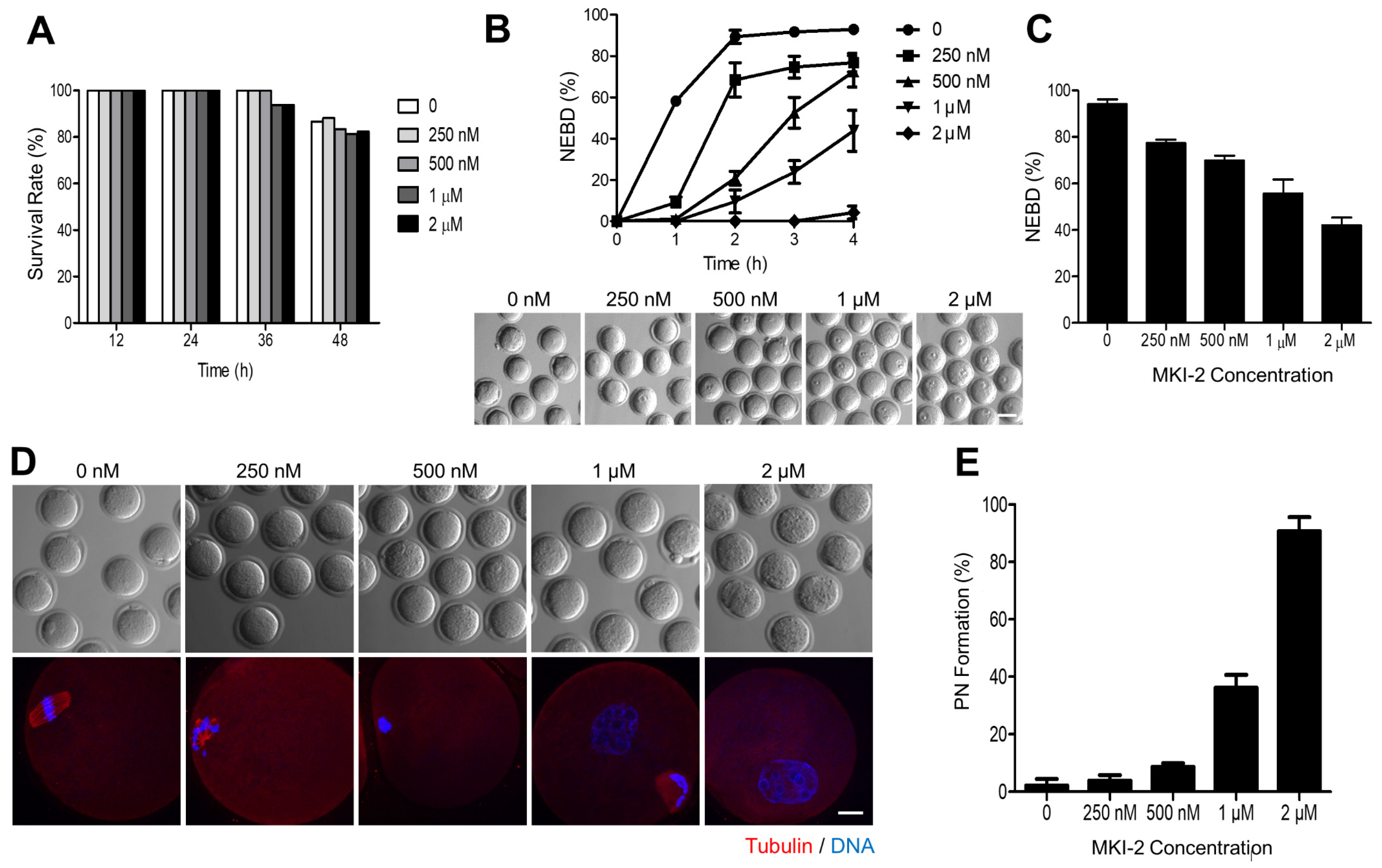
2.4. MKI-2 Treatment Phenocopies the MASTL-Null Oocytes
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. In Silico Screening
4.3. Chemicals and Treatments
4.4. Synthesis of MKI-2
4.4.1. 2-Chloro-N-(5-cyclopropyl-1H-pyrazol-3-yl)quinazolin-4-amine (compound 3)
4.4.2. 2-(4-((4-((5-Cyclopropyl-1H-pyrazol-3-yl)amino)quinazolin-2-yl)amino)phenyl)acetonitrile (MKI-2)
4.5. Immunofluorescence
4.6. HTRF Assay
4.7. In Vitro Kinase Assay
4.8. Cell Viability Assays
4.9. RNA Interference
4.10. Clonogenic and Sphere Formation Assay
4.11. Three-Dimensional Culture
4.12. Invasion and Migration Assay
4.13. Western Blotting Analysis
4.14. PP2A Activity Assay
4.15. GSEA Analysis
4.16. Oocyte Culture and Treatment
4.17. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dominguez-Brauer, C.; Thu, K.L.; Mason, J.M.; Blaser, H.; Bray, M.R.; Mak, T.W. Targeting mitosis in cancer: Emerging strategies. Mol. Cell 2015, 60, 524–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Fleming, S.L.; Williams, B.; Williams, E.V.; Li, Z.; Somma, P.; Rieder, C.L.; Goldberg, M.L. Greatwall kinase: A nuclear protein required for proper chromosome condensation and mitotic progression in Drosophila. J. Cell Biol. 2004, 164, 487–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisteau, X.; Lee, J.; Srinivas, V.; Lee, J.H.S.; Niska-Blakie, J.; Tan, G.; Yap, S.Y.X.; Hom, K.W.; Wong, C.K.; Chae, J.; et al. The Greatwall kinase safeguards the genome integrity by affecting the kinome activity in mitosis. Oncogene 2020, 39, 6816–6840. [Google Scholar] [CrossRef]
- Vigneron, S.; Brioudes, E.; Burgess, A.; Labbe, J.C.; Lorca, T.; Castro, A. Greatwall maintains mitosis through regulation of PP2A. EMBO J. 2009, 28, 2786–2793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, A.; Vigneron, S.; Brioudes, E.; Labbe, J.C.; Lorca, T.; Castro, A. Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proc. Natl. Acad. Sci. USA 2010, 107, 12564–12569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gharbi-Ayachi, A.; Labbe, J.C.; Burgess, A.; Vigneron, S.; Strub, J.M.; Brioudes, E.; Van-Dorsselaer, A.; Castro, A.; Lorca, T. The substrate of Greatwall kinase, Arpp19, controls mitosis by inhibiting protein phosphatase 2A. Science 2010, 330, 1673–1677. [Google Scholar] [CrossRef]
- Mochida, S.; Maslen, S.L.; Skehel, M.; Hunt, T. Greatwall phosphorylates an inhibitor of protein phosphatase 2A that is essential for mitosis. Science 2010, 330, 1670–1673. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Kang, H.; Xu, Y.N.; Heo, Y.T.; Cui, X.S.; Kim, N.H.; Oh, J.S. Greatwall kinase is required for meiotic maturation in porcine oocytes. Biol. Reprod. 2013, 89, 53. [Google Scholar] [CrossRef]
