
Preferential Protection of Low Coordinated Sites in Pt Nanoparticles for Enhancing Durability of Pt/C Catalyst

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Density Functional Theory (DFT) Calculations
2.2. Synthesis of MPTES–Pt/C
2.3. Physical Characterizations of MPTES–Pt/C
2.4. Electrochemical Analysis of MPTES–Pt/C
3. Results and Discussions
3.1. Adsorption Energy of MPTES Calculated by DFT
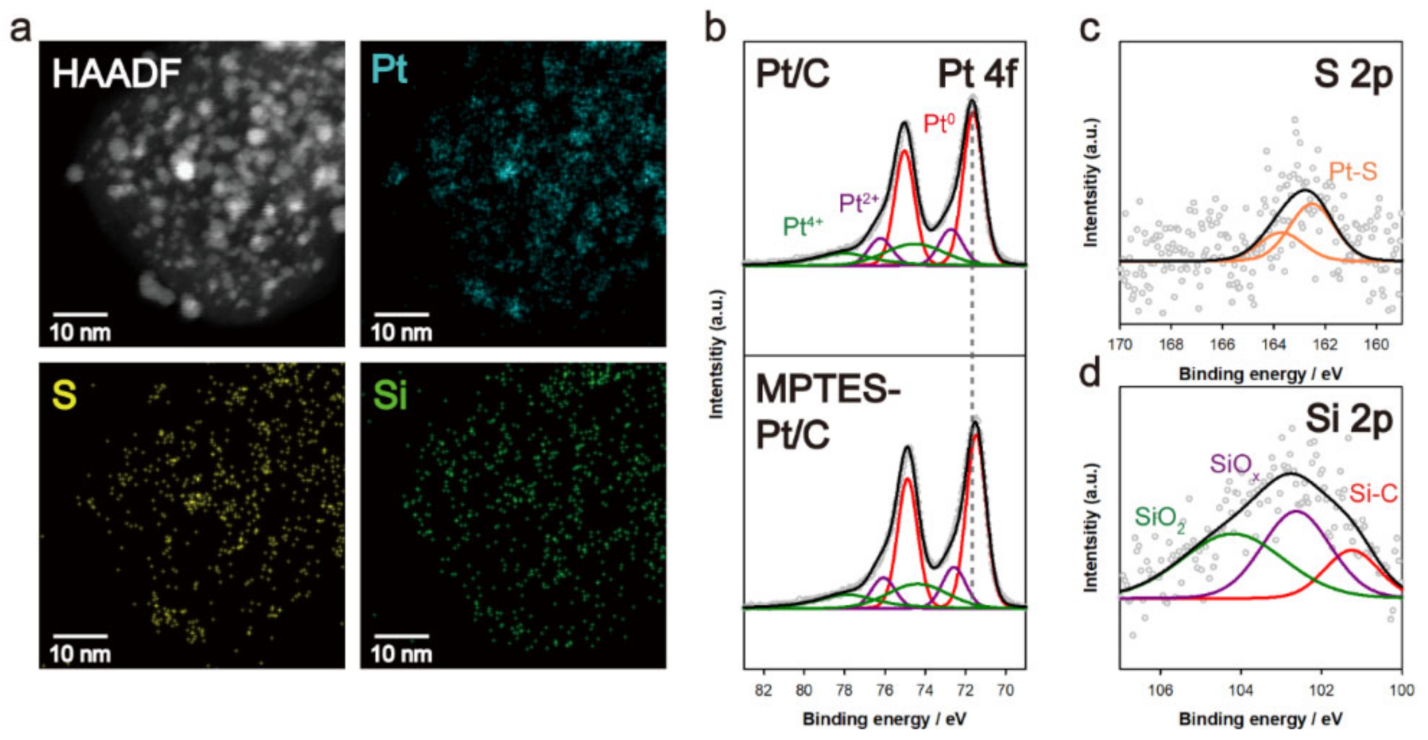
3.2. Elemental Analysis of MPTES–Pt/C
3.3. Electrochemical Analysis of MPTES–Pt/C
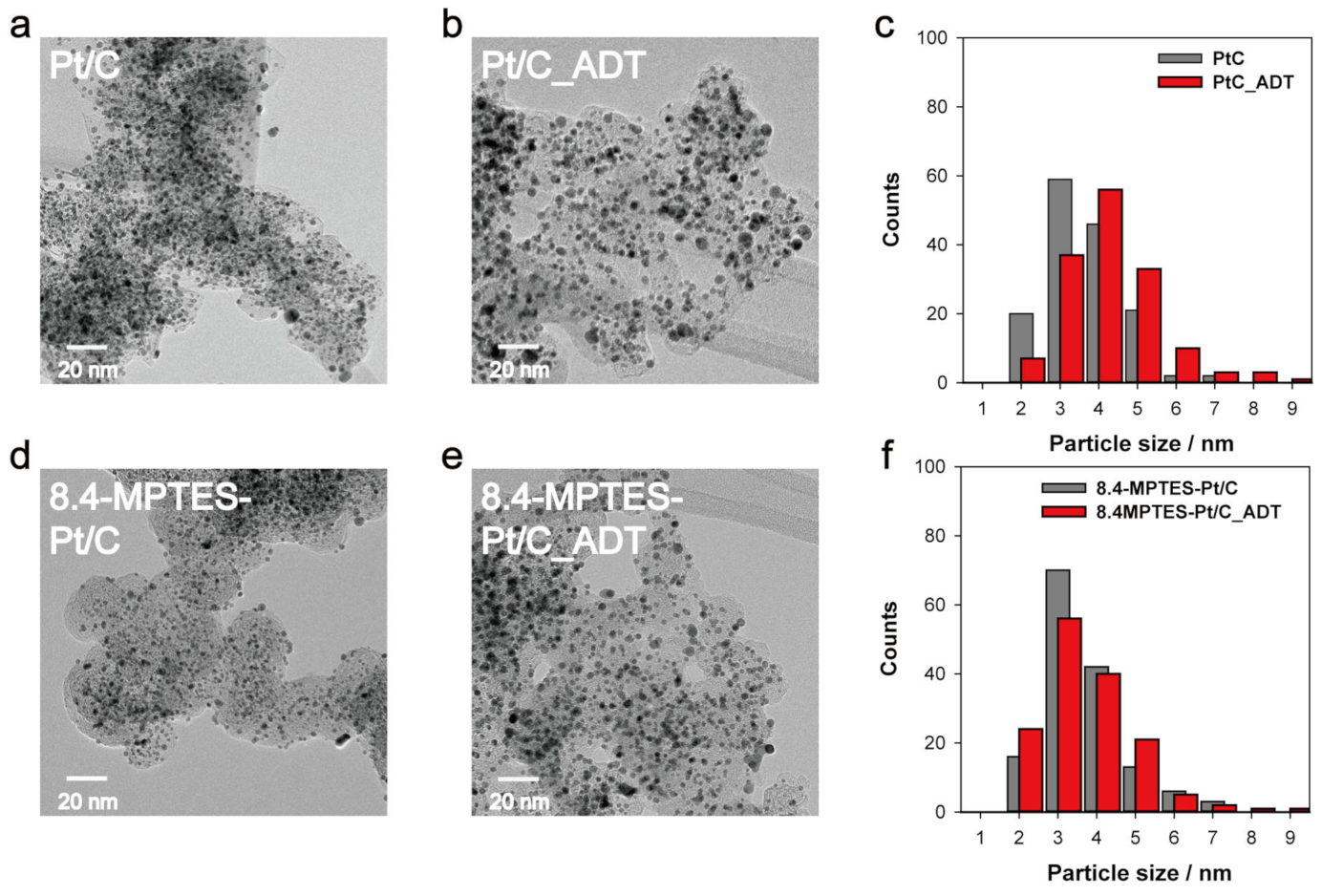
3.4. ADT Results and Post-Mortem Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Steele, B.C.H.; Heinzel, A. Materials for fuel-cell technologies. Nature 2001, 414, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Winter, M.; Brodd, R.J. What Are Batteries, Fuel Cells, and Supercapacitors? Chem. Rev. 2004, 104, 4245–4270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, L.; Liu, F.; Zhao, L.; Yang, W.; Yao, J. Evidence of a Unique Electron Donor−Acceptor Property for Platinum Nanoparticles as Studied by XPS. Langmuir 2006, 22, 4480–4482. [Google Scholar] [CrossRef]
- Yu, K.; Groom, D.J.; Wang, X.; Yang, Z.; Gummalla, M.; Ball, S.C.; Myers, D.J.; Ferreira, P.J. Degradation Mechanisms of Platinum Nanoparticle Catalysts in Proton Exchange Membrane Fuel Cells: The Role of Particle Size. Chem. Mater. 2014, 26, 5540–5548. [Google Scholar] [CrossRef]
- Meier, J.C.; Galeano, C.; Katsounaros, I.; Witte, J.; Bongard, H.J.; Topalov, A.A.; Baldizzone, C.; Mezzavilla, S.; Schüth, F.; Mayrhofer, K.J.J. Design criteria for stable Pt/C fuel cell catalysts. Beilstein J. Nanotechnol. 2014, 5, 44–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.; Andersen, S.M. Quantification on Degradation Mechanisms of Polymer Electrolyte Membrane Fuel Cell Catalyst Layers during an Accelerated Stress Test. ACS Catal. 2018, 8, 3424–3434. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.; Han, B.; Persson, K.; Friesen, C.; He, T.; Sieradzki, K.; Ceder, G. Electrochemical Stability of Nanometer-Scale Pt Particles in Acidic Environments. J. Am. Chem. Soc. 2010, 132, 596–600. [Google Scholar] [CrossRef]
