3.1. The Role of GSK3B Gene in Cholesterol Regulation Involved in Alzheimer’s Disease
GSK3B protein is a growth-signaling kinase regulated by inhibitory phosphorylation downstream of the Wnt pathway [
37]. Studies have attributed this protein to having a role in memory and has been connected to Alzheimer’s disease due to the fact that its activation can lead to tau phosphorylation and amyloid-beta plaques. Two main pathways of GSK3B activation exist, including the Wnt/B-catenin pathway and the PI3K/Akt/mTor pathway (as shown in
Figure 3). Through the Akt pathway, the GSK3B protein inhibits the expression of CREB, which is a binding protein that aids in cell survival and other vital functions [
38]. GSK3B has also been found to phosphorylate various components of this pathway including Akt, RICTOR, TSC1 and 2, PTEN1 and 2, and others [
39]. The Akt pathway is initiated by the activation of PI3K by cell surface receptors including RTK, GPCR, and cytokine receptors. Next, PIP
3-binding proteins stimulate the synthesis of PIP
3 and activate downstream signaling to promote cell growth, proliferation, and metabolism. Akt then phosphorylates and inhibits GSK3B, hindering its kinase activity and promoting cell survival. However, GSK3B directly phosphorylates several components including Akt. On the other hand, GSK3B simultaneously phosphorylates and destroys B-catenin throughout the Wnt pathway [
39]. B-catenin plays a large role in regulating cell differentiation, proliferation, and apoptosis, and therefore has been found to enhance cognitive and memory abilities [
40]. Although this pathway is not completely understood, it is known that GSK3B is associated with Axin and other molecules to create a complex that allows GSK3B to phosphorylate Axin, APC, and B-catenin. GSK3B then phosphorylates B-catenin on Thr41, Ser37, and Ser33, which allows B-TrCP to recognize B-catenin and consequently become degraded [
41]. It has also been found that GSK3B can form a complex with B-catenin, therefore lowering the levels of B-catenin/TCF transcription and inhibiting Wnt signaling. Wnt signaling plays an important role in synaptic plasticity and maintenance in the adult brain. The compromised synaptic signaling is associated with Alzheimer’s disease [
42].
Due to GSK3B’s role in two separate pathways involving cell proliferation, as well as its direct effect on tau phosphorylation [
43] and amyloid-beta production [
44], developments have been made to target GSK3B for the treatment of Alzheimer’s disease. Since the overactivity of GSK3B increases tau phosphorylation, researchers started to develop drugs that inhibit GSK3B and therefore prevent tau tangles from occurring [
43]. A full list of the GSK3B inhibitors that are in preclinical or clinical trials is provided below, in
Table 2. Since this work does not aim to illustrate the mechanisms of each of these inhibitors, the inhibitors of strong clinical interest are briefly illustrated here. The most successful and advanced drug thus far is Tideglusib, which is small-molecule inhibitor of GSK3B that uses the compound thiadiazolidinone to decrease amyloid deposition and lower tau phosphorylation levels [
43]. In preclinical trials, this drug was found to reduce tau phosphorylation, amyloid deposition, neuron loss, and gliosis in mice [
44]. Tideglusib was discontinued for Alzheimer’s treatment in 2012 after Phase 2b due to the fact that the trial missed its primary endpoint and some secondary endpoints [
44]. Another GSK3B inhibitor, AL001, was developed to deliver lithium carbonate while avoiding the harmful effects of lithium [
45]. Lithium inhibits GSK3B by being a direct competitor of magnesium and by decreasing phosphorylation levels through the activation of the Akt pathway [
46]. AL001 is currently in Phase 3 of clinical trials [
47]. Another GSK3B inhibitor that has been developed for Alzheimer’s treatment is SAR502250. This drug was found to prevent the increase in neuronal cell death and improve cognitive deficit in mice [
48].
