
Novel Therapeutic Targets for the Treatment of Right Ventricular Remodeling: Insights from the Pulmonary Artery Banding Model
 ,
,  and
and 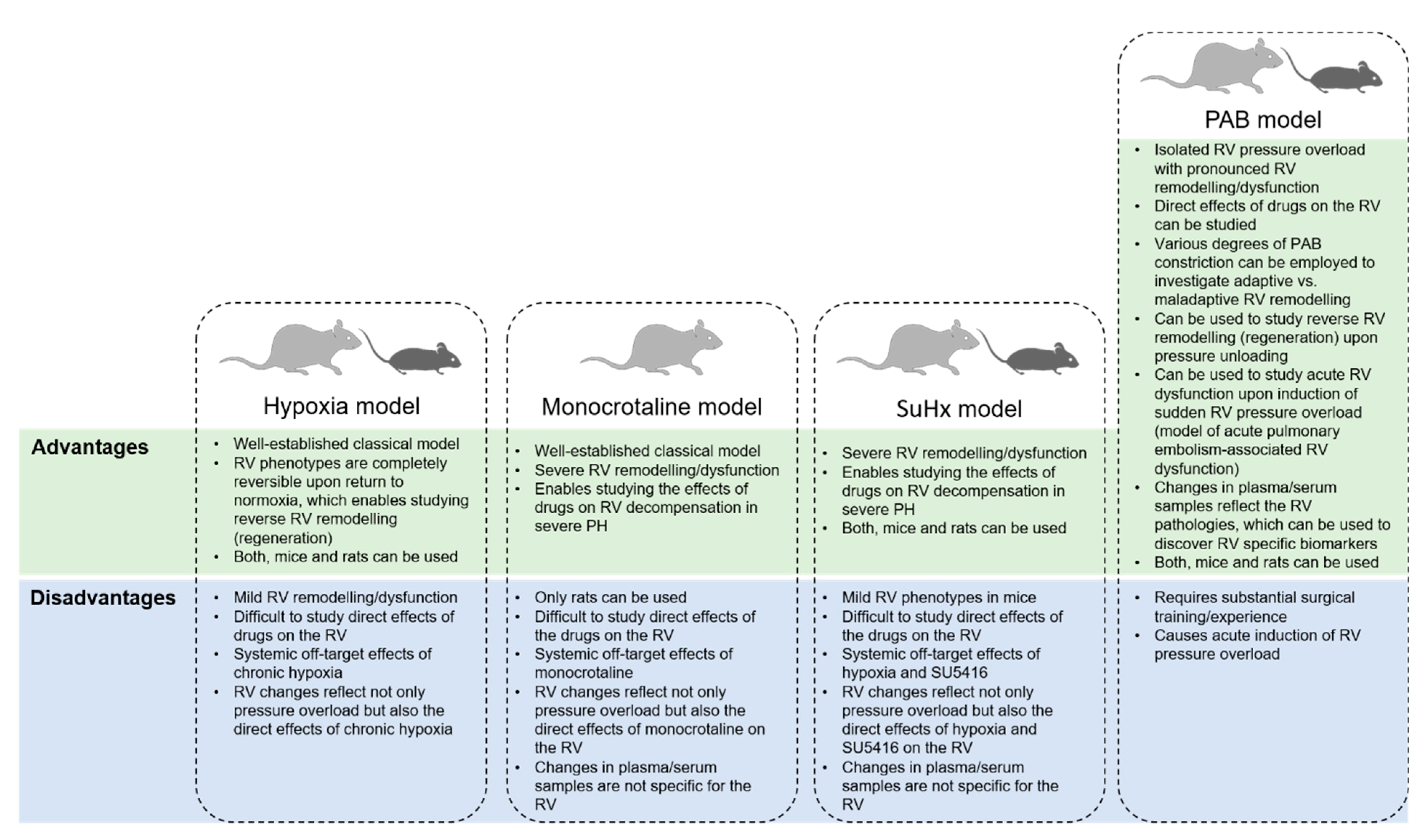{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
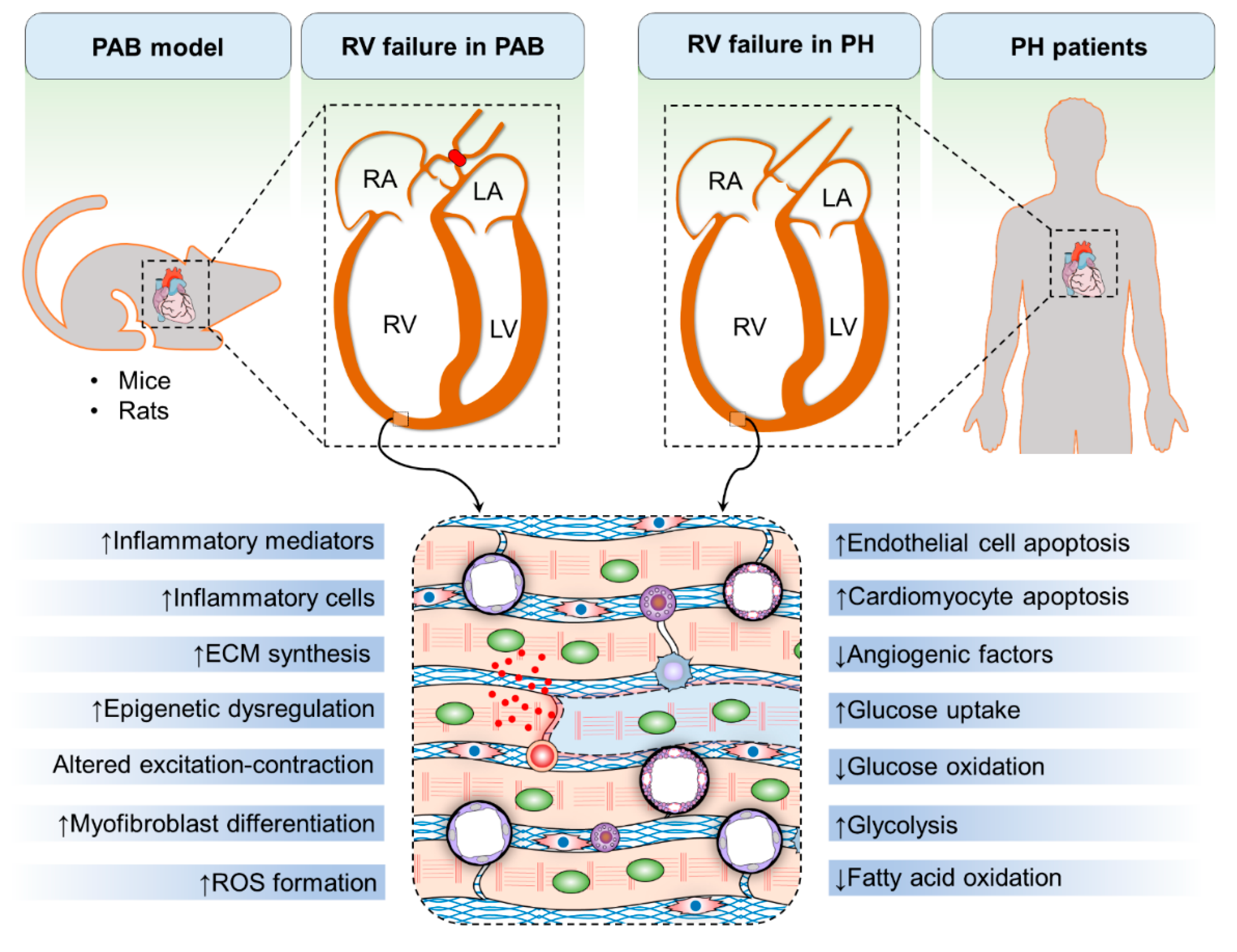
2. Pulmonary Artery Banding as a Model in Right Ventricular-Directed Drug Discovery
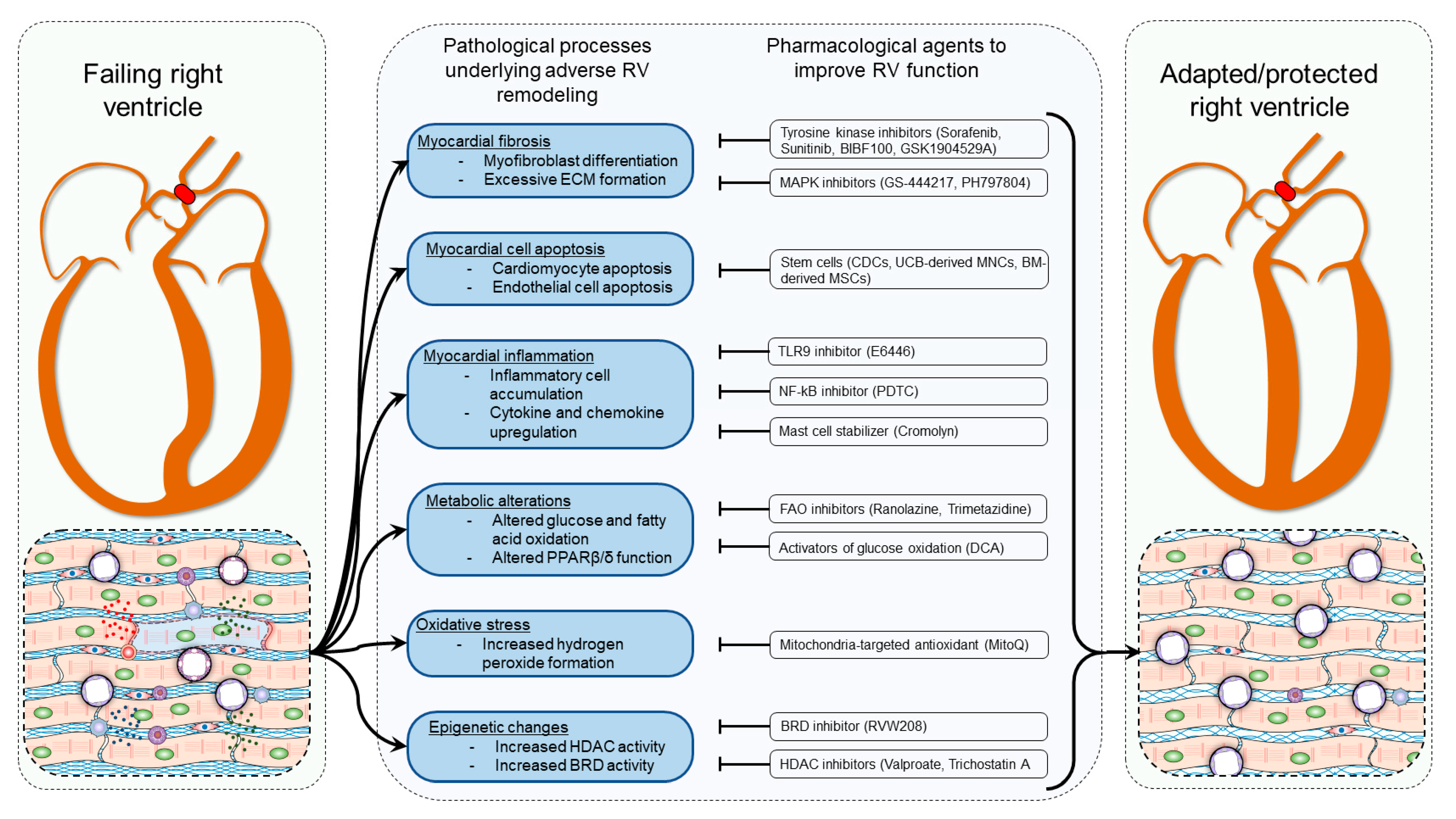
3. Mechanisms of Right Ventricular Hypertrophy and Failure
4. Pharmacotherapies Modulating Myocardial Fibrosis
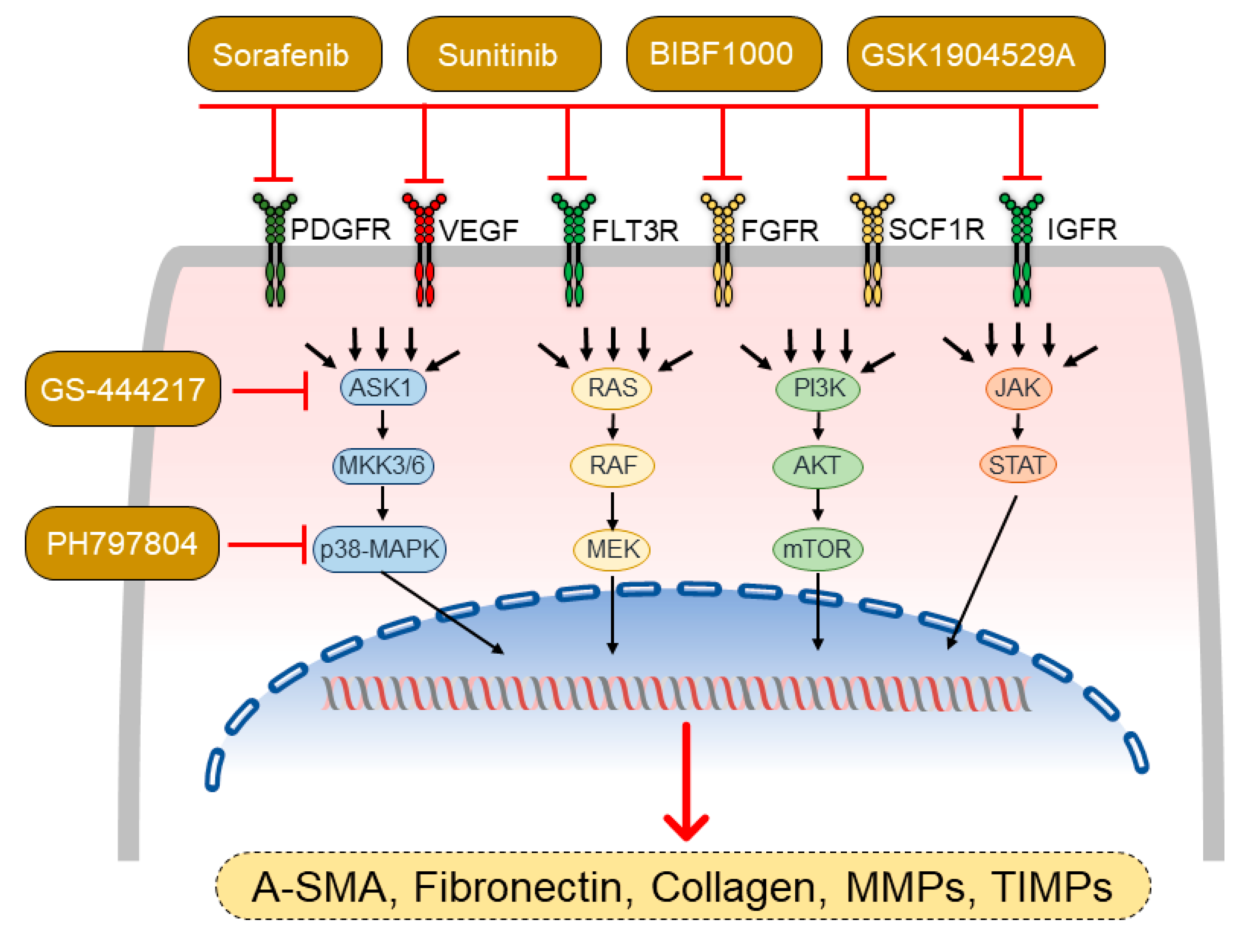
4.1. Modulators of Mitogen-Activated Protein Kinases
4.2. Tyrosine Kinase Inhibitors
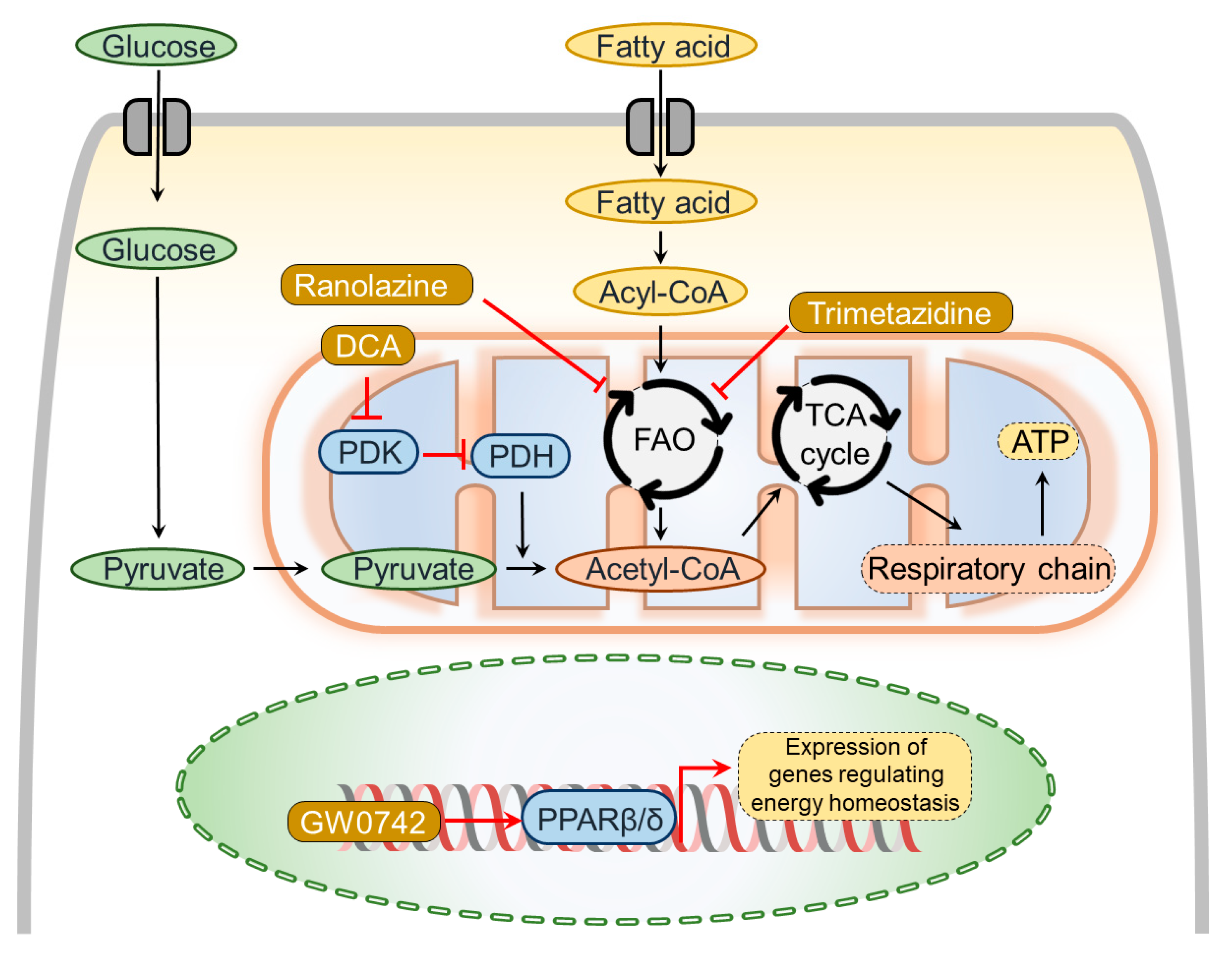
5. Pharmacotherapies Modulating Metabolic Dysregulation
5.1. Peroxisome Proliferator-Activated Receptor Agonists
5.2. Agents Modifying Fatty Acid Metabolism
5.3. Agents Restoring Oxidative Phosphorylation
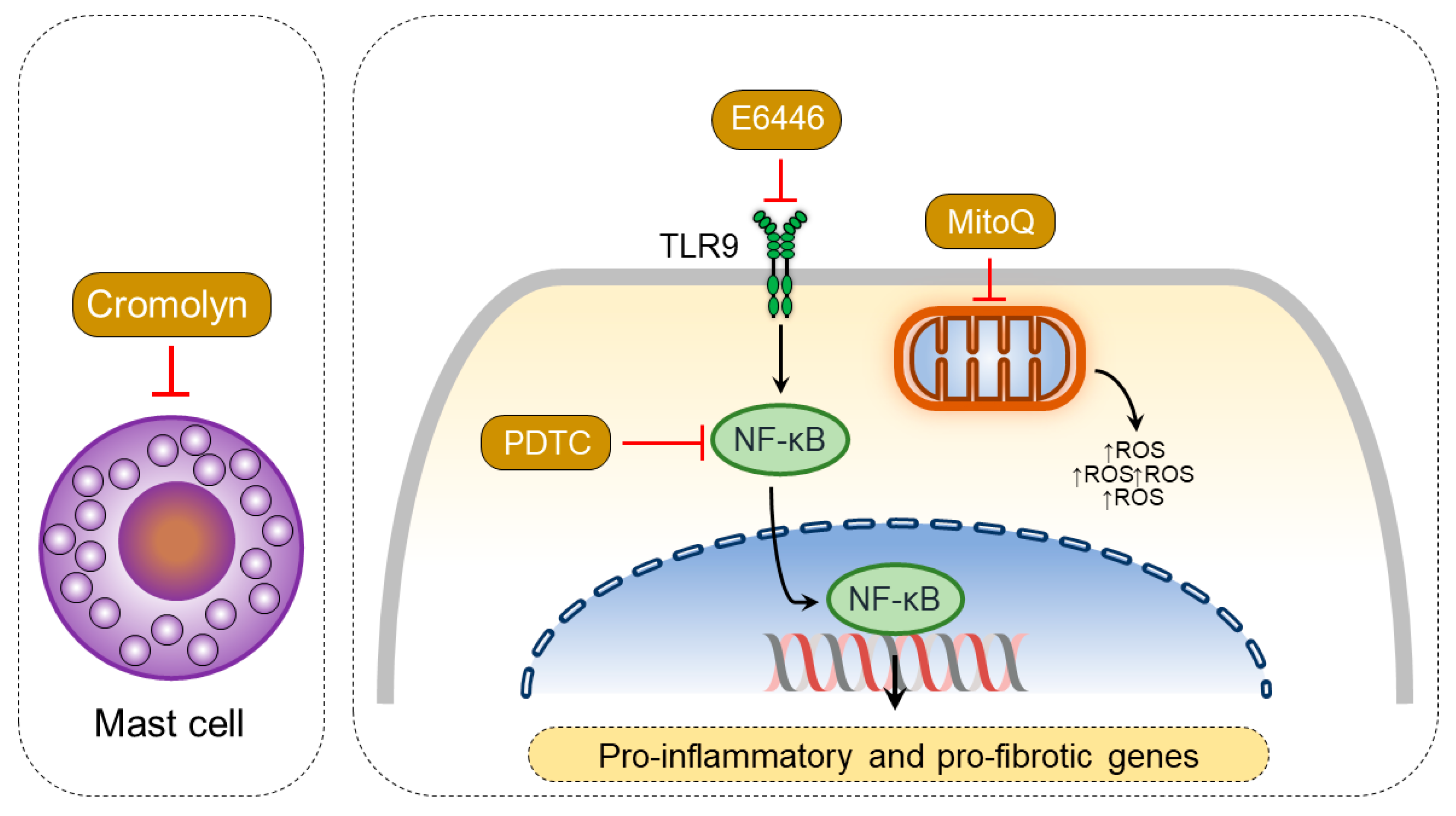
6. Pharmacotherapies Modulating Myocardial Inflammation and ROS
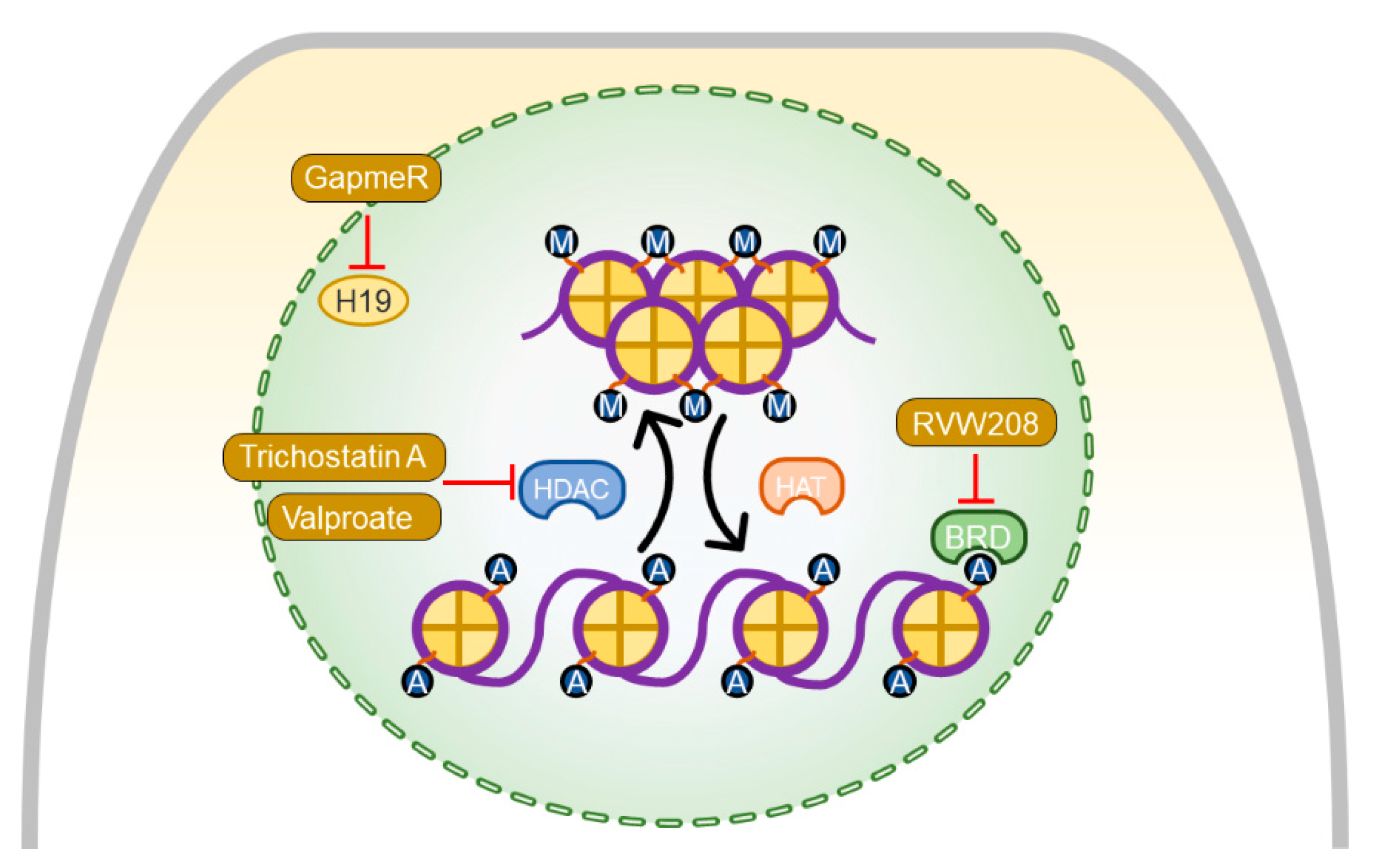
7. Pharmacotherapies Modulating Myocardial Epigenetics
8. Pharmacotherapies Modulating Myocardial Regenerative Mechanisms
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Vonk-Noordegraaf, A.; Haddad, F.; Chin, K.M.; Forfia, P.R.; Kawut, S.M.; Lumens, J.; Naeije, R.; Newman, J.; Oudiz, R.J.; Provencher, S. Right heart adaptation to pulmonary arterial hypertension: Physiology and pathobiology. J. Am. Coll. Cardiol. 2013, 62, D22–D33. [Google Scholar] [CrossRef] [PubMed]
- Sommer, N.; Ghofrani, H.A.; Pak, O.; Bonnet, S.; Provencher, S.; Sitbon, O.; Rosenkranz, S.; Hoeper, M.M.; Kiely, D.G. Current and future treatments of pulmonary arterial hypertension. Br. J. Pharm. 2021, 178, 6–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Bristow, M.R.; Zisman, L.S.; Lowes, B.D.; Abraham, W.T.; Badesch, D.B.; Groves, B.M.; Voelkel, N.F.; Lynch, D.M.; Quaife, R.A. The pressure-overloaded right ventricle in pulmonary hypertension. Chest 1998, 114, 101S–106S. [Google Scholar] [CrossRef] [PubMed]
- Voelkel, N.F.; Quaife, R.A.; Leinwand, L.A.; Barst, R.J.; McGoon, M.D.; Meldrum, D.R.; Dupuis, J.; Long, C.S.; Rubin, L.J.; Smart, F.W. Right ventricular function and failure: Report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation 2006, 114, 1883–1891. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.; Waggoner, A.D.; Dávila-román, V.G.; Barzilai, B.; Trulock, E.P.; Eisenberg, P.R. Echocardiographic characterization of the improvement in right ventricular function in patients with severe pulmonary hypertension after single-lung transplantation. J. Am. Coll. Cardiol. 1993, 22, 1170–1174. [Google Scholar] [CrossRef] [Green Version]
- Selim, A.M.; Wadhwani, L.; Burdorf, A.; Raichlin, E.; Lowes, B.; Zolty, R. Left Ventricular Assist Devices in Pulmonary Hypertension Group 2 With Significantly Elevated Pulmonary Vascular Resistance: A Bridge to Cure. Heart Lung Circ. 2019, 28, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, S.W.; Kapelanski, D.P.; Sakakibara, N.; Manecke, G.R.; Thistlethwaite, P.A.; Kerr, K.M.; Channick, R.N.; Fedullo, P.F.; Auger, W.R. Pulmonary endarterectomy: Experience and lessons learned in 1500 cases. Ann. Thorac. Surg. 2003, 76, 1457–1464. [Google Scholar] [CrossRef]
- Kusunose, K.; Tsutsui, R.S.; Bhatt, K.; Budev, M.M.; Popović, Z.B.; Griffin, B.P.; Bolen, M.A. Prognostic value of RV function before and after lung transplantation. JACC Cardiovasc. Imaging 2014, 7, 1084–1094. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.Y.; Lee, S.Y.; Phan, K.; Celermajer, D.S.; Lal, S. Renin–angiotensin–aldosterone inhibition improves right ventricular function: A meta-analysis. Heart Asia 2018, 10, e010999. [Google Scholar] [CrossRef] [Green Version]
- Maron, B.A.; Leopold, J.A. The role of the renin-angiotensin-aldosterone system in the pathobiology of pulmonary arterial hypertension (2013 Grover Conference series). Pulm. Circ. 2014, 4, 200–210. [Google Scholar] [CrossRef] [Green Version]
- Perros, F.; de Man, F.S.; Bogaard, H.J.; Antigny, F.; Simonneau, G.; Bonnet, S.; Provencher, S.; Galie, N.; Humbert, M. Use of beta-Blockers in Pulmonary Hypertension. Circ. Heart Fail. 2017, 10, e003703. [Google Scholar] [CrossRef]
