
Enabling Conducting Polymer Applications: Methods for Achieving High Molecular Weight in Chemical Oxidative Polymerization in Alkyl- and Ether-Substituted Thiophenes

 , ,
, ,
Abstract
: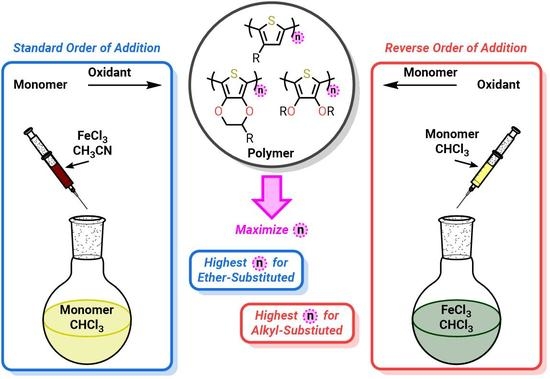
1. Introduction

{kind=link}
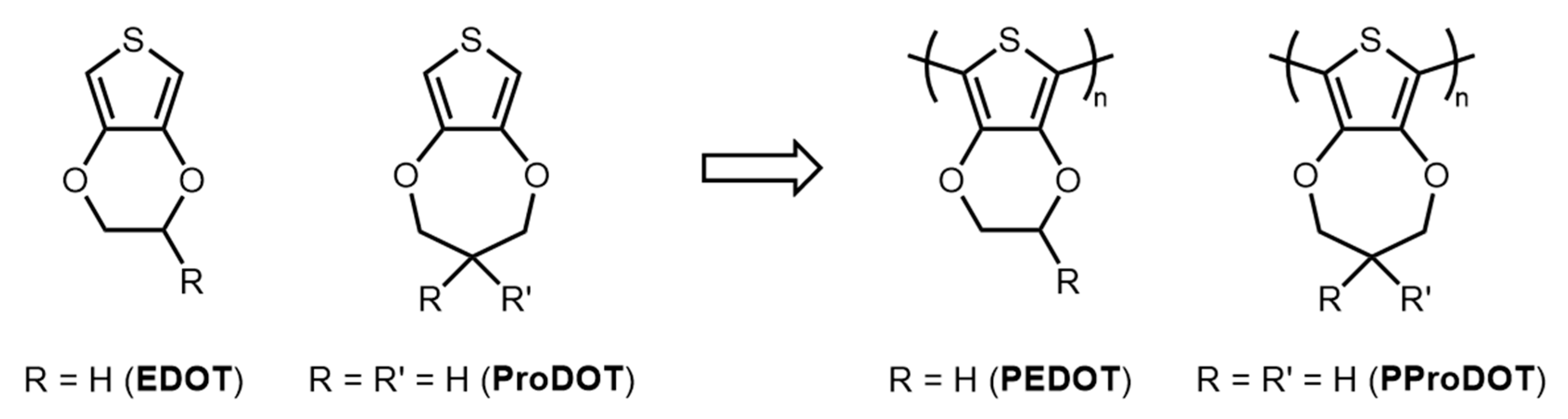{kind=link}
{kind=link}
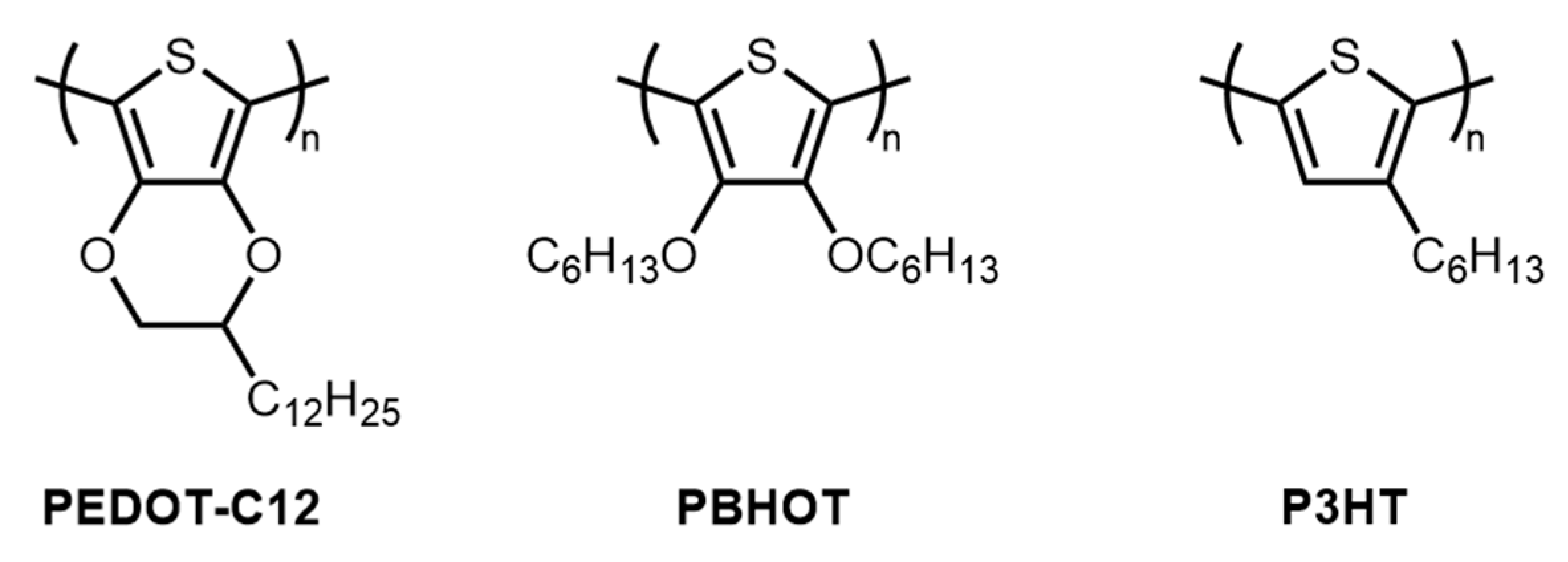{kind=link}
{kind=link}
{kind=link}
| Entry | Structure | Acronym | Equivalents FeCl3 | Mw 2 | Xw 3 | Ref. |
|---|---|---|---|---|---|---|
| 1 |  | P3HT | 2 | 140,000 | 842 | [16] |
| 2 | 4 | 110,700 | 666 | [18] | ||
| 3 | 4 | 411,000 | 2472 | [19] | ||
| 4 |  | P3OT | 4 | 181,440 | 933 | [37] |
| 5 |  | P3DT | 4 | 303,050 | 1362 | [37] |
| 6 |  | PEDOT-C14 | 2 | 11,200 | 33 | [26] |
| 7 | 4 | 22,500 | 67 | [26] | ||
| 8 |  | PBPOT | 4 | 9743 | 38 | [38] |
| 9 |  | PBOOT | 4 | 11,528 | 38 | [38] |
2. Materials and Methods
2.1. General
2.2. Monomer Synthesis
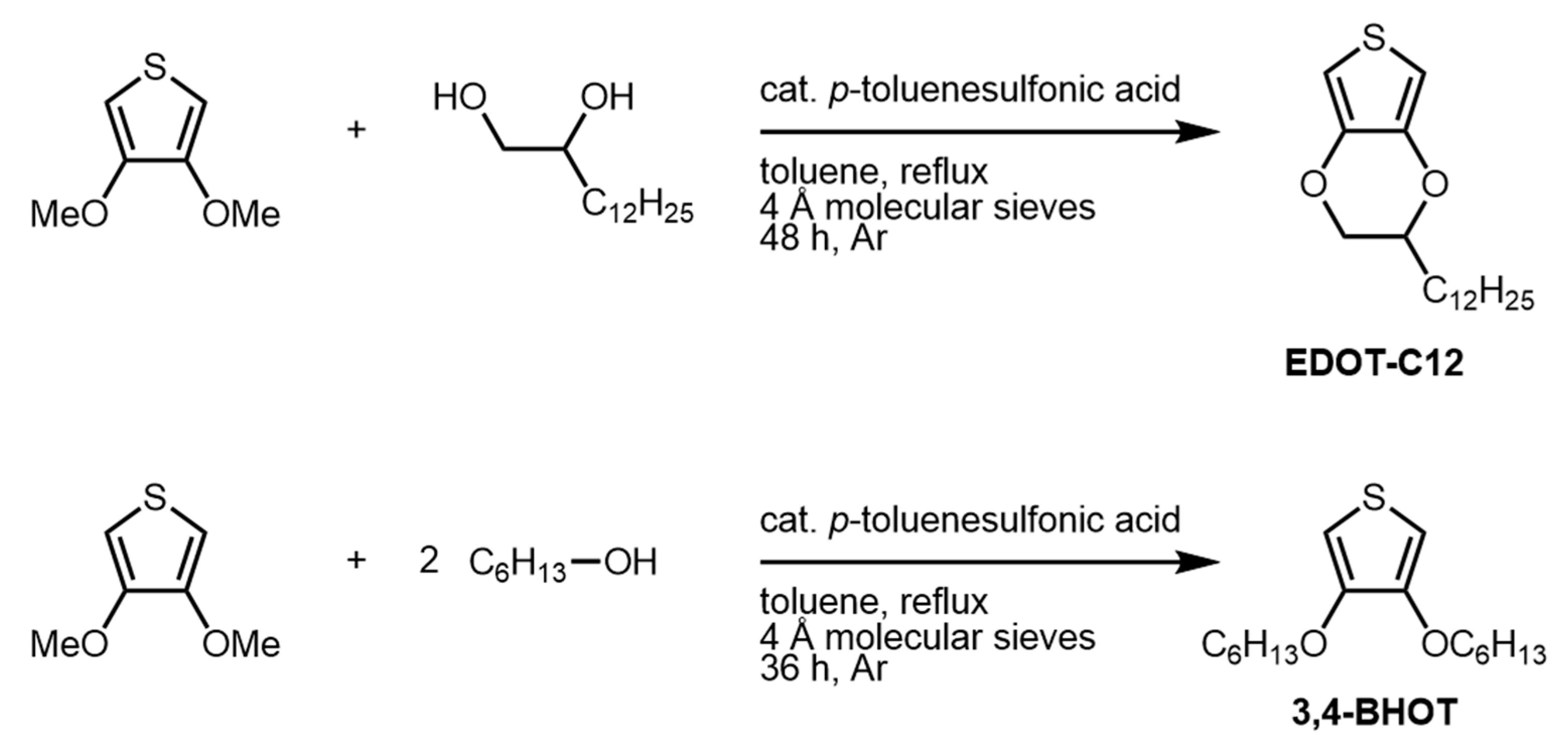
2.2.1. Synthesis of EDOT-C12
2.2.2. Synthesis of 3,4-BHOT
2.3. Polymerizations
2.3.1. General Procedure—Reverse Addition Oxidative Polymerization
2.3.2. PEDOT-C12—Reverse Addition, 2.3 Equivalents FeCl3 (Table 3, Entry 1)
2.3.3. PEDOT-C12—Reverse Addition, 2.3 Equivalents FeCl3 in Chloroform (Table 3, Entry 2)
2.3.4. PBHOT—Reverse Addition, 2.3 Equivalents FeCl3 (Table 3, Entry 5)
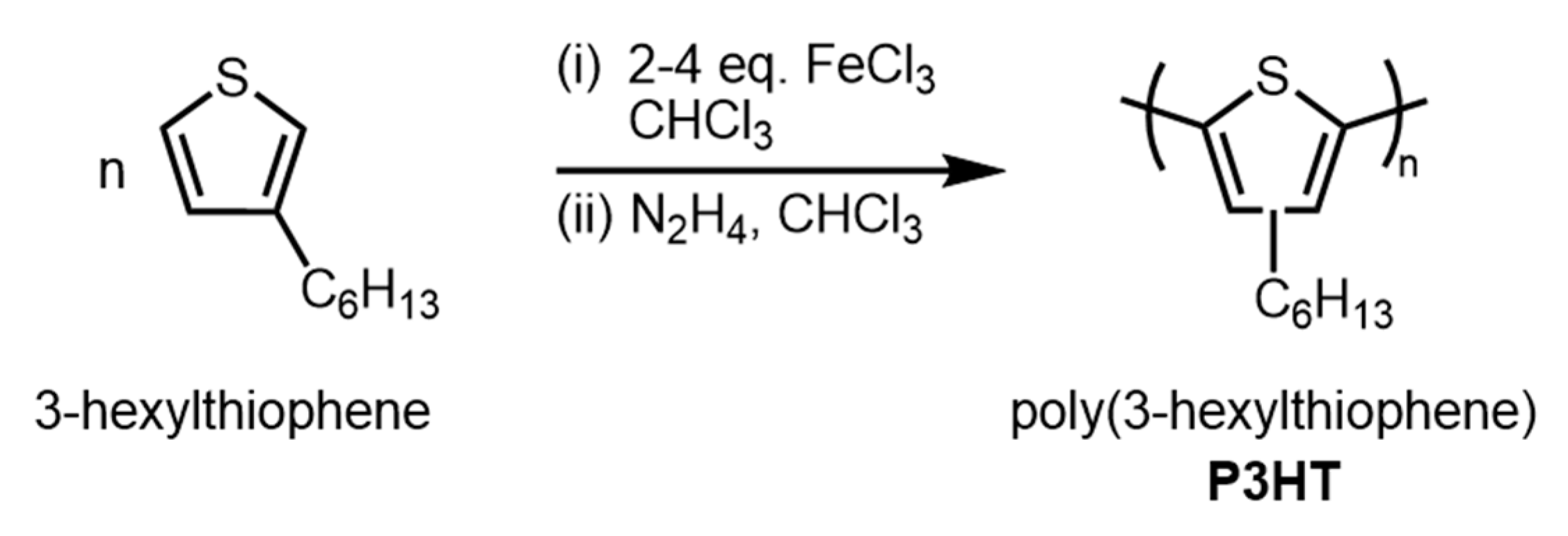
2.3.5. P3HT—Reverse Addition, 2.3 Equivalents FeCl3 (Table 3, Entry 9)
2.3.6. General Procedure—Standard Addition Oxidative Polymerization
2.3.7. PEDOT-C12—Standard Addition, 2.3 Equivalents FeCl3 (Table 3, Entry 3)
2.3.8. PEDOT-C12—Standard Addition, 4 Equivalents FeCl3 (Table 3, Entry 4)
2.3.9. PBHOT—Standard Addition, 2.3 Equivalents FeCl3 (Table 3, Entry 6)
2.3.10. PBHOT—Standard Addition, 4 Equivalents FeCl3 (Table 3, Entry 7)
2.3.11. PBHOT—Standard Addition, 4 Equivalents FeCl3 48 h (Table 3, Entry 8)
2.3.12. P3HT—Standard Addition, 2.3 Equivalents FeCl3 (Table 3, Entry 10)