- Yamamoto, T.M.; Blake-Hodek, K.; Williams, B.C.; Lewellyn, A.L.; Goldberg, M.L.; Maller, J.L. Regulation of Greatwall kinase during Xenopus oocyte maturation. Mol. Biol. Cell 2011, 22, 2157–2164. [Google Scholar] [CrossRef]
- Wlodarchak, N.; Xing, Y. PP2A as a master regulator of the cell cycle. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 162–184. [Google Scholar] [CrossRef]
- Alvarez-Fernandez, M.; Sanz-Flores, M.; Sanz-Castillo, B.; Salazar-Roa, M.; Partida, D.; Zapatero-Solana, E.; Ali, H.R.; Manchado, E.; Lowe, S.; VanArsdale, T.; et al. Therapeutic relevance of the PP2A-B55 inhibitory kinase MASTL/Greatwall in breast cancer. Cell Death Differ. 2017, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Yoon, Y.N.; Choe, M.H.; Jung, K.Y.; Hwang, S.G.; Oh, J.S.; Kim, J.S. MASTL inhibition promotes mitotic catastrophe through PP2A activation to inhibit cancer growth and radioresistance in breast cancer cells. BMC Cancer 2018, 18, 716. [Google Scholar] [CrossRef]
- Marzec, K.; Burgess, A. The oncogenic functions of MASTL Kinase. Front. Cell Dev. Biol. 2018, 6, 162. [Google Scholar] [CrossRef] [PubMed]
- Vera, J.; Lartigue, L.; Vigneron, S.; Gadea, G.; Gire, V.; Del Rio, M.; Soubeyran, I.; Chibon, F.; Lorca, T.; Castro, A. Greatwall promotes cell transformation by hyperactivating AKT in human malignancies. eLife 2015, 4. [Google Scholar] [CrossRef]
- Rogers, S.; McCloy, R.A.; Parker, B.L.; Gallego-Ortega, D.; Law, A.M.K.; Chin, V.T.; Conway, J.R.W.; Fey, D.; Millar, E.K.A.; O’Toole, S.; et al. MASTL overexpression promotes chromosome instability and metastasis in breast cancer. Oncogene 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatima, I.; Singh, A.B.; Dhawan, P. MASTL: A novel therapeutic target for Cancer Malignancy. Cancer Med. 2020, 9, 6322–6329. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Luong, V.Q.; Giannini, P.J.; Peng, A. MASTL kinase, a promising therapeutic target, promotes cancer recurrence. Oncotarget 2014, 5, 11479–11489. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.J.; Li, Y.L.; Wang, L.G.; Liu, L.Q.; Ma, H.; Hou, W.H.; Yu, J.M. Mastl overexpression is associated with epithelial to mesenchymal transition and predicts a poor clinical outcome in gastric cancer. Oncol. Lett. 2017, 14, 7283–7287. [Google Scholar] [CrossRef]
- Cetti, E.; Di Marco, T.; Mauro, G.; Mazzoni, M.; Lecis, D.; Minna, E.; Gioiosa, L.; Brich, S.; Pagliardini, S.; Borrello, M.G.; et al. Mitosis perturbation by MASTL depletion impairs the viability of thyroid tumor cells. Cancer Lett. 2019, 442, 362–372. [Google Scholar] [CrossRef]
