
Phonon Transport and Thermoelectric Properties of Imidazole-Graphyne


1
School of Materials Science and Engineering, Peking University, Beijing 100871, China
2
Center for Applied Physics and Technology, HEPDS, College of Engineering, Peking University, Beijing 100871, China
*
Author to whom correspondence should be addressed.
Materials 2021, 14(19), 5604; https://0-doi-org.brum.beds.ac.uk/10.3390/ma14195604
Submission received: 5 September 2021
/
Revised: 20 September 2021
/
Accepted: 22 September 2021
/
Published: 27 September 2021
(This article belongs to the Special Issue Feature Papers in Materials Simulation and Design)
Abstract
:The pentagon has been proven to be an important structural unit for carbon materials, leading to different physical and chemical properties from those of hexagon-based allotropes. Following the development from graphene to penta-graphene, a breakthrough has very recently been made for graphyne—for example, imidazole-graphyne (ID-GY) was formed by assembling experimentally synthesized pentagonal imidazole molecules and acetylenic linkers. In this work, we study the thermal properties and thermoelectric performance of ID-GY by combining first principle calculations with the Boltzmann transport theory. The calculated lattice thermal conductivity of ID-GY is 10.76 W/mK at 300 K, which is only one tenth of that of γ-graphyne (106.24 W/mK). A detailed analysis of the harmonic and anharmonic properties, including the phonon group velocity, phonon lifetime, atomic displacement parameter, and bond energy curves, reveals that the low lattice thermal conductivity can be attributed to the low Young’s modulus, low Debye temperature, and high Grüneisen parameter. Furthermore, at room temperature, ID-GY can reach a high ZT value of 0.46 with a 5.8 × 1012 cm−2 hole concentration, which is much higher than the value for many other carbon-based materials. This work demonstrates that changing structural units from hexagonal to pentagonal can significantly reduce the lattice thermal conductivity and enhance the thermoelectric performance of carbon-based materials.
1. Introduction
Thermoelectric materials that can convert waste heat to electricity based on the Seebeck effect have aroused great attention in the energy field. The conversion efficiency of thermoelectric materials is evaluated by a dimensionless figure of merit (ZT), ZT = S2σT/(ke + kl), which depends on the synergetic effect of the Seebeck coefficient (S), electrical conductivity (σ), absolute temperature (T), electronic thermal conductivity (ke), and lattice thermal conductivity (kl). However, most commercial thermoelectric materials are based on elements that are relatively scarce and/or toxic, such as Bi2Te3 [1], PbTe [2], and Sb2Te3 [3]. Therefore, there is a need to find other earth-abundant and environmentally friendly materials with a good thermoelectric performance.
For this, carbon-based materials can be candidates because of their nontoxicity, light weight, low cost, and high compatibility. More importantly, the lattice thermal conductivity of carbon materials can vary within a huge range of five orders of magnitude depending on the atomic configuration [4]. Usually, materials with a low lattice thermal conductivity are desirable in thermoelectric applications for energy conversion. It has been found that carbon materials can reach very low lattice thermal conductivities and exhibit a good thermoelectric performance [5,6,7]. For instance, Yan’s group reported that the thermoelectric properties of carbon nanotubes (CNTs) can be significantly enhanced by changing their morphology to CNT bulky papers with Ar plasma treatment. The ZT value of CNT bulky papers is increased from 0.01 for pristine CNTs to 0.4 for Ar plasma-treated CNTs [8]. Chen et al. found that the thermal conductivity of single-walled carbon nanotube (SWNT)/polyaniline (PANI) hybrid film is only 0.43 W/mK and that the ZT value reaches 0.12 at room temperature, remarkably higher than that of either of the individual components of the composite [9]. Meanwhile, for two-dimensional (2D) carbon materials, graphene is a typical representative and exhibits a high electrical conductivity, which is one of the essential requirements for thermoelectric materials. However, graphene possesses an ultra-high lattice thermal conductivity (3151.53 W/mK at 300 K) [10] with a low Seebeck coefficient (about 100 μV/K at 300 K) [11] because of its gapless band structure and strong sp2 covalent bonds, hindering its application in the thermoelectric field. As an allotrope of graphene, the recently synthesized γ-graphyne [12] has provided a new possibility for the application of carbon-based materials in the thermoelectric field due to its high Seebeck coefficient of 690 μV/K [13] and low kl of 106.24 W/mK [14], which are superior to the corresponding values of graphene.
On the other hand, it has been found that changing structural units can not only change the geometrical structures of materials but also significantly change the values of the lattice thermal conductivity. For instance, the lattice thermal conductivity of penta-graphene is found to be 645 W/mK at room temperature [15], much lower than the 3151.53 W/mK of graphene [10]. When going from 2D carbon sheets to one-dimensional (1D) and three-dimensional (3D) carbon structures, a similar trend also exists. For 1D carbon, the lattice thermal conductivity of a pentagon-based nanotube is only 95.87 W/mK, which is less than one tenth of the value of (6,6) a carbon nanotube with a similar tube radius [16]. For penta-diamond, which is a new 3D carbon allotrope consisting of five-membered rings, the lattice thermal conductivity is 490.88 W/mK at room temperature [17], much lower than that of diamond (2664.93 W/mK). These results clearly show that the pentagonal unit can effectively modulate the thermal transport of carbon materials. Very recently, we proposed a 2D pentagon-based derivative of graphyne, imidazole-graphyne [18], named ID-GY, which has a direct band gap of 1.10 eV, a low Young’s modulus, and strong refraction near infrared (IR), with potential applications in nanoelectronics and optical devices. ID-GY could be formed by assembling experimentally synthesized five-membered imidazole molecules with acetylenic linkers, as exhibited in our previous work [18]. In this work, we further study the thermal transport and thermoelectric properties of ID-GY.