2.3.13. P3HT—Standard Addition, 4 Equivalents FeCl3 (Table 3, Entry 11)
2.3.14. P3HT—Standard Addition, 4 Equivalents FeCl3 48 h (Table 3, Entry 12)
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tourillon, G.; Garnier, F. New electrochemically generated organic conducting polymers. J. Electroanal. Chem. 1982, 135, 173–178. [Google Scholar] [CrossRef]
- Kaloni, T.P.; Giesbrecht, P.K.; Schreckenbach, G.; Freund, M.S. Polythiophene: From Fundamental Perspectives to Applications. Chem. Mater. 2017, 29, 10248–10283. [Google Scholar] [CrossRef]
- Zhang, M.; Guo, X.; Ma, W.; Ade, H.; Hou, J. A polythiophene derivative with superior properties for practical application in polymer solar cells. Adv. Mater. 2014, 26, 5880–5885. [Google Scholar] [CrossRef]
- Yu, C.Y.; Ko, B.T.; Ting, C.; Chen, C.P. Two-dimensional regioregular polythiophenes with conjugated side chains for use in organic solar cells. Sol. Energy Mater. Sol. Cells 2009, 93, 613–620. [Google Scholar] [CrossRef]
- Zen, A.; Pflaum, J.; Hirschmann, S.; Zhuang, W.; Jaiser, F.; Asawapirom, U.; Rabe, J.P.; Scherf, U.; Neher, D. Effect of molecular weight and annealing of poly(3-hexylthiophene)s on the performance of organic field-effect transistors. Adv. Funct. Mater. 2004, 14, 757–764. [Google Scholar] [CrossRef]
- Huynh, T.P.; Sharma, P.S.; Sosnowska, M.; D’Souza, F.; Kutner, W. Functionalized polythiophenes: Recognition materials for chemosensors and biosensors of superior sensitivity, selectivity, and detectability. Prog. Polym. Sci. 2015, 47, 1–25. [Google Scholar] [CrossRef]
- Perepichka, I.F.; Perepichka, D.F.; Meng, H.; Wudl, F. Light-emitting polythiophenes. Adv. Mater. 2005, 17, 2281–2305. [Google Scholar] [CrossRef]
- Sun, K.; Zhang, S.; Li, P.; Xia, Y.; Zhang, X.; Du, D.; Isikgor, F.H.; Ouyang, J. Review on application of PEDOTs and PEDOT:PSS in energy conversion and storage devices. J. Mater. Sci. Mater. Electron. 2015, 26, 4438–4462. [Google Scholar] [CrossRef]