3.2. The Role of BACE1 Gene in Cholesterol Regulation Involved in Alzheimer’s Disease
BACE1 is another potential target gene that catalyzes the initial cleavage of APP, which is one of the earliest pathologic events in Alzheimer’s development [
50]. BACE1 has been attributed as the sole initiator of amyloid-beta production, and its activity is elevated in the brains of Alzheimer’s patients [
51]. It also plays a critical role in synaptic development and plasticity through multiple mechanisms such as APP cleavage, neuregulin-1 (Nrg1) cleavage, Sez6 cleavage, and Jagged-1 cleavage [
52]. Nrg1 regulates myelination, the migration of glutamatergic and GABAergic neurons, and synaptic plasticity. The function of Sez6 is not fully known, but it is believed that they may act as receptors at the cell surface and exhibit adhesive and receptor trafficking functions [
52]. Jagged-1 is a BACE1 substrate that regulates astrogenesis and neurogenesis through the Notch signaling pathway, as illustrated in
Figure 4A. BACE1 is involved in one pathway that processes APP via the amyloidogenic pathway that involves the sequential cleavage of APP by beta- and gamma-secretase, which promotes amyloid-beta production. In this pathway, BACE1 cleaves APP in the lipid raft region to release the beta-stubs while also releasing the soluble N-terminus of APP [
53]. Then CTF-beta is cleaved by gamma-secretase to release amyloid-beta into the extracellular space and the cytoplasm. The amyloid-beta released in this pathway can form amyloid plaques. The nonamyloidogenic pathway begins with the cleavage of APP by either alpha-secretase or y-secretase to release two variants of APP, sAPP-
, and sAPP-
, from the endosome or cell membrane [
53]. The C90 fragment is then cleaved by
-secretase, which releases AICD and p3 fragments [
54]. BACE1 also interacts with GSK3B through the NF
B pathway. When this pathway is upregulated, GSK3B induces BACE1 expression by NF
B/p65 nuclear translocation and BACE1 promoter site binding, which increases BACE1 levels [
55] (as illustrated in
Figure 4C). Due to BACE1′s critical role as the initiating enzyme in amyloid-beta production, it has been selected as a prime target for slowing the progression of Alzheimer’s disease by lowering amyloid-beta levels in the brain [
56].
BACE1’s involvement in the amyloidogenic pathway offers the potential to target Alzheimer’s disease. The activity of BACE1 leads to the production of amyloid-beta, so BACE1 inhibitors have been developed to prevent the accumulation of amyloid-beta plaques in the brain of Alzheimer’s patients. BACE1 inhibitors are typically small in size so that they can cross the blood brain barrier easily, which can help reduce the production of amyloid-beta in neurons [
57]. One small-molecule BACE1 inhibitor that has been developed is Verubecestat, which has cellular permeability and good solubility. Preclinical tests of Verubecestat in animals demonstrated no adverse effects. Phase I trials proved that the drug was safe while effective at reducing the amyloid-beta concentration in the cerebrospinal fluid. During Phases II and III trials, participants with mild cognitive deficits were used to determine the effect Verubecestat has on cognitive and functional abilities. While the amyloid-beta plaques in their brains slightly decreased, adverse effects were reported and no improvement in cognitive function was detected. Although trial was terminated in 2018, this drug did prove that BACE1 inhibitors need to be administered several years before symptoms of Alzheimer’s disease are apparent.
Another small-molecule BACE1 inhibitor is Lanabecestat, the inhibition potency of which is as follows: Ki = 0.4 nM for hBACE1, and Ki = 0.8 nM for hBACE2. During Phase I trials, the drug was deemed safe for Alzheimer’s patients with mild cognitive impairment. This drug strongly decreased the amyloid-beta levels and did not result in adverse effects [
57]. During Phases II and III, the main goal was to test the efficacy and safety of the drug while ensuring that amyloid-levels were decreasing while cognitive function was simultaneously improving. In 2018, Phase III was terminated due to the decision that the trial was unlikely to meet the primary endpoint [
58].