- Galie, N.; Manes, A.; Negro, L.; Palazzini, M.; Bacchi-Reggiani, M.L.; Branzi, A. A meta-analysis of randomized controlled trials in pulmonary arterial hypertension. Eur. Heart J. 2009, 30, 394–403. [Google Scholar] [CrossRef] [Green Version]
- Handoko, M.; De Man, F.; Allaart, C.; Paulus, W.; Westerhof, N.; Vonk-Noordegraaf, A. Perspectives on novel therapeutic strategies for right heart failure in pulmonary arterial hypertension: Lessons from the left heart. Eur. Respir. Rev. 2010, 19, 72–82. [Google Scholar] [CrossRef] [Green Version]
- Galiè, N.; Barberà, J.A.; Frost, A.E.; Ghofrani, H.-A.; Hoeper, M.M.; McLaughlin, V.V.; Peacock, A.J.; Simonneau, G.; Vachiery, J.-L.; Grünig, E. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N. Engl. J. Med. 2015, 373, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Sitbon, O.; Jaïs, X.; Savale, L.; Cottin, V.; Bergot, E.; Macari, E.A.; Bouvaist, H.; Dauphin, C.; Picard, F.; Bulifon, S. Upfront triple combination therapy in pulmonary arterial hypertension: A pilot study. Eur. Respir. J. 2014, 43, 1691–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Alto, M.; Badagliacca, R.; Argiento, P.; Romeo, E.; Farro, A.; Papa, S.; Sarubbi, B.; Russo, M.G.; Vizza, C.D.; Golino, P. Risk reduction and right heart reverse remodeling by upfront triple combination therapy in pulmonary arterial hypertension. Chest 2020, 157, 376–383. [Google Scholar] [CrossRef]
- Noordegraaf, A.V.; Bogaard, H.J. Restoring the Right Ventricle. Chest 2020, 157, 251–252. [Google Scholar] [CrossRef] [PubMed]
- Andersen, S.; Schultz, J.G.; Holmboe, S.; Axelsen, J.B.; Hansen, M.S.; Lyhne, M.D.; Nielsen-Kudsk, J.E.; Andersen, A. A Pulmonary Trunk Banding Model of Pressure Overload Induced Right Ventricular Hypertrophy and Failure. J. Vis. Exp. JoVE 2018, 141, e58050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axelsen, J.B.; Andersen, S.; Sun, X.-Q.; Ringgaard, S.; Hyldebrandt, J.A.; Kurakula, K.; Goumans, M.-J.; de Man, F.S.; Nielsen-Kudsk, J.E.; Bogaard, H.-J. Effects of 6-mercaptopurine in pressure overload induced right heart failure. PLoS ONE 2019, 14, e0225122. [Google Scholar] [CrossRef]
- Nagendran, J.; Sutendra, G.; Paterson, I.; Champion, H.C.; Webster, L.; Chiu, B.; Haromy, A.; Rebeyka, I.M.; Ross, D.B.; Michelakis, E.D. Endothelin axis is upregulated in human and rat right ventricular hypertrophy. Circ. Res. 2013, 112, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogaard, H.J.; Natarajan, R.; Henderson, S.C.; Long, C.S.; Kraskauskas, D.; Smithson, L.; Ockaili, R.; McCord, J.M.; Voelkel, N.F. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation 2009, 120, 1951–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akazawa, Y.; Okumura, K.; Ishii, R.; Slorach, C.; Hui, W.; Ide, H.; Honjo, O.; Sun, M.; Kabir, G.; Connelly, K. Pulmonary artery banding is a relevant model to study the right ventricular remodeling and dysfunction that occurs in pulmonary arterial hypertension. J. Appl. Physiol. 2020, 129, 238–246. [Google Scholar] [CrossRef]
- Egemnazarov, B.; Crnkovic, S.; Nagy, B.M.; Olschewski, H.; Kwapiszewska, G. Right ventricular fibrosis and dysfunction: Actual concepts and common misconceptions. Matrix Biol. 2018, 68, 507–521. [Google Scholar] [CrossRef]
- Sydykov, A.; Mamazhakypov, A.; Petrovic, A.; Kosanovic, D.; Sarybaev, A.S.; Weissmann, N.; Ghofrani, H.A.; Schermuly, R.T. Inflammatory Mediators Drive Adverse Right Ventricular Remodeling and Dysfunction and Serve as Potential Biomarkers. Front. Physiol. 2018, 9, 609. [Google Scholar] [CrossRef]
- Frump, A.L.; Bonnet, S.; de Jesus Perez, V.A.; Lahm, T. Emerging role of angiogenesis in adaptive and maladaptive right ventricular remodeling in pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L443–L460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viswanathan, G.; Mamazhakypov, A.; Schermuly, R.T.; Rajagopal, S. The Role of G Protein-Coupled Receptors in the Right Ventricle in Pulmonary Hypertension. Front. Cardiovasc. Med. 2018, 5, 179. [Google Scholar] [CrossRef]
- Ryan, J.J.; Archer, S.L. Emerging concepts in the molecular basis of pulmonary arterial hypertension: Part I: Metabolic plasticity and mitochondrial dynamics in the pulmonary circulation and right ventricle in pulmonary arterial hypertension. Circulation 2015, 131, 1691–1702. [Google Scholar] [CrossRef] [Green Version]