- Dennler, G.; Sariciftci, N.S. Flexible conjugated polymer-based plastic solar cells: From basics to applications. Proc. IEEE 2005, 93, 1429–1439. [Google Scholar] [CrossRef]
- Roncali, J. Conjugated Poly(thiophenes): Synthesis, Functionalization, and Applications. Chem. Rev. 1992, 92, 711–738. [Google Scholar] [CrossRef]
- Sugimoto, R.; Takeda, S.; Gu, H.B.; Yoshino, K. Preparation of Soluble Polythiophene Derivatives Utilizing Transition Metal Halides As Catalysts and Their Property. Chem. Express 1986, 1, 635–638. [Google Scholar]
- Zhai, L.; McCullough, R.D.; Loewe, R.S.; Ewbank, P.; Liu, J. Regioregular, head-to-tail coupled poly(3-alkylthiophenes) made easy by the GRIM method: Investigation of the reaction and the origin of regioselectivity. Macromolecules 2001, 34, 4324–4333. [Google Scholar]
- Rudenko, A.E.; Wiley, C.A.; Stone, S.M.; Tannaci, J.F.; Thompson, B.C. Semi-random P3HT analogs via direct arylation polymerization. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 3691–3697. [Google Scholar] [CrossRef]
- Vangheluwe, M.; Verbiest, T.; Koeckelberghs, G. Influence of the substitution pattern on the chiroptical properties of regioregular poly(3-alkoxythiophene)s. Macromolecules 2008, 41, 1041–1044. [Google Scholar] [CrossRef]
- Andersson, M.R.; Selse, D.; Berggren, M.; Järvinen, H.; Hjertberg, T.; Inganäs, O.; Wennerström, O.; Österholm, J.E. Regioselective Polymerization of 3-(4-Octylphenyl)thiophene with FeCl3. Macromolecules 1994, 27, 6503–6506. [Google Scholar] [CrossRef]
- Hai, T.A.P.; Sugimoto, R. Effect of molar ratio of oxidizer/3-hexylthiophene monomer in chemical oxidative polymerization of poly(3-hexylthiophene). J. Mol. Struct. 2017, 1146, 660–668. [Google Scholar] [CrossRef]
- Hai, T.A.P.; Sugimoto, R. The Catalytic Oxidative Polymerization of 3-Hexylthiophene by Oxidation of Fe2+ to Fe3+. Catal. Lett. 2017, 147, 1955–1965. [Google Scholar] [CrossRef]