2. Computational Methods
Geometry optimization and electronic band structure calculation are carried out using density functional theory (DFT), as implemented in the Vienna ab initio simulation package (VASP) [19], using the projector augmented wave (PAW) method [20,21]. The Perdew–Burke–Ernzerhof (PBE) functional [22] within the generalized gradient approximation (GGA) [23] is used to treat the exchange–correlation interaction of electrons, while the Heyd–Scuseria–Ernzerhof hybrid functional (HSE06) [24] is used for more accurate band-structure calculations. The kinetic energy cutoff of wave function is set to 520 eV, and the Monkhorst–Pack [25] k-point, with a grid density of 2π × 0.02 Å−1, is used to sample the Brillouin zone for integration in the reciprocal space. All atomic positions are fully optimized with convergence thresholds of 10−8 eV and 10−6 eV/Å for the total energy and force component, respectively. During the calculations of geometry optimization and band structure, 2D periodic boundary conditions along the x and y directions are applied to ID-GY, while a vacuum region of 16.59 Å is set along the z direction to exclude the mirror interactions between adjacent images.
The electrical transport properties, including the Seebeck coefficient, electrical conductivity, and electronic thermal conductivity are calculated using BoltzTraP2 software [26]. The lattice thermal conductivity is calculated using the ShengBTE package [27]. The second and third interatomic force constants (IFCs) are obtained based on a 2 × 2 supercell using phonopy software [28] and thirdorder.py code [27], respectively. To calculate the anharmonic IFCs, we include the interactions up to the ninth-nearest neighbor atoms with the cutoff radius of 4.72 Å. The q-grids of 40 × 40 and the thickness of 3.4 Å are chosen to solve the phonon Boltzmann transport equation.
3. Results and Discussion
3.1. Phonon Spectrum and Band Structure
ID-GY crystals in a tetragonal unit cell with the lattice constants of a = b = 12.14 Å containing 32 carbon and 8 nitrogen atoms in the unit cell with the space group symmetry of P4/mbm (No. 127). Chemically nonequivalent atoms are marked in Figure 1a, where C1, C2, and N atoms are in sp2 hybridization while C3, C4, and C5 atoms are in sp hybridization. Unlike γ-graphyne, composed of hexagonal units, ID-GY is composed of pentagonal units connected by acetylenic linkers. Compared with the highly symmetric γ-graphyne, the complex geometric structure and hybridized bonding in ID-GY make it a promising material with a low lattice thermal conductivity, just like graphyne and graphdiyne [29,30].
The phonon dispersion spectrum of ID-GY along the high symmetry k-point path (Γ-X-M-Γ) in the first Brillouin zone is shown in Figure 1c. All the vibrational modes are real in the entire Brillouin zone, confirming that ID-GY is dynamically stable. Since both carbon and nitrogen are light atoms, the highest frequency in the phonon spectrum reaches 67 THz. In addition, one can see that there is a large phonon band gap (about 15 THz) in the high-frequency region, and the corresponding phonon density of states (PhDOS) (Figure 1d), indicate that the high frequency can be attributed to the C3 and C4 atoms. The bond length of C3-C4 is 1.23 Å, showing the characteristics of alkyne bonds. Because the large portion of heat is carried by low-frequency phonons, especially the acoustic phonons, the low-frequency region of the phonon spectrum is magnified, and the acoustic phonon branches are highlighted in red. The longitudinal acoustic (LA) and transverse acoustic (TA) branches of ID-GY are linear when the wave vector q is close to the Γ point, while the out-of-plane acoustic (ZA) branch exhibits parabolic dispersion, which is a characteristic of monolayer 2D materials [31]. The highest frequency of the acoustic phonon is relatively low (<5 THz), lower than that of γ-graphyne (about 8 THz) [14]. The low frequency of the acoustic phonons is associated with a low acoustic Debye temperature, as discussed in the following paragraph. Moreover, there is a strong overlap between the acoustic and low-frequency optical branches. These characteristics indicate that the lattice thermal conductivity of ID-GY might be low.
To examine the mechanical stability of ID-GY, the elastic constants were calculated and are listed in Table 1. It is obvious that ID-GY satisfies the Born–Huang criteria [18,32] for 2D tetragonal materials—namely, C11 > 0, C66 > 0 and C11 > C12. The Young’s modulus Y, Poisson’s ratio ν, bulk modulus B, and shear modulus G were also calculated and are presented in Table 1. It was found that the stiffness (122.20 N/m) of ID-GY is only half that of graphene (342 N/m) [33], owing to weak in-plane bonds. Moreover, the sound velocity, which is usually used to measure the speed of phonons propagating through the lattice, can be determined from bulk modulus B and shear modulus G by the following formulas [34]: longitudinal sound velocity vl = , transverse sound velocity vt = , and average sound velocity vs. = 1/, where ρ is the mass density. Based on the sound velocity, we obtained the Debye temperature using θD = , where N is the number of atoms in the cell and S is the area of the unit cell. Debye temperature measures the temperature above which all modes begin to be excited; therefore, a high θD indicates weak three-phonon scattering and hence a high kl. The calculated Debye temperature of ID-GY is 647 K, which is much lower than the corresponding value of 805 K of γ-graphyne. Consequently, it is natural to expect that ID-GY possesses a lower lattice thermal conductivity than γ-graphyne.