- Piao, L.; Marsboom, G.; Archer, S.L. Mitochondrial metabolic adaptation in right ventricular hypertrophy and failure. J. Mol. Med. 2010, 88, 1011–1020. [Google Scholar] [CrossRef] [Green Version]
- Shults, N.V.; Melnyk, O.; Suzuki, D.I.; Suzuki, Y.J. Redox Biology of Right-Sided Heart Failure. Antioxidants 2018, 7, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, N.A.; Blythe, N.M. Cardiac Fibroblast p38 MAPK: A Critical Regulator of Myocardial Remodeling. J. Cardiovasc. Dev. Dis. 2019, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basilio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-kappaB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pak, O.; Scheibe, S.; Esfandiary, A.; Gierhardt, M.; Sydykov, A.; Logan, A.; Fysikopoulos, A.; Veit, F.; Hecker, M.; Kroschel, F. Impact of the mitochondria-targeted antioxidant MitoQ on hypoxia-induced pulmonary hypertension. Eur. Respir. J. 2018, 51, 1701024. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Janocha, A.J.; Erzurum, S.C. Metabolism in Pulmonary Hypertension. Annu. Rev. Physiol. 2021, 83, 551–576. [Google Scholar] [CrossRef] [PubMed]
- Chelladurai, P.; Boucherat, O.; Stenmark, K.; Kracht, M.; Seeger, W.; Bauer, U.M.; Bonnet, S.; Pullamsetti, S.S. Targeting histone acetylation in pulmonary hypertension and right ventricular hypertrophy. Br. J. Pharmacol. 2021, 178, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Andersen, S.; Nielsen-Kudsk, J.E.; Vonk Noordegraaf, A.; de Man, F.S. Right ventricular fibrosis: A pathophysiological factor in pulmonary hypertension? Circulation 2019, 139, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Elimban, V.; Nijjar, M.S.; Gupta, S.K.; Dhalla, N.S. Role of mitogen-activated protein kinase in cardiac hypertrophy and heart failure. Exp. Clin. Cardiol. 2003, 8, 173. [Google Scholar]
- Petrich, B.G.; Wang, Y. Stress-activated MAP kinases in cardiac remodeling and heart failure: New insights from transgenic studies. Trends Cardiovasc. Med. 2004, 14, 50–55. [Google Scholar] [CrossRef]
- Budas, G.R.; Boehm, M.; Kojonazarov, B.; Viswanathan, G.; Tian, X.; Veeroju, S.; Novoyatleva, T.; Grimminger, F.; Hinojosa-Kirschenbaum, F.; Ghofrani, H.A. ASK1 inhibition halts disease progression in preclinical models of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2018, 197, 373–385. [Google Scholar] [CrossRef]
- Kojonazarov, B.; Novoyatleva, T.; Boehm, M.; Happe, C.; Sibinska, Z.; Tian, X.; Sajjad, A.; Luitel, H.; Kriechling, P.; Posern, G. p38 MAPK inhibition improves heart function in pressure-loaded right ventricular hypertrophy. Am. J. Respir. Cell Mol. Biol. 2017, 57, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, S.; Feldman, J.; McLaughlin, V.; Rischard, F.; White, J.; Ebrahimi, R.; Brooks, G.; Satler, C.; Frantz, R.; Lange, T. The ARROW study: A phase 2, prospective, randomized, double-blind, placebo-controlled study of selonsertib in subjects with pulmonary arterial hypertension. Eur. Respir. J. 2017, 50, OA1983. [Google Scholar]
- Paul, M.K.; Mukhopadhyay, A.K. Tyrosine kinase—Role and significance in Cancer. Int. J. Med. Sci. 2004, 1, 101–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, A.; Boehm, M.; Egemnazarov, B.; Grimminger, F.; Savai Pullamsetti, S.; Kwapiszewska, G.; Schermuly, R.T. Kinases as potential targets for treatment of pulmonary hypertension and right ventricular dysfunction. Br. J. Pharmacol. 2021, 178, 31–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.H.; Kerkelä, R.; Force, T. Mechanisms of cardiac dysfunction associated with tyrosine kinase inhibitor cancer therapeutics. Circulation 2008, 118, 84–95. [Google Scholar] [CrossRef]
- Kojonazarov, B.; Sydykov, A.; Pullamsetti, S.S.; Luitel, H.; Dahal, B.K.; Kosanovic, D.; Tian, X.; Majewski, M.; Baumann, C.; Evans, S. Effects of multikinase inhibitors on pressure overload-induced right ventricular remodeling. Int. J. Cardiol. 2013, 167, 2630–2637. [Google Scholar] [CrossRef]