Furthermore, Atabecestat was developed by Shionogi and later entered a collaboration with Janssen. Administering Atabecestat, at doses ranging from 5 to 150 mg daily for a duration of up to 14 days in both elderly and young healthy individuals, led to a substantial and consistent decrease of Aβ levels. The reduction was up to 90% in the group receiving 90 mg, observed in both blood plasma and cerebrospinal fluid (CSF) [
59]. Phase I studies demonstrated a significant decrease in amyloid-beta deposits in patients in the early stages of Alzheimer’s disease. Phase II trials proved that Atabecestat was able to reduce amyloid-beta levels in both plasma and the cerebrospinal fluid. However, the trial was discontinued in 2018 because of the unfavorable benefit-risk ratio, specifically regarding the increase in liver enzymes and abnormal liver function found in certain patients [
57].
Elenbecestat is another small-molecule BACE1 inhibitor that was found to decrease amyloid-beta levels in plasma and CSF. A Phase I clinical study (NCT01294540) has revealed that a one-time dose of 50 mg, administered to 73 healthy individuals (both genders, aged 30 to 85 years, across six distinct groups), was both safe and well-received. In an investigation using a single oral dose increasing from 5 to 800 mg, as well as a 14-day multiple ascending-dose study in the range of 25–400 mg, it was established that elenbecestat could significantly lower the Aβ levels in either the blood plasma or cerebrospinal fluid (CSF) by a substantial margin of up to 92%. Specifically, the plasma Aβ(1-X) levels demonstrated a 52% reduction at a 5 mg dose and an impressive 92% decrease at the 800 mg dosage, when compared to baseline levels [
60]. A few adverse effects were reported throughout Phases I and II. Elenbecestat was found to delay clinical symptoms of mild dementia in Alzheimer’s patients during a Phase II trial. In June 2018, Biogen declared that the phase II study, which lasted for 18 months, demonstrated not only a substantial decrease in Aβ levels, as verified by amyloid PET imaging, but also less deterioration in cognitive function in patients with a mild to moderate form of Alzheimer’s Disease. Nonetheless, they also reported certain side effects, which included upper respiratory tract infections, unusual dreams and nightmares, contact dermatitis, headaches, diarrhea, and incidents of falling [
61]. In 2019, a Phase III trial named MISSION was ended due to the unfavorable benefit-risk ratio [
62]. Another drug, CNP520, was terminated in 2020 during Phase III due to safety issues.
Some therapeutic strategies to increase the efficacy of BACE1 inhibitors are being researched, one of which includes binding BACE1 to a transferrin receptor antibody, which would allow the complex to travel through the BBB better and reduce amyloid-beta levels in the brain [
63]. This approach is still being developed and is currently in preclinical phases. The full list of BACE1 inhibitors that are in preclinical/clinical trials is provided below, in
Table 3.
3.3. The Role of SREBP2 Gene in Cholesterol Regulation Involved in Alzheimer’s Disease
SREBP2 is a transcription factor that regulates the synthesis and uptake of both cholesterol and fatty acids and is the main regulator of cholesterol metabolism [
18]. Specifically, SREBP2 activates HMGCR, which is the rate limiting enzyme in cholesterol synthesis [
64]. There are about thirty consecutive reactions that contribute to the metabolization of cholesterol, and SREBP2 plays an important role specifically in the mevalonate pathway, which directly links the regulation of cholesterol to amyloid-beta accumulation. During de novo synthesis of cholesterol, SREBP2 binds to SCAP and transfers the complex from the endoplasmic reticulum to the Golgi apparatus, where it is then processed to enter the nucleus and induce transcription for many genes such as HMGCR, mevalonate kinase, and squalene monooxygenase [
6]. SREBP2 also binds to the promoter region of ABCA1 and inhibits its transcription, therefore releasing more cholesterol [
65]. Multiple signaling pathways can regulate SREBP2 activation during cholesterol synthesis, such as p53, androgen, and Akt, which was a major pathway for the GSK3B target [
66]. The p53 pathway can inhibit SREPB2 and reduce the transcription of mevalonate pathway genes (
Figure 5A). On the other hand, androgen is a hormone that can enhance SREBP2 activation. Also, the Akt pathway activates SREBP2, which induces the genes involved in cholesterol synthesis (
Figure 5B). The Akt pathway is required for the transfer of SREBP2 to the Golgi apparatus. It was found that Akt inhibition leads to the inactivation of SREBP2 and, as a result, cholesterol synthesis is inhibited [
18]. Not only is SREBP2 related to the progression of Alzheimer’s disease through the production of cholesterol, it is also directly involved in the regulation of certain genes that contribute to amyloid-beta and tau protein generation. For example, SREBP2 was found to be involved in the regulation of BACE1. SREBP2 activation due to high cholesterol results in the hyperexpression of BACE1. It was determined that blocking SREBP2 expression completely blocked up-regulation of BACE1, which, as previously mentioned, contributes to the production of amyloid-beta [
64].