To study the electrical transport property, we calculated the band structure of ID-GY. As shown in Figure 1b, ID-GY exhibits semiconducting electronic features with a direct bandgap value of 1.10 eV. Compared with the bandgap (0.47 eV) of γ-graphyne [14], the larger bandgap can effectively overcome the high-temperature bipolar conduction problem, benefitting thermoelectric performance [35]. Moreover, the sharp conduction band and valence band around the Γ point suggest low carrier effective masses and a possible large carrier mobility. It is worth noting that the valence band maximum (VBM) is doubly degenerated, leading to a sharp density of states (DOS). The high degeneracy of the valence band and the sharp DOS would enhance the Seebeck coefficient of p-type ID-GY, as is the case with bilayer MoS2 [36].
3.2. Thermal Transport Properties
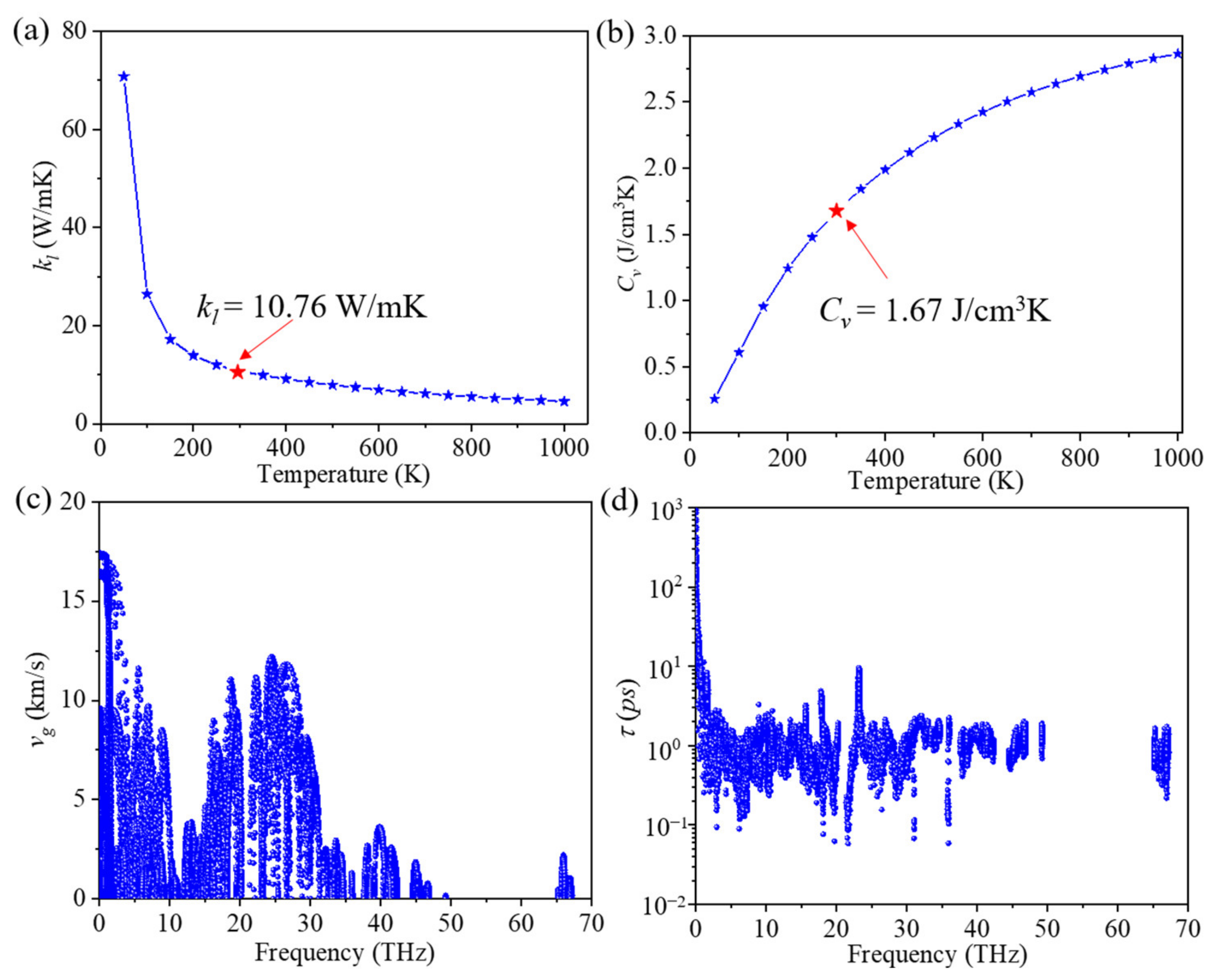
The lattice thermal conductivity (kl) of ID-GY was calculated for different temperatures. As shown in Figure 2a, the lattice thermal conductivity of ID-GY is 10.76 W/mK at 300 K, which is two orders of magnitude lower than that of graphene (3151.53 W/mK) [10] and much lower than that of many other 2D carbon hexagonal structures, including α-graphyne (21.11 W/mK) [14], β-graphyne (22.3 W/mK) [14], γ-graphyne (106.24 W/mK) [14], and graphdiyne (22.3 W/mK) [29], at the same temperature. This shows the importance of structural units in affecting the thermal conductivity of a material. We fitted the relationship of kl with temperature and found that kl is proportional to 1/T1.05, indicating that the three-phonon scattering is dominant in ID-GY, as is the case with graphene [38] and penta-graphene [15]. This was further confirmed by comparing the scattering rates of three-phonon scattering with those of the isotopic scattering process. The calculated results for these two scattering processes are plotted in Figure S1 in the Supplementary Materials. We found that the three-phonon scattering rates are nearly 100 times larger than the isotopic scattering ones. The calculated cumulative kl as a function of frequency is plotted in Figure S2, which shows that the phonons with frequencies lower than 20 THz contribute about 85% to the lattice thermal conductivity. Therefore, we focus on the low-frequency phonon branches (<20 THz) in the following discussion.