- De Raaf, M.A.; Herrmann, F.E.; Schalij, I.; de Man, F.S.; Vonk-Noordegraaf, A.; Guignabert, C.; Wollin, L.; Bogaard, H.J. Tyrosine kinase inhibitor BIBF1000 does not hamper right ventricular pressure adaptation in rats. Am. J. Physiol. Circ. Physiol. 2016, 311, H604–H612. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Kojonazarov, B.; Elgheznawy, A.; Popp, R.; Dahal, B.K.; Böhm, M.; Pullamsetti, S.S.; Ghofrani, H.-A.; Gödecke, A.; Jungmann, A. miR-223–IGF-IR signalling in hypoxia-and load-induced right-ventricular failure: A novel therapeutic approach. Cardiovasc. Res. 2016, 111, 184–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamagami, K.; Oka, T.; Wang, Q.; Ishizu, T.; Lee, J.-K.; Miwa, K.; Akazawa, H.; Naito, A.T.; Sakata, Y.; Komuro, I. Pirfenidone exhibits cardioprotective effects by regulating myocardial fibrosis and vascular permeability in pressure-overloaded hearts. Am. J. Physiol. Circ. Physiol. 2015, 309, H512–H522. [Google Scholar] [CrossRef] [Green Version]
- Crnkovic, S.; Egemnazarov, B.; Damico, R.; Marsh, L.M.; Nagy, B.M.; Douschan, P.; Atsina, K.; Kolb, T.M.; Mathai, S.C.; Hooper, J.E. Disconnect between fibrotic response and right ventricular dysfunction. Am. J. Respir. Crit. Care Med. 2019, 199, 1550–1560. [Google Scholar] [CrossRef]
- Andersen, S.; Birkmose Axelsen, J.; Ringgaard, S.; Randel Nyengaard, J.; Holm Nielsen, S.; Genovese, F.; Asser Karsdal, M.; Adler Hyldebrandt, J.; Brandt Sørensen, C.; de Man, F.S. Pressure overload induced right ventricular remodeling is not attenuated by the anti-fibrotic agent pirfenidone. Pulm. Circ. 2019, 9, 2045894019848659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pena, A.; Kobir, A.; Goncharov, D.; Goda, A.; Kudryashova, T.V.; Ray, A.; Vanderpool, R.; Baust, J.; Chang, B.; Mora, A.L.; et al. Pharmacological Inhibition of mTOR Kinase Reverses Right Ventricle Remodeling and Improves Right Ventricle Structure and Function in Rats. Am. J. Respir. Cell Mol. Biol. 2017, 57, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Stephens, O.R.; Weiss, K.; Frimel, M.; Rose, J.A.; Sun, Y.; Asosingh, K.; Farha, S.; Highland, K.B.; Prasad, S.V.N.; Erzurum, S.C. Interdependence of hypoxia and beta-adrenergic receptor signaling in pulmonary arterial hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L369–L380. [Google Scholar] [CrossRef]
- Ingwall, J.S. Energy metabolism in heart failure and remodelling. Cardiovasc. Res. 2009, 81, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Fillmore, N.; Mori, J.; Lopaschuk, G. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br. J. Pharmacol. 2014, 171, 2080–2090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, L.; Wu, D.; Dasgupta, A.; Chen, K.-H.; Mewburn, J.; Potus, F.; Lima, P.D.; Hong, Z.; Zhao, Y.-Y.; Hindmarch, C.C. Epigenetic metabolic reprogramming of right ventricular fibroblasts in pulmonary arterial hypertension: A pyruvate dehydrogenase kinase-dependent shift in mitochondrial metabolism promotes right ventricular fibrosis. Circ. Res. 2020, 126, 1723–1745. [Google Scholar] [CrossRef]
- Semba, H.; Takeda, N.; Isagawa, T.; Sugiura, Y.; Honda, K.; Wake, M.; Miyazawa, H.; Yamaguchi, Y.; Miura, M.; Jenkins, D.M. HIF-1α-PDK1 axis-induced active glycolysis plays an essential role in macrophage migratory capacity. Nat. Commun. 2016, 7, 11635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundgrin, E.L.; Park, M.M.; Sharp, J.; Tang, W.W.; Thomas, J.D.; Asosingh, K.; Comhair, S.A.; DiFilippo, F.P.; Neumann, D.R.; Davis, L. Fasting 2-deoxy-2-[18F] fluoro-D-glucose positron emission tomography to detect metabolic changes in pulmonary arterial hypertension hearts over 1 year. Ann. Am. Thorac. Soc. 2013, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.-H.; Piao, L.; Hong, Z.; Toth, P.T.; Marsboom, G.; Bache-Wiig, P.; Rehman, J.; Archer, S.L. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: Exploiting Randle’s cycle. J. Mol. Med. 2012, 90, 31–43. [Google Scholar] [CrossRef] [Green Version]