The overproduction of cholesterol has been linked to increase the amyloid-beta production, which offers the potential of SREBP2 to be targeted for Alzheimer’s treatment. One development in SREBP2 inhibition for Alzheimer’s treatment is osmotin. Osmotin is a homolog of adiponectin whose treatment significantly induces AMPK/SIRT1 activation and reduces SREBP2 expression, therefore diminishing amyloidogenic amyloid-beta production [
67]. Osmotin treatment has been found to protect against memory impairment, synaptic dysfunction, and neurodegeneration. This treatment also significantly decreased the LDL levels and increased the HDL levels, and in turn also reduced the levels of soluble amyloid-beta in the brain. The use of osmotin also reduces BACE1 expression and alleviates hyperphosphorylation by inhibiting GSK3B through the PI3K/Akt pathway [
68]. Although treatment of Alzheimer’s disease using osmotin is still being researched, some small-molecule inhibitors of SREBP2 have been developed and demonstrate promising results in decreasing cholesterol levels.
Betulin is a drug that inhibits the SREBP2 pathway and decreases the biosynthesis of cholesterol [
69]. This drug inhibits SREBP2 by binding to SCAP and promoting the interaction between SCAP and Insig. While statins are typically used to treat hypercholesterolemia, they often activate SREBP2 in the process. Betulin manages to decrease cholesterol levels while also directly inhibiting SREBP2, which makes this drug a promising treatment for Alzheimer’s disease. Although Betulin is often used to treat diabetes and atherosclerosis, the ability of this drug to decrease cholesterol levels and the fact that betulinic acid has been found to prevent Alzheimer’s-induced neurodegenerative issues offer the potential of this SREBP2 inhibitor to benefit Alzheimer’s patients [
70]. Other SREBP2 inhibitors, such as Curcumin and Fatostatin, both inhibit SREBP2 activation by binding to SCAP and decreasing cholesterol levels, which demonstrate potential for future Alzheimer’s treatment [
70,
71]. The full list of SREBP2 inhibitors that are in preclinical/clinical trials is provided below, in
Table 4.
3.4. The Role of CYP46A1 Gene in Cholesterol Regulation Involved in Alzheimer’s Disease
CYP46A1 is a protein coding gene that is also the rate-limiting enzyme for cholesterol degradation in the brain [
72]. Modification of CYP46A1 has been found to improve memory either through isoprenoid synthesis or the production of 24-OH through the modulation of NMDAR [
73]. During cholesterol biosynthesis, as illustrated in
Figure 1, CYP46A1 leads to the conversion of cholesterol in 24S-OHC, which can cross the blood brain barrier and eliminate cerebral cholesterol. Neurons carry CYP46A1 in order to convert excess cholesterol in the endoplasmic reticulum into 24S-OHC, which forms a major export pathway for excess cholesterol from the brain and metabolizes the excess cholesterol in the liver [
6]. Throughout this pathway, the activation of CYP46A1 has been found to increase the expression of SREBP2, which can lead to an increase in cholesterol synthesis [
72]. Decreased expression of CYP46A1 in mice was found to increase the amount of cholesterol in neurons, which led to apoptotic death and cognitive deficits [
74]. When CYP46A1 is inhibited or mutated, the levels of 24S-OHC significantly decrease, therefore contributing to an accumulation of cholesterol in the brain since 24S-OHC is not available to extrude excess cholesterol from the brain. Studies have found that CYP46A1 activation helps reduce cholesterol levels in the brain, not by slowing the rate of synthesis of cholesterol but instead by increasing the rate at which cholesterol is expelled from the brain. CYP46A1 activation also leads to a decreased production of both amyloid-beta and tau proteins [
73]. The downregulation of CYP46A1 was found to increase amyloid-beta levels through the recruitment of APP in lipid rafts and eventually resulted in neuronal death [
74]. Studies indicate that a few gene variations in CYP46A1, including the CYP46A1 T allele, are possible genetic risk factors of Alzheimer’s disease [
75]. Due to its role in amyloid-beta and 24S-OHC production, as well as its possible involvement as a genetic risk factor, the activation of CYP46A1 has the potential to treat Alzheimer’s disease.