It is important to understand the reasons for the low kl of ID-GY. kl can be expressed in the following form through the summation of the contribution of all of the phonon modes λ(q, j) with the wave vector q and branch index j:
where α and β denote the three directions (x, y, or z) and λ is the phonon mode consisting of both wave vector q and branch index j. , , and represent the phonon volumetric-specific heat, group velocity, and phonon lifetime, respectively.
Our calculated phonon specific heat value of ID-GY is 1.67 J/cm3K at 300 K. The variation in the phonon volumetric-specific heat with temperature is plotted in Figure 2b. It is worth mentioning that the phonon-specific heat value usually is not different from one material to another [39]. For instance, the phonon-specific heat value of γ-GY is 1.68 J/cm3K [29], which is almost same as that of ID-GY. The change in group velocity of ID-GY with frequency is given in Figure 2c, which shows that at the long-wavelength limit, the group velocity reaches the highest value of 17.5 km/s, close to that of γ-GY (~17.9 km/s) [14]. In the low-frequency region (below 20 THz), the overall group velocity is only slightly lower than that of γ-GY. The variation in phonon lifetime with frequency at room temperature is plotted in Figure 2d. The lifetime for most low-frequency phonon modes (0~20 THz) is about 2 ps, while that for γ-GY is larger than 10 ps [14]. Therefore, the short phonon lifetime in ID-GY is the main reason for the low lattice thermal conductivity.
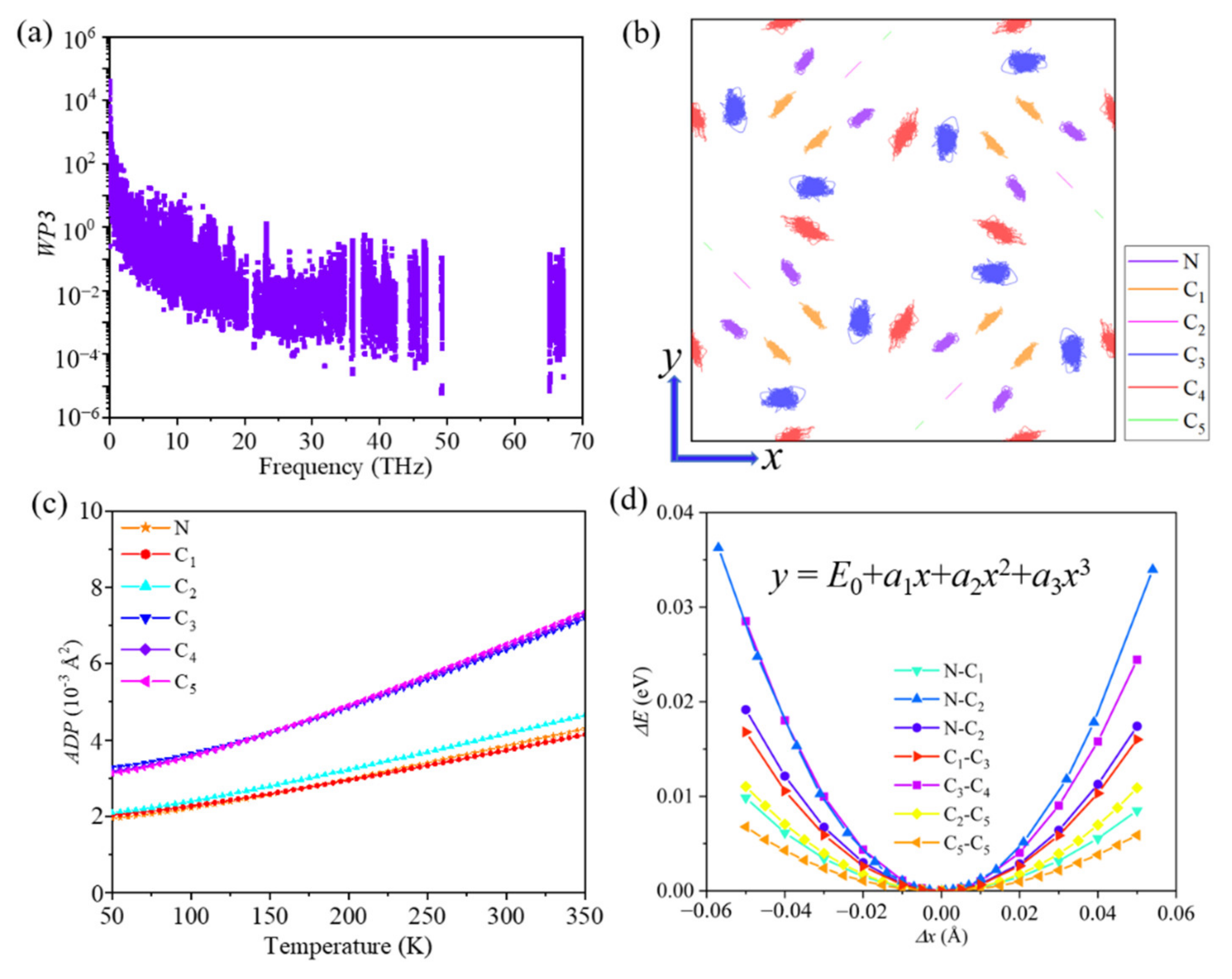
A short phonon lifetime is usually associated with strong phonon scattering. Therefore, we studied the phonon scatterings by carrying out additional calculations based on the scattering mechanism of phonon modes. Three-phonon scattering is the dominant process in the heat transport process and usually depends on two factors: the number of existing scattering channels and the strength of each scattering channel, which can be described by weighted phase space (WP3) and mode Grüneisen parameter (γ). The weighted phase space of each phonon mode with respect to phonon frequency is plotted in Figure 3a. We found that the value of the WP3 is relatively large, which results from the large number of atoms per unit cell and the complex geometric structure of ID-GY. Moreover, the Grüneisen parameter, which quantifies the intensity of anharmonic interactions between the phonon branches, is another key factor in determining the phonon lifetime of a system. The average Grüneisen parameter of ID-GY was calculated to be 1.51, which is comparable to that of traditional thermoelectric material PbTe (1.65) [40], suggesting the existence of a strong anharmonicity in ID-GY.