- Piao, L.; Fang, Y.-H.; Cadete, V.J.; Wietholt, C.; Urboniene, D.; Toth, P.T.; Marsboom, G.; Zhang, H.J.; Haber, I.; Rehman, J. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: Resuscitating the hibernating right ventricle. J. Mol. Med. 2010, 88, 47–60. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Gilde, A.J.; van der Lee, K.A.; Willemsen, P.H.; Chinetti, G.; van der Leij, F.R.; van der Vusse, G.J.; Staels, B.; van Bilsen, M. Peroxisome proliferator-activated receptor (PPAR) α and PPARβ/δ, but not PPARγ, modulate the expression of genes involved in cardiac lipid metabolism. Circ. Res. 2003, 92, 518–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Ding, G.; Qin, Q.; Huang, Y.; Lewis, W.; He, N.; Evans, R.M.; Schneider, M.D.; Brako, F.A.; Xiao, Y. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-δ deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat. Med. 2004, 10, 1245–1250. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, P.; Luo, J.; Huang, Y.; He, L.; Yang, H.; Li, Q.; Wu, S.; Zhelyabovska, O.; Yang, Q. Peroxisome proliferator-activated receptor β/δ activation in adult hearts facilitates mitochondrial function and cardiac performance under pressure-overload condition. Hypertension 2011, 57, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Harrington, L.S.; Moreno, L.; Reed, A.; Wort, S.J.; Desvergne, B.; Garland, C.; Zhao, L.; Mitchell, J.A. The PPARβ/δagonist GW0742 relaxes pulmonary vessels and limits right heart hypertrophy in rats with hypoxia-induced pulmonary hypertension. PLoS ONE 2010, 5, e9526. [Google Scholar] [CrossRef] [Green Version]
- Kojonazarov, B.; Luitel, H.; Sydykov, A.; Dahal, B.K.; Paul-Clark, M.J.; Bonvini, S.; Reed, A.; Schermuly, R.T.; Mitchell, J.A. The peroxisome proliferator–activated receptor β/δ agonist GW0742 has direct protective effects on right heart hypertrophy. Pulm. Circ. 2013, 3, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Lopaschuk, G.D.; Barr, R.; Thomas, P.D.; Dyck, J.R. Beneficial effects of trimetazidine in ex vivo working ischemic hearts are due to a stimulation of glucose oxidation secondary to inhibition of long-chain 3-ketoacyl coenzyme a thiolase. Circ. Res. 2003, 93, e33–e37. [Google Scholar] [CrossRef] [Green Version]
- Kantor, P.F.; Lucien, A.; Kozak, R.; Lopaschuk, G.D. The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long-chain 3-ketoacyl coenzyme A thiolase. Circ. Res. 2000, 86, 580–588. [Google Scholar] [CrossRef] [Green Version]
- Tuunanen, H.; Engblom, E.; Naum, A.; Någren, K.; Scheinin, M.; Hesse, B.; Airaksinen, K.J.; Nuutila, P.; Iozzo, P.; Ukkonen, H. Trimetazidine, a metabolic modulator, has cardiac and extracardiac benefits in idiopathic dilated cardiomyopathy. Circulation 2008, 118, 1250–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciapponi, A.; Pizarro, R.; Harrison, J. Trimetazidine for stable angina. Cochrane Database Syst. Rev. 2005, 4, CD003614. [Google Scholar]
- Marzilli, M.; Klein, W.W. Efficacy and tolerability of trimetazidine in stable angina: A meta-analysis of randomized, double-blind, controlled trials. Coron. Artery Dis. 2003, 14, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Fraser, H.; Lloyd, S.G.; McVeigh, J.J.; Belardinelli, L.; Chatham, J.C. A comparison between ranolazine and CVT-4325, a novel inhibitor of fatty acid oxidation, on cardiac metabolism and left ventricular function in rat isolated perfused heart during ischemia and reperfusion. J. Pharmacol. Exp. Ther. 2007, 321, 213–220. [Google Scholar] [CrossRef] [Green Version]
- MacInnes, A.; Fairman, D.A.; Binding, P.; Rhodes, J.a.; Wyatt, M.J.; Phelan, A.; Haddock, P.S.; Karran, E.H. The antianginal agent trimetazidine does not exert its functional benefit via inhibition of mitochondrial long-chain 3-ketoacyl coenzyme A thiolase. Circ. Res. 2003, 93, e26–e32. [Google Scholar] [CrossRef] [Green Version]
- Zannad, F.; Ferreira, J.P.; Pocock, S.J.; Anker, S.D.; Butler, J.; Filippatos, G.; Brueckmann, M.; Ofstad, A.P.; Pfarr, E.; Jamal, W.; et al. SGLT2 inhibitors in patients with heart failure with reduced ejection fraction: A meta-analysis of the EMPEROR-Reduced and DAPA-HF trials. Lancet 2020, 396, 819–829. [Google Scholar] [CrossRef]
- Szekeres, Z.; Toth, K.; Szabados, E. The Effects of SGLT2 Inhibitors on Lipid Metabolism. Metabolites 2021, 11, 87. [Google Scholar] [CrossRef]