- Cherevko, S.; Kulyk, N.; Mayrhofer, K.J.J. Durability of platinum-based fuel cell electrocatalysts: Dissolution of bulk and nanoscale platinum. Nano Energy 2016, 29, 275–298. [Google Scholar] [CrossRef]
- Calle-Vallejo, F.; Tymoczko, J.; Colic, V.; Vu, Q.H.; Pohl, M.D.; Morgenstern, K.; Loffreda, D.; Sautet, P.; Schuhmann, W.; Bandarenka, A.S. Finding optimal surface sites on heterogeneous catalysts by counting nearest neighbors. Science 2015, 350, 185–189. [Google Scholar] [CrossRef]
- Lopes, P.P.; Strmcnik, D.; Tripkovic, D.; Connell, J.G.; Stamenkovic, V.; Markovic, N.M. Relationships between Atomic Level Surface Structure and Stability/Activity of Platinum Surface Atoms in Aqueous Environments. ACS Catal. 2016, 6, 2536–2544. [Google Scholar] [CrossRef]
- Subramanian, N.P.; Greszler, T.A.; Zhang, J.; Gu, W.; Makharia, R. Pt-Oxide Coverage-Dependent Oxygen Reduction Reaction (ORR) Kinetics. J. Electrochem. Soc. 2012, 159, B531–B540. [Google Scholar] [CrossRef]
- Chung, D.Y.; Shin, H.; Yoo, J.M.; Lee, K.-S.; Lee, N.-S.; Kang, K.; Sung, Y.-E. Functional link between surface low-coordination sites and the electrochemical durability of Pt nanoparticles. J. Power Sources 2016, 334, 52–57. [Google Scholar] [CrossRef]
- Sandbeck, D.J.S.; Brummel, O.; Mayrhofer, K.J.J.; Libuda, J.; Katsounaros, I.; Cherevko, S. Dissolution of Platinum Single Crystals in Acidic Medium. ChemPhysChem 2019, 20, 2997–3003. [Google Scholar] [CrossRef] [Green Version]
- Stephens, I.E.L.; Bondarenko, A.S.; Grønbjerg, U.; Rossmeisl, J.; Chorkendorff, I. Understanding the electrocatalysis of oxygen reduction on platinum and its alloys. Energy Environ. Sci. 2012, 5, 6744–6762. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Chiba, H.; Kato, T.; Endo, S.; Hayashi, T.; Todoroki, N.; Wadayama, T. Oxygen reduction reaction activity and structural stability of Pt–Au nanoparticles prepared by arc-plasma deposition. Phys. Chem. Chem. Phys. 2015, 17, 18638–18644. [Google Scholar] [CrossRef]
- Cai, J.; Zhang, J.; Cao, K.; Gong, M.; Lang, Y.; Liu, X.; Chu, S.; Shan, B.; Chen, R. Selective Passivation of Pt Nanoparticles with Enhanced Sintering Resistance and Activity toward CO Oxidation via Atomic Layer Deposition. ACS Appl. Nano Mater. 2018, 1, 522–530. [Google Scholar] [CrossRef]