To find the origin of the strong anharmonicity from the point of view of geometric structure, we plotted the simulated trajectory of atoms in the xy-plane at 800 K during an ab initio molecular dynamics (AIMD) simulation. As shown in Figure 3b, the C3 and C4 atoms vibrate strongly around their equilibrium positions. The atomic trajectory shows that -C3C4- pairs in acetylenic linkers are weakly bonded to the pentagonal rings. These conclusions are further confirmed by the atomic displacement parameter (ADP) and the bond energy curve, which can provide a visualization of the anharmonicity. As shown in Figure 3c, the ADPs of the C3 and C4 atoms are much larger. The bond energy curve shows the vibration of a relative energy change ΔE (in eV/per atom) when the bond length changes and reveals the phonon anharmonicity. By fitting the bond energy curve (Figure 3d), we obtain the anharmonic parameters (a3) for different type of bonds in ID-GY. The data in Table 2 show that the strong anharmonicity in ID-GY mainly originates from the -C3C4- pairs, which are weakly bonded to the pentagonal rings. The single N-C4 and C1-C3 bonds are too weak to yield an inefficient thermal transport by lattice vibration. The inhomogeneous bond environment and large lattice vibrational mismatch between the pentagonal rings and the acetylenic linkers hinder the transport of heat.
3.3. Electrical Transport Properties
ID-GY possesses an appropriate band gap and a low lattice thermal conductivity; therefore, it could be a high-performance thermoelectric material. To study the thermoelectric performance of ID-GY, we calculated its electrical transport properties. To this end, the Seebeck coefficient (S) was calculated using the BoltzTraP2 software [26]. As shown in Figure 4a, the S can reach a peak of 1150 μV/K at 300 K, which is much larger than that of γ-graphyne (690 μV/K) [13] owing to the larger bandgap and doubly degenerate valence bands of ID-GY. To obtain the electronic conductivity, the carrier relaxation time (τ) is necessary, which can be expressed as:
where μ is the carrier mobility, C is the in-plane elastic constant, and E1 is the deformation-potential constant. The effective mass of carrier m* was calculated from the curvature of the conduction band minimum or valence band maximum by the parabolic fitting of the band edge using the formula m* = . These calculated results are summarized in Table 3.
As expected from the band structure, the effective mass of the carrier would be very low. The calculated value is indeed only 0.11~0.25 me, indicating that ID-GY could have a considerably high carrier mobility. The obtained carrier mobilities of ID-GY are 7933 and 1826 cm2/Vs for electrons and holes, respectively; accordingly, the electron relaxation time (τe) is much longer than the hole relaxation time (τh). It is worth noting that although the deformation potential approximation [41] has been widely used for predicting the carrier mobility of new thermoelectric materials [42,43], the carrier mobility is usually overestimated as compared to the experimental result due to the neglect of scattering between the carrier and either the defect or the substrate [44]. Conversely, these scattering processes can also reduce the thermal conductivity. Thus, the overestimated electrical conductivity and overestimated thermal conductivity may cancel each other out to some extent, resulting in a more reliable prediction.
Based on the carrier relaxation time, the electrical conductivity (σ) is obtained and presented in Figure 4b. The electronic conductivity of n-type ID-GY is higher than that of p-type ID-GY due to the longer electron relaxation time. We note that the absolute values of the Seebeck coefficient and electrical conductivity show opposite trends when the chemical potential changes. Thus, to obtain a good power factor (PF = S2σ), an optimum chemical potential is needed. The maximum PF value for p-type ID-GY is 22.14 mW/mK2 at 300 K, while that for p-type ID-GY is 20.18 mW/mK2.
Thermal conductivity is the sum of the lattice thermal conductivity and electronic thermal conductivity. The former is described in Equation (1) and the latter follows the Wiedemann–Frranz law ke = LσT, where the Lorenz number L is equal to 2.44 × 10−8 WΩ/K2 [45]. As shown in Figure 4d, electronic thermal conductivity has a similar tendency to electrical conductivity.
Finally, the thermoelectric performance of ID-GY was evaluated using the ZT value. The variations in our calculated ZT values with chemical potential μ at different temperatures are plotted in Figure 5. At 300 K, the optimized ZT values of ID-GY can reach 0.46 and 0.38 for p-type and n-type with the hole- and electron-doping concentrations of 5.8 × 1012 and 1.75 × 1012 cm−2, respectively, which are higher than the ZT values of many other 2D carbon materials [30], including graphene (0.01), α-graphyne (0.03), β-graphyne (0.12), γ-graphyne (0.17), and 6,6,12-graphyne (0.05). While at 800 K, the ZT values are 2.20 and 2.21 for p-type and n-type ID-GY.