- Uppada, S.B.; Gowrikumar, S.; Ahmad, R.; Kumar, B.; Szeglin, B.; Chen, X.; Smith, J.J.; Batra, S.K.; Singh, A.B.; Dhawan, P. MASTL induces Colon Cancer progression and Chemoresistance by promoting Wnt/beta-catenin signaling. Mol. Cancer 2018, 17, 111. [Google Scholar] [CrossRef] [Green Version]
- Anania, M.; Gasparri, F.; Cetti, E.; Fraietta, I.; Todoerti, K.; Miranda, C.; Mazzoni, M.; Re, C.; Colombo, R.; Ukmar, G.; et al. Identification of thyroid tumor cell vulnerabilities through a siRNA-based functional screening. Oncotarget 2015, 6, 34629–34648. [Google Scholar] [CrossRef]
- Kim, A.Y.; Yoon, Y.N.; Leem, J.; Lee, J.Y.; Jung, K.Y.; Kang, M.; Ahn, J.; Hwang, S.G.; Oh, J.S.; Kim, J.S. MKI-1, a Novel small-molecule inhibitor of MASTL, exerts antitumor and radiosensitizer activities through PP2A activation in breast cancer. Front. Oncol. 2020, 10, 571601. [Google Scholar] [CrossRef] [PubMed]
- Ocasio, C.A.; Rajasekaran, M.B.; Walker, S.; Le Grand, D.; Spencer, J.; Pearl, F.M.; Ward, S.E.; Savic, V.; Pearl, L.H.; Hochegger, H.; et al. A first generation inhibitor of human Greatwall kinase, enabled by structural and functional characterisation of a minimal kinase domain construct. Oncotarget 2016, 7, 71182–71197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ammarah, U.; Kumar, A.; Pal, R.; Bal, N.C.; Misra, G. Identification of new inhibitors against human Great wall kinase using in silico approaches. Sci. Rep. 2018, 8, 4894. [Google Scholar] [CrossRef]
- Yap, T.A.; Walton, M.I.; Grimshaw, K.M.; Te Poele, R.H.; Eve, P.D.; Valenti, M.R.; de Haven Brandon, A.K.; Martins, V.; Zetterlund, A.; Heaton, S.P.; et al. AT13148 is a novel, oral multi-AGC kinase inhibitor with potent pharmacodynamic and antitumor activity. Clin. Cancer Res. 2012, 18, 3912–3923. [Google Scholar] [CrossRef] [Green Version]
- Pearce, L.R.; Komander, D.; Alessi, D.R. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 2010, 11, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yu, D.; Feng, C.; Deng, X.; Wu, D.; Jin, M.; Wang, E.; Wang, X.; Yu, B. Role of Greatwall kinase in release of mouse oocytes from diplotene arrest. Dev. Growth Differ. 2014, 56, 669–678. [Google Scholar] [CrossRef]