- Jiang, Z.-Z.; Wang, Z.-B.; Gu, D.-M.; Smotkin, E.S. Carbon riveted Pt/C catalyst with high stability prepared by in situ carbonized glucose. Chem. Commun. 2010, 46, 6998–7000. [Google Scholar] [CrossRef]
- Lee, H.; Sung, Y.-E.; Choi, I.; Lim, T.; Kwon, O.J. Novel synthesis of highly durable and active Pt catalyst encapsulated in nitrogen containing carbon for polymer electrolyte membrane fuel cell. J. Power Sources 2017, 362, 228–235. [Google Scholar] [CrossRef]
- Chung, D.Y.; Jun, S.W.; Yoon, G.; Kwon, S.G.; Shin, D.Y.; Seo, P.; Yoo, J.M.; Shin, H.; Chung, Y.-H.; Kim, H.; et al. Highly Durable and Active PtFe Nanocatalyst for Electrochemical Oxygen Reduction Reaction. J. Am. Chem. Soc. 2015, 137, 15478–15485. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Choun, M.; Jeong, B.; Lee, J.K.; Lee, J. Atomic layer deposition of ultrathin layered TiO2 on Pt/C cathode catalyst for extended durability in polymer electrolyte fuel cells. J. Energy Chem. 2016, 25, 258–264. [Google Scholar] [CrossRef]
- Cheng, N.; Banis, M.N.; Liu, J.; Riese, A.; Li, X.; Li, R.; Ye, S.; Knights, S.; Sun, X. Extremely Stable Platinum Nanoparticles Encapsulated in a Zirconia Nanocage by Area-Selective Atomic Layer Deposition for the Oxygen Reduction Reaction. Adv. Mater. 2015, 27, 277–281. [Google Scholar] [CrossRef]
- Takenaka, S.; Matsumori, H.; Nakagawa, K.; Matsune, H.; Tanabe, E.; Kishida, M. Improvement in the Durability of Pt Electrocatalysts by Coverage with Silica Layers. J. Phys. Chem. C 2007, 111, 15133–15136. [Google Scholar] [CrossRef]
- Takenaka, S.; Miyamoto, H.; Utsunomiya, Y.; Matsune, H.; Kishida, M. Catalytic Activity of Highly Durable Pt/CNT Catalysts Covered with Hydrophobic Silica Layers for the Oxygen Reduction Reaction in PEFCs. J. Phys. Chem. C 2014, 118, 774–783. [Google Scholar] [CrossRef]
- Aoki, N.; Inoue, H.; Kawasaki, H.; Daimon, H.; Doi, T.; Inaba, M. Durability Improvement of Pd Core-Pt Shell Structured Catalyst by Porous SiO2 Coating. J. Electrochem. Soc. 2018, 165, F737–F747. [Google Scholar] [CrossRef]