4. Conclusions
In this work, based on first principle calculations, we investigated the thermal transport and thermoelectric properties of ID-GY, a new pentagon-based 2D material that is constructed by assembling an experimentally synthesized pentagonal imidazole molecule and acetylenic linkers. We showed that the thermoelectric properties of graphyne are significantly improved by changing its structural unit to a pentagonal imidazole molecule. The lattice thermal conductivity was decreased from 106.24 W/mK for γ-graphyne to 10.76 W/mK for ID-GY at 300 K, while the Seebeck coefficient was increased to 1150 V/K for ID-GY from 690 μV/K for γ-graphyne at room temperature. The mechanism of the low lattice thermal conductivity was further studied by analyzing the group velocity, scattering rate, weighted phase space, and bond energy curve. The mismatched lattice vibration between the pentagonal rings and the linkers in ID-GY resulted in a strong anharmonicity. Moreover, as compared to γ-graphyne, the doubly degenerated VBM and larger band gap resulted in a higher Seebeck coefficient in ID-GY. The calculated thermoelectric figure of merit at 300 K was 0.46, suggesting the effectiveness of using the pentagonal structural unit for enhancing the thermoelectric performance of carbon-based materials.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/ma14195604/s1, Figure S1: the isotopic scattering rates of ID-GY, Figure S2: viaration of the normalized cumulative lattice thermal conductivity with frequency for ID-GY.
Author Contributions
Calculation, Y.C.; formal analysis, Y.C., J.S. and W.K.; writing—original draft preparation, Y.C.; writing—review and editing, Q.W. and J.S.; supervision, Q.W.; project administration, Q.W.; funding acquisition, Q.W. and W.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by grants from the National Key Research and Development Program of the Ministry of Science and technology of China (Grant No. 2017YFA0205003) and the National Natural Science Foundation of China (Grants Nos. NSFC-11974028 and NSFC-21773004). It is also supported by the high-performance computing platform of Peking University, China.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
This work is partially supported by grants from the National Key Research and Development Program of the Ministry of Science and Technology of China (Grant No. 2017YFA0205003) and the National Natural Science Foundation of China (Grants Nos. NSFC-11974028 and NSFC-21773004). It is also supported by the high-performance computing platform of Peking University, China.
Conflicts of Interest
There are no conflicts to declare.
References
- Poudel, B.; Hao, Q.; Ma, Y.; Lan, Y.; Minnich, A.; Yu, B.; Yan, X.; Wang, D.; Muto, A.; Vashaee, D.; et al. High-thermoelectric performance of nanostructured bismuth antimony telluride bulk alloys. Science 2008, 320, 634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heremans, J.P.; Jovovic, V.; Toberer, E.S.; Saramat, A.; Kurosaki, K.; Charoenphakdee, A.; Yamanaka, S.; Snyder, G.J. Enhancement of thermoelectric efficiency in PbTe by distortion of the electronic density of states. Science 2008, 321, 554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudryashov, A.A.; Kytin, V.G.; Lunin, R.A.; Kulbachinskii, V.A.; Banerjee, A. Effect of thallium doping on the mobility of electrons in Bi2Se3 and holes in Sb2Te3. Semic 2016, 50, 869–875. [Google Scholar] [CrossRef]
- Balandin, A.A. Thermal properties of graphene and nanostructured carbon materials. Nat. Mater. 2011, 10, 569–581. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, Q.; Chen, G. Carbon and carbon composites for thermoelectric applications. Carbon Energy 2020, 2, 408–436. [Google Scholar] [CrossRef]
- Cho, C.; Wallace, K.L.; Tzeng, P.; Hsu, J.; Yu, C.; Grunlan, J.C. Outstanding low temperature thermoelectric power factor from completely organic thin films enabled by multidimensional conjugated nanomaterials. Adv. Energy Mater. 2016, 6, 1502168. [Google Scholar] [CrossRef]
- Blackburn, J.L.; Ferguson, A.J.; Cho, C.; Grunlan, J.C. Carbon-nanotube-based thermoelectric materials and devices. Adv. Mater. 2018, 30, 1704386. [Google Scholar] [CrossRef]
- Zhao, W.; Fan, S.; Xiao, N.; Liu, D.; Tay, Y.Y.; Yu, C.; Sim, D.; Hng, H.H.; Zhang, Q.; Boey, F.; et al. Flexible carbon nanotube papers with improved thermoelectric properties. Energy Environ. Sci. 2012, 5, 5364–5369. [Google Scholar] [CrossRef]
- Yao, Q.; Wang, Q.; Wang, L.; Chen, L. Abnormally enhanced thermoelectric transport properties of SWNT/PANI hybrid films by the strengthened PANI molecular ordering. Energy Environ. Sci. 2014, 7, 3801–3807. [Google Scholar] [CrossRef]
- Yue, S.; Qin, G.; Zhang, X.; Sheng, X.; Su, G.; Hu, M. Thermal transport in novel carbon allotropes with sp2 or sp3 hybridization: An ab initio study. Phys. Rev. B 2017, 95, 085207. [Google Scholar] [CrossRef]
- Zuev, Y.M.; Chang, W.; Kim, P. Thermoelectric and magnetothermoelectric transport measurements of graphene. Phys. Rev. Lett. 2009, 102, 096807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Li, Y.; Chen, Y.; Wu, L.; Yang, C.; Cui, X. Synthesis of γ-graphyne by mechanochemistry and its electronic structure. Carbon 2018, 136, 248–254. [Google Scholar] [CrossRef]