- Amou, S.; Haba, O.; Shirato, K.; Hayakawa, T.; Ueda, M.; Takeuchi, K.; Asai, M. Head-to-tail regioregularity of poly(3-hexylthiophene) in oxidative coupling polymerization with FeCl3. J. Polym. Sci. Part A Polym. Chem. 1999, 37, 1943–1948. [Google Scholar] [CrossRef]
- Liu, Y.; Nishiwaki, N.; Saigo, K.; Sugimoto, R. Polymerization of 3-hexylthiophene with FeCl3 in aromatic solvents. Polym. Bull. 2015, 72, 1817–1826. [Google Scholar] [CrossRef] [Green Version]
- McCullough, R.D. The Chemistry of Conducting Polythiophenes. Adv. Mater. 1998, 10, 93–116. [Google Scholar] [CrossRef]
- Pomerantz, M.; Tseng, J.J.; Zhu, H.; Sproull, S.J.; Reynolds, J.R.; Uitz, R.; Arnott, H.J.; Haider, M.I. Processable polymers and copolymers of 3-alkylthiophenes and their blends. Synth. Met. 1991, 41, 825–830. [Google Scholar] [CrossRef]
- Dey, T.; Invernale, M.A.; Ding, Y.; Buyukmumcu, Z.; Sotzing, G.A. Poly(3,4-propylenedioxythiophene)s as a single platform for full color realization. Macromolecules 2011, 44, 2415–2417. [Google Scholar] [CrossRef]
- Wolfs, M.; Darmanin, T.; Guittard, F. Versatile superhydrophobic surfaces from a bioinspired approach. Macromolecules 2011, 44, 9286–9294. [Google Scholar] [CrossRef]
- Dong, L.; Zhang, L.; Duan, X.; Mo, D.; Xu, J.; Zhu, X. Synthesis and characterization of chiral PEDOT enantiomers bearing chiral moieties in side chains: Chiral recognition and its mechanism using electrochemical sensing technology. RSC Adv. 2016, 6, 11536–11545. [Google Scholar] [CrossRef]
- Dufen, H.; Baoyang, L.; Xuemin, D.; Jingkun, X.; Long, Z.; Kaixin, Z.; Shimin, Z.; Shijie, Z. Synthesis of novel chiral L-leucine grafted PEDOT derivatives with excellent electrochromic performances. RSC Adv. 2014, 4, 35597–35608. [Google Scholar] [CrossRef]
- Kumar, A.; Reynolds, J.R. Soluble alkyl-substituted poly(ethylenedioxythiophenes) as electrochromic materials. Macromolecules 1996, 29, 7629–7630. [Google Scholar] [CrossRef]
- Hai, W.; Goda, T.; Takeuchi, H.; Yamaoka, S.; Horiguchi, Y.; Matsumoto, A.; Miyahara, Y. Specific Recognition of Human Influenza Virus with PEDOT Bearing Sialic Acid-Terminated Trisaccharides. ACS Appl. Mater. Interfaces 2017, 9, 14162–14170. [Google Scholar] [CrossRef]