- Choi, J.; Jang, J.-H.; Roh, C.-W.; Yang, S.; Kim, J.; Lim, J.; Yoo, S.J.; Lee, H. Gram-scale synthesis of highly active and durable octahedral PtNi nanoparticle catalysts for proton exchange membrane fuel cell. Appl. Catal. B Environ. 2018, 225, 530–537. [Google Scholar] [CrossRef] [Green Version]
- Bu, L.; Feng, Y.; Yao, J.; Guo, S.; Guo, J.; Huang, X. Facet and dimensionality control of Pt nanostructures for efficient oxygen reduction and methanol oxidation electrocatalysts. Nano Res. 2016, 9, 2811–2821. [Google Scholar] [CrossRef]
- Turchanin, A.; El-Desawy, M.; Gölzhäuser, A. High thermal stability of cross-linked aromatic self-assembled monolayers: Nanopatterning via selective thermal desorption. Appl. Phys. Lett. 2007, 90, 053102. [Google Scholar] [CrossRef]
- Chung, D.Y.; Lee, M.J.; Kim, M.; Shin, H.; Kim, M.-J.; Yoo, J.M.; Park, S.; Sung, Y.-E. CO electro-oxidation reaction on Pt nanoparticles: Understanding peak multiplicity through thiol derivative molecule adsorption. Catal. Today 2017, 293–294, 2–7. [Google Scholar] [CrossRef]
- Dablemont, C.; Lang, P.; Mangeney, C.; Piquemal, J.-Y.; Petkov, V.; Herbst, F.; Viau, G. FTIR and XPS Study of Pt Nanoparticle Functionalization and Interaction with Alumina. Langmuir 2008, 24, 5832–5841. [Google Scholar] [CrossRef]
- Genorio, B.; Strmcnik, D.; Subbaraman, R.; Tripkovic, D.; Karapetrov, G.; Stamenkovic, V.R.; Pejovnik, S.; Marković, N.M. Selective catalysts for the hydrogen oxidation and oxygen reduction reactions by patterning of platinum with calix[4]arene molecules. Nat. Mater. 2010, 9, 998–1003. [Google Scholar] [CrossRef]
- Wang, S.; Yang, F.; Jiang, S.P.; Chen, S.; Wang, X. Tuning the electrocatalytic activity of Pt nanoparticles on carbon nanotubes via surface functionalization. Electrochem. Commun. 2010, 12, 1646–1649. [Google Scholar] [CrossRef]
- Kim, T.-J.; Kwon, G.; Kim, Y.-T. Anomalously increased oxygen reduction reaction activity with accelerated durability test cycles for platinum on thiolated carbon nanotubes. Chem. Commun. 2014, 50, 596–598. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, P.M.; Glamsch, S.; Ehlert, C.; Lippitz, A.; Kulak, N.; Unger, W.E.S. Synchrotron-radiation XPS analysis of ultra-thin silane films: Specifying the organic silicon. Appl. Surf. Sci. 2016, 363, 406–411. [Google Scholar] [CrossRef]