- Jiang, P.H.; Liu, H.J.; Cheng, L.; Fan, D.D.; Zhang, J.; Wei, J.; Liang, J.H.; Shi, J. Thermoelectric properties of γ-graphyne from first-principles calculations. Carbon 2017, 113, 108–113. [Google Scholar] [CrossRef]
- Yang, X.; Dai, Z.; Zhao, Y.; Meng, S. Phonon thermal transport in a class of graphene allotropes from first principles. Phys. Chem. Chem. Phys. 2018, 20, 15980–15985. [Google Scholar] [CrossRef]
- Wang, F.Q.; Yu, J.; Wang, Q.; Kawazoe, Y.; Jena, P. Lattice thermal conductivity of penta-graphene. Carbon 2016, 105, 424–429. [Google Scholar] [CrossRef]
- Sun, J.; Chen, Y.; Wang, Q. Low lattice thermal conductivity of a 5-8-peanut-shaped carbon nanotube. Phys. Chem. Chem. Phys. 2021, 23, 5460–5466. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, J.; Li, T.; Wang, Q. Low lattice thermal conductivity of pentadiamond. J. Appl. Phys. 2021, 129, 215107. [Google Scholar] [CrossRef]
- Zhou, W.; Guo, Y.; Shen, Y.; Wang, Q.; Jena, P. Imidazole-graphyne: A new 2D carbon nitride with a direct bandgap and strong IR refraction. Phys. Chem. Chem. Phys. 2021, 23, 10274–10280. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.F.; Storey, C. Efficient generalized conjugate gradient algorithms, part 2: Implementation. J. Optim. Theory Appl. 1991, 69, 139–152. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Madsen, G.K.H.; Carrete, J.; Verstraete, M.J. BoltzTraP2, a program for interpolating band structures and calculating semi-classical transport coefficients. Comput. Phys. Commun. 2018, 231, 140–145. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Carrete, J.; AKatcho, N.; Mingo, N. ShengBTE: A solver of the Boltzmann transport equation for phonons. Comput. Phys. Commun. 2014, 185, 1747–1758. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.; Shao, H.; Hu, T.; Liu, G.; Jiang, J.; Jiang, H. High thermoelectric performance in two-dimensional graphyne sheets predicted by first-principles calculations. Phys. Chem. Chem. Phys. 2015, 17, 22872–22881. [Google Scholar] [CrossRef]
- Sevinçli, H.; Sevik, C. Electronic, phononic, and thermoelectric properties of graphyne sheets. Appl. Phys. Lett. 2014, 105, 223108. [Google Scholar] [CrossRef] [Green Version]
- Şahin, H.; Cahangirov, S.; Topsakal, M.; Bekaroglu, E.; Akturk, E.; Senger, R.T.; Ciraci, S. Monolayer honeycomb structures of group-IV elements and III-V binary compounds: First-principles calculations. Phys. Rev. B 2009, 80, 155453. [Google Scholar] [CrossRef] [Green Version]
- Born, M.; Huang, K. Dynamical Theory of Crystal Lattices; Oxford University Press: Oxford, UK, 1954. [Google Scholar]
- Peng, B.; Zhang, H.; Shao, H.; Xu, Y.; Ni, G.; Zhang, R.; Zhu, H. Phonon transport properties of two-dimensional group-IV materials from ab initio calculations. Phys. Rev. B 2016, 94, 245420. [Google Scholar] [CrossRef] [Green Version]
- Zhuo, Y.; Mansouri Tehrani, A.; Oliynyk, A.O.; Duke, A.C.; Brgoch, J. Identifying an efficient, thermally robust inorganic phosphor host via machine learning. Nat. Commun. 2018, 9, 4377. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.Q.; Zhang, S.; Yu, J.; Wang, Q. Thermoelectric properties of single-layered SnSe sheet. Nanoscale 2015, 7, 15962–15970. [Google Scholar] [CrossRef]
- Hong, J.; Lee, C.; Park, J.-S.; Shim, J.H. Control of valley degeneracy in MoS2 by layer thickness and electric field and its effect on thermoelectric properties. Phys. Rev. B 2016, 93, 035445. [Google Scholar] [CrossRef] [Green Version]
- Asadpour, M.; Malakpour, S.; Faghihnasiri, M.; Taghipour, B. Mechanical properties of two-dimensional graphyne sheet, analogous system of BN sheet and graphyne-like BN sheet. Solid State Commun. 2015, 212, 46–52. [Google Scholar] [CrossRef]
- Choudhry, U.; Yue, S.; Liao, B. Origins of significant reduction of lattice thermal conductivity in graphene allotropes. Phys. Rev. B 2019, 100, 165401. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Varshney, V.; Lee, J.; Zhang, T.; Wohlwend, J.L.; Roy, A.K.; Luo, T. Hydrogenation of penta-graphene leads to unexpected large improvement in thermal conductivity. Nano Lett. 2016, 16, 3925–3935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Chang, C.; Pei, Y.; Wu, D.; Peng, K.; Zhou, X.; Gong, S.; He, J.; Zhang, Y.; Zeng, Z.; et al. Origin of low thermal conductivity in SnSe. Phys. Rev. B 2016, 94, 125203. [Google Scholar] [CrossRef]
- Bardeen, J.; Shockley, W. Deformation potentials and mobilities in non-polar crystals. Phys. Rev. 1950, 80, 72–80. [Google Scholar] [CrossRef]
- Gao, Z.; Wang, J. Thermoelectric penta-silicene with a high room-temperature figure of merit. ACS Appl. Mater. Interfaces 2020, 12, 14298–14307. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Chen, X.; Hu, C.; Cheng, Y.; Chen, Q. Penta-PdX2 (X = S, Se, Te) monolayers: Promising anisotropic thermoelectric materials. J. Mater. Chem. A 2019, 7, 11134–11142. [Google Scholar] [CrossRef]
- Poljak, M.; Suligoj, T.; Wang, K.L. Influence of substrate type and quality on carrier mobility in graphene nanoribbons. J. Appl. Phys. 2013, 114, 053701. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Li, Z.; Duan, W. Thermal and thermoelectric properties of graphene. Small 2014, 10, 2182–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
(a) Geometric structure, (b) electronic band structure, (c) phonon spectrum, and (d) partial phonon density of states (PhDOS) of ID-GY.