- Donahue, M.J.; Sanchez-Sanchez, A.; Inal, S.; Qu, J.; Owens, R.M.; Mecerreyes, D.; Malliaras, G.G.; Martin, D.C. Tailoring PEDOT properties for applications in bioelectronics. Mater. Sci. Eng. R Rep. 2020, 140, 100546. [Google Scholar] [CrossRef] [Green Version]
- Heywang, G.; Jonas, F. Poly(alkylenedioxythiophene)s—New, very stable conducting polymers. Adv. Mater. 1992, 4, 116–118. [Google Scholar] [CrossRef]
- Groenendaal, L.; Zotti, G.; Aubert, P.H.; Waybright, S.M.; Reynolds, J.R. Electrochemistry of poly(3,4-alkylenedioxythiophene) derivatives. Adv. Mater. 2003, 15, 855–879. [Google Scholar] [CrossRef]
- Koeckelberghs, G.; Vangheluwe, M.; Samyn, C.; Persoons, A.; Verbiest, T. Regioregular poly(3-alkoxythiophene)s: Toward soluble, chiral conjugated polymers with a stable oxidized state. Macromolecules 2005, 38, 5554–5559. [Google Scholar] [CrossRef]
- Ouhib, F.; Dkhissi, A.; Iratçabal, P.; Hiorns, R.C.; Khoukh, A.; Desbrières, J.; Pouchan, C.; Dagron-Lartigau, C. Electronic Structure and Optical Properties of Poly[3 -(4-octylphenoxy)thiophene]: Experimental and Theoretical Studies. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 7505–7516. [Google Scholar] [CrossRef]
- Qu, S.; Yao, Q.; Yu, B.; Zeng, K.; Shi, W.; Chen, Y.; Chen, L. Optimizing the Thermoelectric Performance of Poly(3-hexylthiophene) through Molecular-Weight Engineering. Chem. Asian J. 2018, 13, 3246–3253. [Google Scholar] [CrossRef]
- Trznadel, M.; Pron, A.; Zagorska, M.; Chrzaszcz, R.; Pielichowski, J. Effect of molecular weight on spectroscopic and spectroelectrochemical properties of regioregular poly(3-hexylthiophene). Macromolecules 1998, 31, 5051–5058. [Google Scholar] [CrossRef]
- Kline, R.J.; McGehee, M.D.; Kadnikova, E.N.; Liu, J.; Fréchet, J.M.J. Controlling the field-effect mobility of regioregular polythiophene by changing the molecular weight. Adv. Mater. 2003, 15, 1519–1522. [Google Scholar] [CrossRef]