- Chowdhury, B.; Luu, A.Z.; Luu, V.Z.; Kabir, M.G.; Pan, Y.; Teoh, H.; Quan, A.; Sabongui, S.; Al-Omran, M.; Bhatt, D.L.; et al. The SGLT2 inhibitor empagliflozin reduces mortality and prevents progression in experimental pulmonary hypertension. Biochem. Biophys. Res. Commun. 2020, 524, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Van de Veerdonk, M.C.; Bogaard, H.J.; Voelkel, N.F. The right ventricle and pulmonary hypertension. Heart Fail. Rev. 2016, 21, 259–271. [Google Scholar] [CrossRef]
- Michelakis, E.D.; Gurtu, V.; Webster, L.; Barnes, G.; Watson, G.; Howard, L.; Cupitt, J.; Paterson, I.; Thompson, R.B.; Chow, K.; et al. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci. Transl. Med. 2017, 9, eaao4583. [Google Scholar] [CrossRef] [Green Version]
- Welsh, P.; Grassia, G.; Botha, S.; Sattar, N.; Maffia, P. Targeting inflammation to reduce cardiovascular disease risk: A realistic clinical prospect? Br. J. Pharmacol. 2017, 174, 3898–3913. [Google Scholar] [CrossRef] [Green Version]
- Adamo, L.; Rocha-Resende, C.; Prabhu, S.D.; Mann, D.L. Reappraising the role of inflammation in heart failure. Nat. Rev. Cardiol. 2020, 17, 269–285. [Google Scholar] [CrossRef]
- Frieler, R.A.; Mortensen, R.M. Immune cell and other noncardiomyocyte regulation of cardiac hypertrophy and remodeling. Circulation 2015, 131, 1019–1030. [Google Scholar] [CrossRef] [Green Version]
- Mamazhakypov, A.; Viswanathan, G.; Lawrie, A.; Schermuly, R.T.; Rajagopal, S. The role of chemokines and chemokine receptors in pulmonary arterial hypertension. Br. J. Pharmacol. 2021, 178, 72–89. [Google Scholar] [CrossRef]
- Luitel, H.; Sydykov, A.; Schymura, Y.; Mamazhakypov, A.; Janssen, W.; Pradhan, K.; Wietelmann, A.; Kosanovic, D.; Dahal, B.K.; Weissmann, N. Pressure overload leads to an increased accumulation and activity of mast cells in the right ventricle. Physiol. Rep. 2017, 5, e13146. [Google Scholar] [CrossRef]
- Sydykov, A.; Luitel, H.; Mamazhakypov, A.; Wygrecka, M.; Pradhan, K.; Pak, O.; Petrovic, A.; Kojonazarov, B.; Weissmann, N.; Seeger, W. Genetic Deficiency and Pharmacological Stabilization of Mast Cells Ameliorate Pressure Overload-Induced Maladaptive Right Ventricular Remodeling in Mice. Int. J. Mol. Sci. 2020, 21, 9099. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Abe, K.; Ishikawa, M.; Saku, K.; Shinoda-Sakamoto, M.; Ishikawa, T.; Watanabe, T.; Oka, M.; Sunagawa, K.; Tsutsui, H. Inhibition of TLR9-NF-κB-mediated sterile inflammation improves pressure overload-induced right ventricular dysfunction in rats. Cardiovasc. Res. 2019, 115, 658–668. [Google Scholar] [CrossRef]
- Harston, R.K.; McKillop, J.C.; Moschella, P.C.; Van Laer, A.; Quinones, L.S.; Baicu, C.F.; Balasubramanian, S.; Zile, M.R.; Kuppuswamy, D. Rapamycin treatment augments both protein ubiquitination and Akt activation in pressure-overloaded rat myocardium. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1696–H1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Li, Y.; Heims-Waldron, D.; Bezzerides, V.; Guatimosim, S.; Guo, Y.; Gu, F.; Zhou, P.; Lin, Z.; Ma, Q. Mitochondrial cardiomyopathy caused by elevated reactive oxygen species and impaired cardiomyocyte proliferation. Circ. Res. 2018, 122, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Redout, E.M.; Wagner, M.J.; Zuidwijk, M.J.; Boer, C.; Musters, R.J.; van Hardeveld, C.; Paulus, W.J.; Simonides, W.S. Right-ventricular failure is associated with increased mitochondrial complex II activity and production of reactive oxygen species. Cardiovasc. Res. 2007, 75, 770–781. [Google Scholar] [CrossRef]
- He, J.; Li, X.; Luo, H.; Li, T.; Zhao, L.; Qi, Q.; Liu, Y.; Yu, Z. Galectin-3 mediates the pulmonary arterial hypertension–induced right ventricular remodeling through interacting with NADPH oxidase 4. J. Am. Soc. Hypertens. 2017, 11, 275–289.e2. [Google Scholar] [CrossRef]
- Schlüter, K.-D.; Kutsche, H.S.; Hirschhäuser, C.; Schreckenberg, R.; Schulz, R. Review on chamber-specific differences in right and left heart reactive oxygen species handling. Front. Physiol. 2018, 9, 1799. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Hu, J.; Sun, K.; Xu, D. The role and molecular mechanism of epigenetics in cardiac hypertrophy. Heart Fail. Rev. 2020, 1–10, Online ahead of print. [Google Scholar] [CrossRef]