The most successful and advanced drug for Alzheimer’s disease that activates CYP46A1 is Efavirenz. This drug is an anti-HIV medication that is used to treat patients with mild cognitive impairment or early dementia due to Alzheimer’s disease [
76]. The activation of CYP46A1 due to Efavirenz resulted in the enhancement of brain cholesterol turnover and behavior improvements. Efavirenz interacts with CYP46A1 by binding to the P450 active site and activating it. Studies have found that this drug successfully reduces the amyloid-beta levels, increases cholesterol elimination, and improves cognitive behavior. Small doses of Efavirenz were found to be safe in Alzheimer’s patients and even have some advantages over other potential options for Alzheimer’s treatment. For example, Efavirenz increases the production of 24S-OHC and promotes the elimination of cellular cholesterol [
77]. Also, CYP46A1 activation will only affect the central nervous system, whereas synthetic LXR agonists may affect multiple systems of the body (more detail to be given in the next section).
Some other drugs that target CYP46A1 for Alzheimer’s treatment are huperzine and galantamine. Galantamine found modest but consistent cognitive benefits over a short period of time and has the potential to treat Alzheimer’s throughout long-term treatment [
78]. Huperzine A is a Chinese herb extract that has been found to improve cognitive function in participants with Alzheimer’s disease, but some issues were reported throughout trials regarding poor methodological quality [
79]. Studies have also found that adeno-associated virus gene therapy can be used to overexpress CYP46A1 in order to increase 24S-OHC in the brain [
80]. Using this form of therapy, CYP46A1 overexpression before the onset of amyloid plaques aided in the improvement of cognitive function in mice, as well as the reduction of microgliosis and astrogliosis. However, further studies need to be done to determine the long-term effects of selective CYP46A1 overexpression. The full list of CYP46A1 inducers that are in preclinical/clinical trials is provided below, in
Table 5.
3.5. The Role of ABAC1 Gene in Cholesterol Regulation Involved in Alzheimer’s Disease
ABCA1 is an ATP binding cassette that mediates cellular cholesterol efflux (
Figure 6). ABCA1 is expressed with the highest levels in liver hepatocytes but is also found in the central nervous system through the neurons, astrocytes, and microglia [
81]. It is an integral membrane protein that utilizes ATP to transport cholesterol to APP, and its main function is to maintain lipid homeostasis by controlling the transport of cholesterol [
82]. ABCA1 also interacts with ApoE1 to develop HDL, which aids in the efflux of cholesterol from the periphery to the liver. ABCA1 may also protect cells from the cytotoxic effects of excess cholesterol. ABCA1 plays a vital role in central nervous system cholesterol transport through the reverse cholesterol transport pathway. ApoE is mainly synthesized in astrocytes, where ApoE then receives cholesterol and phospholipids from ABCA1. The cholesterol and phospholipids are then secreted into the brain as lipoproteins, and excess cholesterol is internalized by LRP1. It is believed that CYP46A1 utilizes ABCA1 to convert cholesterol into 24-OHC and consequently remove cholesterol from the brain [