- Gadient, J.; Groch, R.; Lind, C. An in depth study of solvent effects on yield and average molecular weight in poly(3-hexylthiophene). Polymer 2017, 115, 21–27. [Google Scholar] [CrossRef]
- Leclerc, M.; Diaz, F.M.; Wegner, G. Structural analysis of poly(3-alkylthiophene)s. Die Makromol. Chem. 1989, 190, 3105–3116. [Google Scholar] [CrossRef]
- Qi, Z.J.; Wei, B.; Sun, Y.M.; Wang, X.M.; Kang, F.; Hong, M.X.; Tang, L.L. Comparative study of photoelectric properties of regiosymmetrical poly(3,4-dialkoxythiophene)s. Polym. Bull. 2011, 66, 905–915. [Google Scholar] [CrossRef]
- Niemi, V.M.; Knuuttila, P.; Österholm, J.E.; Korvola, J. Polymerization of 3-alkylthiophenes with FeCl3. Polymer 1992, 33, 1559–1562. [Google Scholar] [CrossRef]
- Fukumoto, H.; Omori, Y.; Yamamoto, T. Effects of solvent and temperature on regioregularity of poly(3-hexylthiophene-2,5-diyl) prepared by chemical oxidative polymerization. Polym. J. 2013, 45, 462–465. [Google Scholar] [CrossRef] [Green Version]
- Olinga, T.; François, B. Kinetics of polymerization of thiophene by FeCl3 in choloroform and acetonitrile. Synth. Met. 1995, 69, 297–298. [Google Scholar] [CrossRef]
- Jiang, C.; Chen, G.; Wang, X. High-conversion synthesis of poly(3,4-ethylenedioxythiophene) by chemical oxidative polymerization. Synth. Met. 2012, 162, 1968–1971. [Google Scholar] [CrossRef]
- Barbarella, G.; Zambianchi, M.; Di Toro, R.; Colonna, M.; Iarossi, D.; Goldoni, F.; Bongini, A. Regioselective oligomerization of 3-(alkylsulfanyl)thiophenes with ferric chloride. J. Org. Chem. 1996, 61, 8285–8292. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.M.; Chan, H.S.O.; Ng, S.C.; Chung, T.S. Polymers synthesized from (3-alkylthio)thiophenes by the FeCl3 oxidation method. Synth. Met. 2002, 132, 63–69. [Google Scholar] [CrossRef]