- Gillette, T.G.; Hill, J.A. Readers, writers, and erasers: Chromatin as the whiteboard of heart disease. Circ. Res. 2015, 116, 1245–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.-P.; Han, P. Epigenetic and lncRNA regulation of cardiac pathophysiology. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2016, 1863, 1767–1771. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, D.; Li, Y. LncRNAs in cardiac hypertrophy: From basic science to clinical application. Cell. Mol. Med. 2020, 24, 11638–11645. [Google Scholar] [CrossRef]
- Omura, J.; Habbout, K.; Shimauchi, T.; Wu, W.-H.; Breuils-Bonnet, S.; Tremblay, E.; Martineau, S.; Nadeau, V.; Gagnon, K.; Mazoyer, F. Identification of Long Noncoding RNA H19 as a New Biomarker and Therapeutic Target in Right Ventricular Failure in Pulmonary Arterial Hypertension. Circulation 2020, 142, 1464–1484. [Google Scholar] [CrossRef]
- Backs, J.; Olson, E.N. Control of cardiac growth by histone acetylation/deacetylation. Circ. Res. 2006, 98, 15–24. [Google Scholar] [CrossRef]
- Kong, Y.; Tannous, P.; Lu, G.; Berenji, K.; Rothermel, B.A.; Olson, E.N.; Hill, J.A. Suppression of class I and II histone deacetylases blunts pressure-overload cardiac hypertrophy. Circulation 2006, 113, 2579. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, C.M.; Luo, Y.; Yin, Z.; Zhang, M.; Zhu, W.; Wang, T.; Floss, T.; Goettlicher, M.; Noppinger, P.R.; Wurst, W. Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3β activity. Nat. Med. 2007, 13, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Bogaard, H.J.; Mizuno, S.; Hussaini, A.A.A.; Toldo, S.; Abbate, A.; Kraskauskas, D.; Kasper, M.; Natarajan, R.; Voelkel, N.F. Suppression of histone deacetylases worsens right ventricular dysfunction after pulmonary artery banding in rats. Am. J. Respir. Crit. Care Med. 2011, 183, 1402–1410. [Google Scholar] [CrossRef]
- Cho, Y.K.; Eom, G.H.; Kee, H.J.; Kim, H.-S.; Choi, W.-Y.; Nam, K.-I.; Ma, J.S.; Kook, H. Sodium valproate, a histone deacetylase inhibitor, but not captopril, prevents right ventricular hypertrophy in rats. Circ. J. 2010, 74, 760–770. [Google Scholar] [CrossRef] [Green Version]
- Borck, P.C.; Guo, L.-W.; Plutzky, J. BET epigenetic reader proteins in cardiovascular transcriptional programs. Circ. Res. 2020, 126, 1190–1208. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-Y.; Chiang, C.-M. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J. Biol. Chem. 2007, 282, 13141–13145. [Google Scholar] [CrossRef] [Green Version]
- Spiltoir, J.I.; Stratton, M.S.; Cavasin, M.A.; Demos-Davies, K.; Reid, B.G.; Qi, J.; Bradner, J.E.; McKinsey, T.A. BET acetyl-lysine binding proteins control pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 2013, 63, 175–179. [Google Scholar] [CrossRef] [Green Version]
- Meloche, J.; Lampron, M.-C.; Nadeau, V.; Maltais, M.; Potus, F.; Lambert, C.; Tremblay, E.; Vitry, G.; Breuils-Bonnet, S.; Boucherat, O. Implication of inflammation and epigenetic readers in coronary artery remodeling in patients with pulmonary arterial hypertension. Arter. Thromb. Vasc. Biol. 2017, 37, 1513–1523. [Google Scholar] [CrossRef] [Green Version]
- Van der Feen, D.E.; Kurakula, K.; Tremblay, E.; Boucherat, O.; Bossers, G.P.; Szulcek, R.; Bourgeois, A.; Lampron, M.-C.; Habbout, K.; Martineau, S. Multicenter preclinical validation of BET inhibition for the treatment of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2019, 200, 910–920. [Google Scholar] [CrossRef]
- Tompkins, B.A.; Balkan, W.; Winkler, J.; Gyöngyösi, M.; Goliasch, G.; Fernández-Avilés, F.; Hare, J.M. Preclinical studies of stem cell therapy for heart disease. Circ. Res. 2018, 122, 1006–1020. [Google Scholar] [CrossRef] [PubMed]
- Sanganalmath, S.K.; Bolli, R. Cell therapy for heart failure: A comprehensive overview of experimental and clinical studies, current challenges, and future directions. Circ. Res. 2013, 113, 810–834. [Google Scholar] [CrossRef]
- Vagnozzi, R.J.; Maillet, M.; Sargent, M.A.; Khalil, H.; Johansen, A.K.Z.; Schwanekamp, J.A.; York, A.J.; Huang, V.; Nahrendorf, M.; Sadayappan, S.; et al. An acute immune response underlies the benefit of cardiac stem cell therapy. Nature 2020, 577, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Van Berlo, J.H.; Kanisicak, O.; Maillet, M.; Vagnozzi, R.J.; Karch, J.; Lin, S.C.; Middleton, R.C.; Marban, E.; Molkentin, J.D. c-kit+ cells minimally contribute cardiomyocytes to the heart. Nature 2014, 509, 337–341. [Google Scholar] [CrossRef]