83]. ABCA1 is involved in many cell-signaling pathways, but two pathways in particular are cholesterol-dependent. Through de novo synthesis, cholesterol in cells can be converted into oxysterols. Oxysterols increase ABCA1 expression through the activation of the liver X receptor pathway, which then forms a complex with retinoid X receptor (RXR) and together form a transcription factor that binds to ABCA1 to increase its expression [
82]. The LXR/RXR pathway can be activated to increase ABCA1 expression, which would increase the efflux of cholesterol out of the brain by inversely decreasing BACE1 expression. By activating this pathway, ABCA1 can be activated and BACE1 can be inhibited, which decreases the cholesterol and amyloid-beta levels in the brain [
84]. The other cholesterol-dependent pathway that ABCA1 is involved in is the SREBP2 pathway. Unlike the LXR/RXR pathway explained above, the activation of the SREBP2 pathway decreases the expression of ABCA1. As mentioned in previous sections, this pathway is activated by the binding of SCAP to SREBP2, which promotes the transcription of many genes including SREBP2 itself. MiR-33a is co-transcribed with SREBP2, which has been found to inhibit ABCA1 expression. Therefore, activation of the LXR/RXR or inactivation of the SREBP2 pathway by inhibiting miR-33a both offer possible therapeutic possibilities for Alzheimer’s disease, by increasing the expression of ABCA1.
The activation of ABCA1 has been the target for multiple studies involved in Alzheimer’s treatment and has been found to offer positive results in decreasing cholesterol levels. Synthetic LXR agonists such as T0901317 have been proven to increase ABCA1 expression while simultaneously reducing the soluble amyloid-beta levels in the brain [
85]. One study with mice even found that T0901317 reversed memory deficits. Synthetic LXR agonists have also been found to stimulate the enzymatic degradation of amyloid-beta by microglia. Synthetic RXR agonists such as bexarotene have more conflicting results, but studies have found this type of treatment to enhance the clearance of amyloid-beta plaques and improve cognitive performance. However, both LXR and RXR agonists led to unfavorable effects in the liver and therefore few developments have reached clinical trials. Small molecule inducers have been developed to enhance ABCA1 expression while avoiding the hepatoxic side effects of LXR agonists. Antagonists of P2X7, such as AZ-1 and AZ-2, resulted in the enhancement of ABAC1 enhancement and activity, which significantly increased cholesterol efflux [
86]. These compounds were also found to activate the LXR pathway indirectly. Also, AZ-1 and AZ-2 significantly increased ABAC1 protein levels in CNS cells such as astrocytes, microglia, and vascular pericytes. Other nonlipogenic ABCA1 inducing compounds have been researched to selectively activate ABAC1 while keeping the liver unaffected. A new small molecule ABCA1 inducer called E17241 dose-dependently upregulates ABCA1 expression and increases cholesterol efflux [
87]. Administered at two different doses, this drug enhances ABAC1 expression without provoking any adverse effects on the liver. Compared to the other therapeutic developments mentioned above, E17241 offers antiatherogenic effects upon oral administration without the need for nanoparticles. The full list of ABCA1 inducers that are in preclinical/clinical trials is provided below, in
Table 6.