- Cai, T.; Zhou, Y.; Wang, E.; Hellström, S.; Zhang, F.; Xu, S.; Inganäs, O.; Andersson, M.R. Low bandgap polymers synthesized by FeCl3 oxidative polymerization. Sol. Energy Mater. Sol. Cells 2010, 94, 1275–1281. [Google Scholar] [CrossRef] [Green Version]
- Andersson, M.R.; Mammo, W.; Olinga, T.; Svensson, M.; Theander, M.; Inganäs, O. Synthesis of regioregular phenyl substituted polythiophenes with FeCl3. Synth. Met. 1999, 101, 11–12. [Google Scholar] [CrossRef]
- Guzinski, M.; Jarvis, J.M.; D’Orazio, P.; Izadyar, A.; Pendley, B.D.; Lindner, E. Solid-Contact pH Sensor without CO2 Interference with a Superhydrophobic PEDOT-C14 as Solid Contact: The Ultimate “water Layer” Test. Anal. Chem. 2017, 89, 8468–8475. [Google Scholar] [CrossRef]
- Bhardwaj, D.; Shahjad; Gupta, S.; Yadav, P.; Bhargav, R.; Patra, A. All Conjugated Poly (3-hexylthiophene)-block-poly(hexyl-3,4-ethylenedioxythiophene) Copolymers. ChemistrySelect 2017, 2, 9557–9562. [Google Scholar] [CrossRef]
- Yiğit, D.; Aykan, M.; Güllü, M. Substituent effect on supercapacitive performances of conducting polymer-based redox electrodes: Poly(3′,4′-bis(alkyloxy) 2,2′:5′,2″-terthiophene) derivatives. J. Polym. Sci. Part A Polym. Chem. 2018, 56, 480–495. [Google Scholar] [CrossRef]
- MacHui, F.; Langner, S.; Zhu, X.; Abbott, S.; Brabec, C.J. Determination of the P3HT:PCBM solubility parameters via a binary solvent gradient method: Impact of solubility on the photovoltaic performance. Sol. Energy Mater. Sol. Cells 2012, 100, 138–146. [Google Scholar] [CrossRef]
- Daoust, G.; Leclerc, M. Structure-Property Relationships in Alkoxy-Substituted Polythiophenes. Macromolecules 1991, 24, 455–459. [Google Scholar] [CrossRef]
- Somanathan, N.; Wegner, G. Comparative studies on poly(3-cyclohexylthiophene) with polyalkylthiophenes. Synth. Met. 1995, 75, 123–126. [Google Scholar] [CrossRef]
- Ochieng, M.A.; Ponder, J.F.; Reynolds, J.R. Effects of linear and branched side chains on the redox and optoelectronic properties of 3,4-dialkoxythiophene polymers. Polym. Chem. 2020, 11, 2173–2181. [Google Scholar] [CrossRef]