- Darmakkolla, S.R.; Tran, H.; Gupta, A.; Rananavare, S.B. A method to derivatize surface silanol groups to Si-alkyl groups in carbon-doped silicon oxides. RSC Adv. 2016, 6, 93219–93230. [Google Scholar] [CrossRef]
- Wadayama, T.; Yoshida, H.; Ogawa, K.; Todoroki, N.; Yamada, Y.; Miyamoto, K.; Iijima, Y.; Sugawara, T.; Arihara, K.; Sugawara, S.; et al. Outermost Surface Structures and Oxygen Reduction Reaction Activities of Co/Pt(111) Bimetallic Systems Fabricated Using Molecular Beam Epitaxy. J. Phys. Chem. C 2011, 115, 18589–18596. [Google Scholar] [CrossRef]
- Park, I.-S.; Tong, Y.Y.J. Sulfide–Adsorption-Enhanced Oxygen Reduction Reaction on Carbon-Supported Pt Electrocatalyst. Electrocatalysis 2013, 4, 117–122. [Google Scholar] [CrossRef]
- Ferreira, P.J.; la O’, G.J.; Shao-Horn, Y.; Morgan, D.; Makharia, R.; Kocha, S.; Gasteiger, H.A. Instability of Pt ∕ C Electrocatalysts in Proton Exchange Membrane Fuel Cells: A Mechanistic Investigation. J. Electrochem. Soc. 2005, 152, A2256–A2271. [Google Scholar] [CrossRef] [Green Version]
- Meier, J.C.; Galeano, C.; Katsounaros, I.; Topalov, A.A.; Kostka, A.; Schüth, F.; Mayrhofer, K.J.J. Degradation Mechanisms of Pt/C Fuel Cell Catalysts under Simulated Start–Stop Conditions. ACS Catal. 2012, 2, 832–843. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, D.W.; Yuk, S.; Choi, S.; Lee, D.-H.; Doo, G.; Hyun, J.; Kwen, J.; Kim, J.Y.; Kim, H.-T. Preferential Protection of Low Coordinated Sites in Pt Nanoparticles for Enhancing Durability of Pt/C Catalyst. Energies 2021, 14, 1419. https://0-doi-org.brum.beds.ac.uk/10.3390/en14051419
Lee DW, Yuk S, Choi S, Lee D-H, Doo G, Hyun J, Kwen J, Kim JY, Kim H-T. Preferential Protection of Low Coordinated Sites in Pt Nanoparticles for Enhancing Durability of Pt/C Catalyst. Energies. 2021; 14(5):1419. https://0-doi-org.brum.beds.ac.uk/10.3390/en14051419
Chicago/Turabian StyleLee, Dong Wook, Seongmin Yuk, Sungyu Choi, Dong-Hyun Lee, Gisu Doo, Jonghyun Hyun, Jiyun Kwen, Jun Young Kim, and Hee-Tak Kim. 2021. "Preferential Protection of Low Coordinated Sites in Pt Nanoparticles for Enhancing Durability of Pt/C Catalyst" Energies 14, no. 5: 1419. https://0-doi-org.brum.beds.ac.uk/10.3390/en14051419