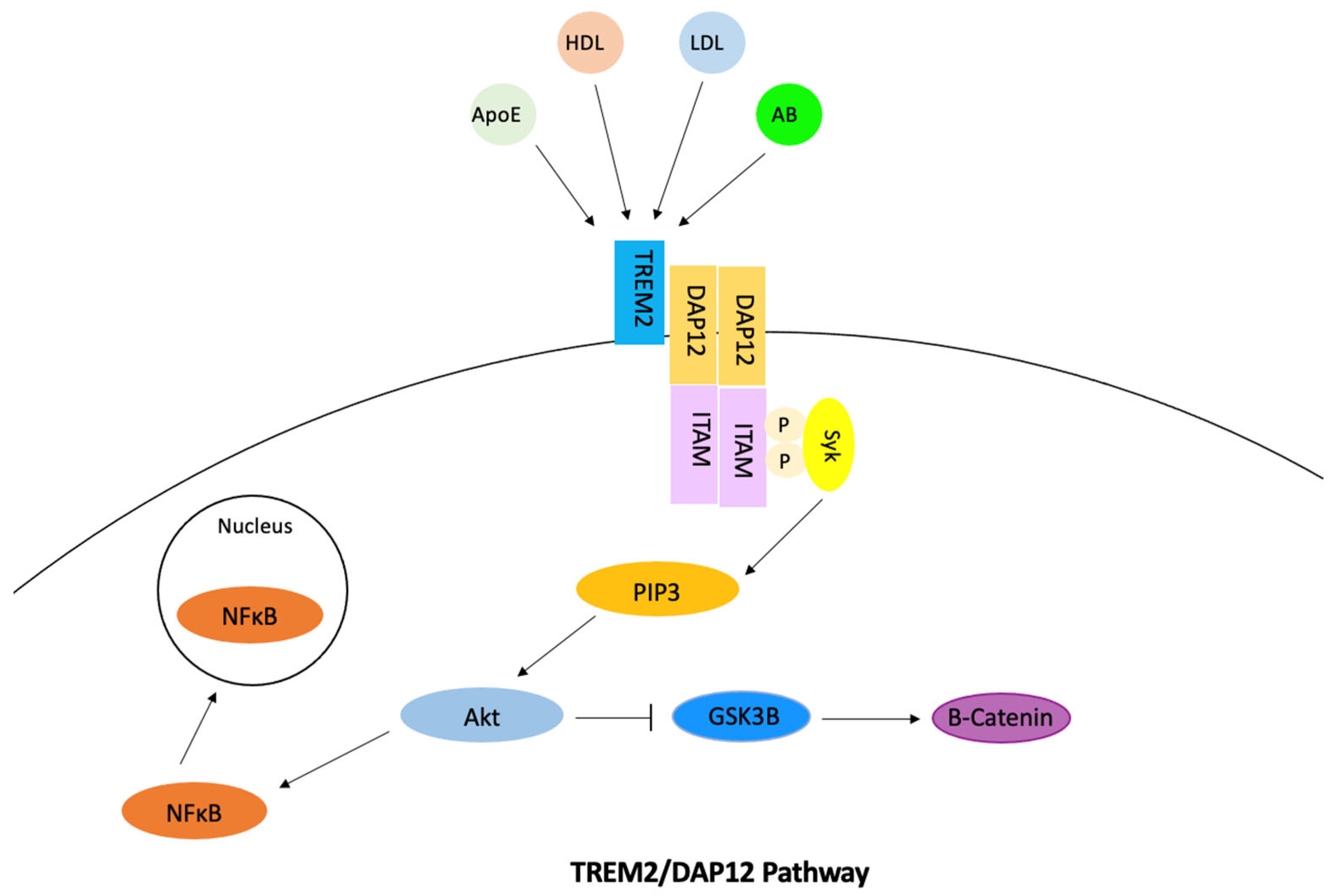
3.6. The Role of TREM2 Gene in Cholesterol Regulation Involved in Alzheimer’s Disease
TREM2 is a transmembrane immune receptor that is expressed by microglia in the brain and enhances the rate of phagocytosis. This gene also has anti-inflammatory properties and modulates inflammatory signaling [
88]. In the microglia, signaling of the TREM2/DAP12 pathway was found to promote cholesterol efflux and therefore reduce how much intracellular cholesterol is stored as cholesterol esters (
Figure 7). Extracellular ligand binding activates TREM2, which then activates DAP12 [
89]. ITAM then becomes phosphorylated, which causes the Syk kinase to activate downstream signaling molecules including PI3K, ERK, PLCy, and Vav [
90]. The cholesterol is then transported out of the microglia via ApoE-containing lipoproteins. Phosphorylation of DAP12 often affects other pathways and has the ability to inhibit the RAS/MEK/ERK pathway. This inhibition may decrease the secretion of pro-inflammatory cytokines. The phosphorylation of DAP12 also has a direct effect on the uptake of amyloid-beta through its involvement with SHIP1 and the SHIP1/CD2A/RIN3/BIN1 complex. TREM2 also mediates proliferation and survival of myeloid cells by the alteration of the Wnt/B-catenin pathway. Studies found that TREM2 stabilized B-catenin by inhibiting its degradation through the Akt/GSK3B pathway [
91]. TREM2 is also involved in mTOR signaling through the same pathway, which has the ability to alter microglial survival. In relation to Alzheimer’s disease, studies have found TREM2 expression leads to an increase in tau phosphorylation, but this relation has not been studied as much as TREM’s connection to amyloid-beta. TREM2 is thought to have a time-dependent effect on amyloid-beta accumulation. The signaling of TREM2 was found to increase amyloid-beta accumulation in early stages of Alzheimer’s disease and decrease amyloid-beta accumulation in later stages [
92]. Other studies have found TREM2 overexpression to reduce amyloid-beta accumulation [
93]. Through the activation of the NF
B pathway, BACE1 expression increases and TREM2 expression decreases due to regulation of miRNA-34a, which causes amyloid-beta accumulation [
94]. Mutations of TREM2 contribute to late-onset Alzheimer’s disease because these variants disturb TREM2 signaling and its functions [
88].
The increased expression of TREM2 in Alzheimer’s patients has been researched as a possible therapeutic strategy. Most strategies to treat Alzheimer’s disease by targeting TREM2 found it most effective to stimulate TREM2 in the early stages of the disease. The soluble form of TREM2 (sTREM2) has reached preclinical trials due to its involvement in microglial regulation and tau phosphorylation levels [