- Adhikari, D.; Diril, M.K.; Busayavalasa, K.; Risal, S.; Nakagawa, S.; Lindkvist, R.; Shen, Y.; Coppola, V.; Tessarollo, L.; Kudo, N.R.; et al. Mastl is required for timely activation of APC/C in meiosis I and Cdk1 reactivation in meiosis II. J. Cell Biol. 2014, 206, 843–853. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Fernandez, M.; Sanchez-Martinez, R.; Sanz-Castillo, B.; Gan, P.P.; Sanz-Flores, M.; Trakala, M.; Ruiz-Torres, M.; Lorca, T.; Castro, A.; Malumbres, M. Greatwall is essential to prevent mitotic collapse after nuclear envelope breakdown in mammals. Proc. Natl. Acad. Sci. USA 2013, 110, 17374–17379. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.O.; Choe, M.H.; Yoon, Y.N.; Ahn, J.; Yoo, M.; Jung, K.Y.; An, S.; Hwang, S.G.; Oh, J.S.; Kim, J.S. Antihelminthic drug niclosamide inhibits CIP2A and reactivates tumor suppressor protein phosphatase 2A in non-small cell lung cancer cells. Biochem Pharmacol 2017, 144, 78–89. [Google Scholar] [CrossRef]
- Kim, J.S.; Kim, E.J.; Oh, J.S.; Park, I.C.; Hwang, S.G. CIP2A modulates cell-cycle progression in human cancer cells by regulating the stability and activity of Plk1. Cancer Res. 2013, 73, 6667–6678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.S.; Chang, J.W.; Yun, H.S.; Yang, K.M.; Hong, E.H.; Kim, D.H.; Um, H.D.; Lee, K.H.; Lee, S.J.; Hwang, S.G. Chloride intracellular channel 1 identified using proteomic analysis plays an important role in the radiosensitivity of HEp-2 cells via reactive oxygen species production. Proteomics 2010, 10, 2589–2604. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.G.; Yoon, Y.N.; Choi, H.S.; Kim, J.; Seol, H.; Lee, J.K.; Seong, M.; Park, I.C.; Kim, K.I.; Kim, H.; et al. Breast cancer stem cells in HER2-negative breast cancer cells contribute to HER2-mediated radioresistance and molecular subtype conversion: Clinical implications for serum HER2 in recurrent HER2-negative breast cancer. Oncotarget 2018, 9, 5628–5639. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, M.; Kim, C.; Leem, J.; Kim, Y.-h.; Kwon, Y.-j.; Yoon, Y.N.; Chae, C.H.; Ahn, J.; Jung, K.-Y.; Oh, J.S.; et al. Discovery and Characterization of a Novel MASTL Inhibitor MKI-2 Targeting MASTL-PP2A in Breast Cancer Cells and Oocytes. Pharmaceuticals 2021, 14, 647. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14070647
Kang M, Kim C, Leem J, Kim Y-h, Kwon Y-j, Yoon YN, Chae CH, Ahn J, Jung K-Y, Oh JS, et al. Discovery and Characterization of a Novel MASTL Inhibitor MKI-2 Targeting MASTL-PP2A in Breast Cancer Cells and Oocytes. Pharmaceuticals. 2021; 14(7):647. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14070647
Chicago/Turabian StyleKang, Minsung, Chijung Kim, Jiyeon Leem, Ye-hyun Kim, Young-ju Kwon, Yi Na Yoon, Chong Hak Chae, Jiyeon Ahn, Kwan-Young Jung, Jeong Su Oh, and et al. 2021. "Discovery and Characterization of a Novel MASTL Inhibitor MKI-2 Targeting MASTL-PP2A in Breast Cancer Cells and Oocytes" Pharmaceuticals 14, no. 7: 647. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14070647