- Ashur, C.; Frishman, W.H. Cardiosphere-derived cells and ischemic heart failure. Cardiol. Rev. 2018, 26, 8–21. [Google Scholar] [CrossRef]
- Malliaras, K.; Zhang, Y.; Seinfeld, J.; Galang, G.; Tseliou, E.; Cheng, K.; Sun, B.; Aminzadeh, M.; Marbán, E. Cardiomyocyte proliferation and progenitor cell recruitment underlie therapeutic regeneration after myocardial infarction in the adult mouse heart. EMBO Mol. Med. 2013, 5, 191–209. [Google Scholar] [CrossRef]
- Gallet, R.; Dawkins, J.; Valle, J.; Simsolo, E.; De Couto, G.; Middleton, R.; Tseliou, E.; Luthringer, D.; Kreke, M.; Smith, R.R. Exosomes secreted by cardiosphere-derived cells reduce scarring, attenuate adverse remodelling, and improve function in acute and chronic porcine myocardial infarction. Eur. Heart J. 2017, 38, 201–211. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, M.N.; Bolli, R.; Hare, J.M. Clinical studies of cell therapy in cardiovascular medicine: Recent developments and future directions. Circ. Res. 2018, 123, 266–287. [Google Scholar] [CrossRef]
- Haller, C.; Friedberg, M.K.; Laflamme, M.A. The role of regenerative therapy in the treatment of right ventricular failure: A literature review. Stem Cell Res. Ther. 2020, 11, 502. [Google Scholar] [CrossRef] [PubMed]
- Wehman, B.; Sharma, S.; Pietris, N.; Mishra, R.; Siddiqui, O.T.; Bigham, G.; Li, T.; Aiello, E.; Murthi, S.; Pittenger, M. Mesenchymal stem cells preserve neonatal right ventricular function in a porcine model of pressure overload. Am. J. Physiol. Circ. Physiol. 2016, 310, H1816–H1826. [Google Scholar] [CrossRef] [Green Version]
- Oommen, S.; Yamada, S.; Peral, S.C.; Campbell, K.A.; Bruinsma, E.S.; Terzic, A.; Nelson, T.J. Human umbilical cord blood-derived mononuclear cells improve murine ventricular function upon intramyocardial delivery in right ventricular chronic pressure overload. Stem Cell Res. Ther. 2015, 6, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Davies, B.; Elwood, N.J.; Li, S.; Cullinane, F.; Edwards, G.A.; Newgreen, D.F.; Brizard, C.P. Human cord blood stem cells enhance neonatal right ventricular function in an ovine model of right ventricular training. Ann. Thorac. Surg. 2010, 89, 585–593.e584. [Google Scholar] [CrossRef] [PubMed]
- Wehman, B.; Pietris, N.; Bigham, G.; Siddiqui, O.; Mishra, R.; Li, T.; Aiello, E.; Jack, G.; Wang, W.; Murthi, S. Cardiac progenitor cells enhance neonatal right ventricular function after pulmonary artery banding. Ann. Thorac. Surg. 2017, 104, 2045–2053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bittle, G.J.; Morales, D.; Pietris, N.; Parchment, N.; Parsell, D.; Peck, K.; Deatrick, K.B.; Rodriguez-Borlado, L.; Smith, R.R.; Marbán, L. Exosomes isolated from human cardiosphere–derived cells attenuate pressure overload–induced right ventricular dysfunction. J. Thorac. Cardiovasc. Surg. 2020. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mamazhakypov, A.; Sommer, N.; Assmus, B.; Tello, K.; Schermuly, R.T.; Kosanovic, D.; Sarybaev, A.S.; Weissmann, N.; Pak, O. Novel Therapeutic Targets for the Treatment of Right Ventricular Remodeling: Insights from the Pulmonary Artery Banding Model. Int. J. Environ. Res. Public Health 2021, 18, 8297. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph18168297
Mamazhakypov A, Sommer N, Assmus B, Tello K, Schermuly RT, Kosanovic D, Sarybaev AS, Weissmann N, Pak O. Novel Therapeutic Targets for the Treatment of Right Ventricular Remodeling: Insights from the Pulmonary Artery Banding Model. International Journal of Environmental Research and Public Health. 2021; 18(16):8297. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph18168297
Chicago/Turabian StyleMamazhakypov, Argen, Natascha Sommer, Birgit Assmus, Khodr Tello, Ralph Theo Schermuly, Djuro Kosanovic, Akpay Sh. Sarybaev, Norbert Weissmann, and Oleg Pak. 2021. "Novel Therapeutic Targets for the Treatment of Right Ventricular Remodeling: Insights from the Pulmonary Artery Banding Model" International Journal of Environmental Research and Public Health 18, no. 16: 8297. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph18168297