Figure 1.
(a) Geometric structure, (b) electronic band structure, (c) phonon spectrum, and (d) partial phonon density of states (PhDOS) of ID-GY.

Figure 2.
(a) Lattice thermal conductivity (kl), (b) phonon volumetric-specific heat (Cv), (c) group velocity (vg), and (d) phonon lifetime (τ) of ID-GY.
Figure 2.
(a) Lattice thermal conductivity (kl), (b) phonon volumetric-specific heat (Cv), (c) group velocity (vg), and (d) phonon lifetime (τ) of ID-GY.

Figure 3.
(a) Weighted phase space (WP3), (b) trajectory of the atoms in the xy-plane from ab initio molecular dynamics simulations at 800 K, (c) atomic displacement parameter (ADP), and (d) bond energy curves of ID-GY.
Figure 3.
(a) Weighted phase space (WP3), (b) trajectory of the atoms in the xy-plane from ab initio molecular dynamics simulations at 800 K, (c) atomic displacement parameter (ADP), and (d) bond energy curves of ID-GY.

Figure 4.
(a) Seebeck coefficient (S), (b) electrical conductivity (σ), (c) power factor (PF), and (d) electronic thermal conductivity (ke) of ID-GY as a function of chemical potential μ, respectively.
Figure 4.
(a) Seebeck coefficient (S), (b) electrical conductivity (σ), (c) power factor (PF), and (d) electronic thermal conductivity (ke) of ID-GY as a function of chemical potential μ, respectively.

Figure 5.
Variation in the thermoelectric figure of merit (ZT) of ID-GY with the chemical potential (μ).
Figure 5.
Variation in the thermoelectric figure of merit (ZT) of ID-GY with the chemical potential (μ).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Calculated elastic coefficients Cij (in N/m), Young’s modulus Y (in N/m), Poisson’s ratio ν, bulk modulus B (in N/m), shear modulus G (in N/m), longitudinal sound velocity vl (in km/s), transverse sound velocity vt (in km/s), average sound velocity vs. (in km/s), and Debye temperature θD (in K) for ID-GY. For comparison, the corresponding values for γ-graphyne (γ-GY) are also listed here.
Table 1.
Calculated elastic coefficients Cij (in N/m), Young’s modulus Y (in N/m), Poisson’s ratio ν, bulk modulus B (in N/m), shear modulus G (in N/m), longitudinal sound velocity vl (in km/s), transverse sound velocity vt (in km/s), average sound velocity vs. (in km/s), and Debye temperature θD (in K) for ID-GY. For comparison, the corresponding values for γ-graphyne (γ-GY) are also listed here.
| C11 | C12 | C66 | Y | ν | B | G | vl | vt | vs | θD | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ID-GY | 164.26 | 83.12 | 12.79 | 122.20 | 0.51 | 124.69 | 40.46 | 17.17 | 8.5 | 4.59 | 647 |
| γ-GY | - | - | - | - | 0.41 * | 122.73 * | 77.04 * | 18.53 | 11.51 | 5.50 | 805 |
* Data from ref. [37].
Table 2.
Fitted anharmonic parameters (a3) of the bond energy curves for ID-GY.
| C3-C4 | N-C4 | N-C1 | C1-C3 | C5-C5 | N-C2 | C2-C5 | |
|---|---|---|---|---|---|---|---|
| a3 | 15.41 | 7.13 | 5.90 | 4.28 | 3.28 | 0.68 | 0.60 |
Table 3.
In-plane elastic constant C (in N/m), deformation-potential constant E1 (in eV), effective mass of carrier m* (in me), carrier mobility μ (in cm2/Vs), and carrier relaxation time τ (in 10−14 s) of ID-GY at 300 K.
Table 3.
In-plane elastic constant C (in N/m), deformation-potential constant E1 (in eV), effective mass of carrier m* (in me), carrier mobility μ (in cm2/Vs), and carrier relaxation time τ (in 10−14 s) of ID-GY at 300 K.
| Carrier Type | C | E1 | m* | μ | τ |
|---|---|---|---|---|---|
| electron | 164.26 | 4.94 | 0.11 | 7932.54 | 49.56 |
| hole | 164.26 | 4.53 | 0.25 | 1826.31 | 25.93 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chen, Y.; Sun, J.; Kang, W.; Wang, Q. Phonon Transport and Thermoelectric Properties of Imidazole-Graphyne. Materials 2021, 14, 5604. https://0-doi-org.brum.beds.ac.uk/10.3390/ma14195604
AMA Style
Chen Y, Sun J, Kang W, Wang Q. Phonon Transport and Thermoelectric Properties of Imidazole-Graphyne. Materials. 2021; 14(19):5604. https://0-doi-org.brum.beds.ac.uk/10.3390/ma14195604
Chicago/Turabian StyleChen, Yanyan, Jie Sun, Wei Kang, and Qian Wang. 2021. "Phonon Transport and Thermoelectric Properties of Imidazole-Graphyne" Materials 14, no. 19: 5604. https://0-doi-org.brum.beds.ac.uk/10.3390/ma14195604
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.