95]. Soluble TREM2 is released into the extracellular space when TREM2 is cleaved and has been found to promote cell survival. Increased levels of sTREM2 are found in the cerebrospinal fluid of those with Alzheimer’s disease. Studies found that an increase in sTREM2 leads to a decrease in amyloid-beta levels as well as diminished cognitive decline [
96]. Although sTREM2 drugs remain in preclinical trials, other therapeutic strategies such as anti-TREM2 antibodies have reached clinical trials. AL002 is an anti-human TREM2 antibody that binds to TREM2 and activates signaling. It was found to cause microglia to express more pro-inflammatory and repair genes, while significantly decreasing amyloid-beta accumulation [
97]. Phase 1 clinical trials proved that AL002 was able to reduce sTREM2 levels in the brain without causing any adverse effects. A Phase 2 study is currently recruiting participants and is expected to be completed in January 2024 [
98].
On the other hand, AL003 is a monoclonal antibody that targets the receptor CD33 and as a result inhibits TREM2. Phase 1 clinical trials demonstrated conflicting results from this drug. Participants from the multiple-ascending-dose phase demonstrated an increase in soluble CD33 levels in Cerebrospinal fluid (CSF) with no adverse effects. During the phase of single ascending doses, 38 healthy individuals were given infusions with increasing dosages from one of eight levels. The evaluation parameters included safety, tolerability, pharmacokinetics, pharmacodynamics, and immunogenicity. It was observed that in healthy volunteers, the drug was well received up to a dosage of 15 mg/kg. However, one participant developed hip inflammation while another experienced severe hypersensitivity, and both were hospitalized [
99]. In June 2022, AL003 was terminated without any justified reason. Another agnostic antibody drug that has been developed is TREM2 TVD-Ig, which is an agnostic antibody that activates TREM2. This drug aims at enhancing microglia functions and reducing amyloid pathology [
100]. During preclinical development, this drug was found to enhance microglial phagocytosis of amyloid plaques. Future experiments on this drug aim at focusing on the microglia activation phenotype, which is necessary to understand the mechanism of this drug [
101]. An antibody transport vehicle named DNL919 was developed to activate TREM2 and analyze the effects this drug has on CSF1R [
102]. This drug was put on hold by the FDA in January 2022 but is currently recruiting participants and is set to finish Phase 1 clinical trials in July 2023 [
103]. The full list of TREM2 inducers that are in preclinical/clinical trials is provided below, in
Table 7.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}