- Roncali, J.; Garreau, R.; Yassar, A.; Marque, P.; Garnier, F.; Lemaire, M. Effects of steric factors on the electrosynthesis and properties of conducting poly(3-alkylthiophenes). J. Phys. Chem. 1987, 91, 6706–6714. [Google Scholar] [CrossRef]
- Sankaran, B.; Reynolds, J.R. High-contrast electrochromic polymers from alkyl-derivatized poly(3,4-ethylenedioxythiophenes). Macromolecules 1997, 30, 2582–2588. [Google Scholar] [CrossRef]
- Szkurlat, A.; Palys, B.; Mieczkowski, J.; Skompska, M. Electrosynthesis and spectroelectrochemical characterization of poly(3,4-dimethoxy-thiophene), poly(3,4-dipropyloxythiophene) and poly(3,4-dioctyloxythiophene) films. Electrochim. Acta 2003, 48, 3665–3676. [Google Scholar] [CrossRef]





| Reaction Parameter | Effect | Mechanism | Ref. |
|---|---|---|---|
| Reduced Temperature | Improvement of Ð with slight decrease in yield. | Suppression of active (oxidized) monomers in favor of dimers/oligomers. | [18,40] |
| Improved Solvent | Increased molecular weight and improved regioregularity. | Improved solvation of polymer. | [40] |
| Reduced Monomer Concentration | Increased molecular weight and improved regioregularity. | Suppression of dimer/oligomer couplings and improved solvation of polymer. | [18] |
| Decreased Oxidant/Monomer Ratio | Yields are severely decreased, molecular weight sharply decreases (sub-stoichiometric ratio). | Overall reduction in number of oxidized species present. | [16] |
| Entry | Structure | Acronym | Order of Addition | Equivalents FeCl3 | Reaction Time | Yield (%) | Mw (g/mol) | Xw |
|---|---|---|---|---|---|---|---|---|
| 1 |  | PEDOT-C12 | Reverse | 2.3 | 24 h | 73 | 6300 | 20 |
| 2 | Reverse 1 | 2.3 | 24 h | 80 | <5200 3 | <17 | ||
| 3 | Standard | 2.3 | 24 h | 26 | 20,000 | 65 | ||
| 4 | Standard | 4 | 24 h | 58 | 231,000 | 747 | ||
| 5 |  | PBHOT | Reverse | 2.3 | 24 h | 42 | <5200 3 | <18 |
| 6 | Standard | 2.3 | 24 h | 78 2 | <5200 3 | <18 | ||
| 7 | Standard | 4 | 24 h | 11 | 28,000 | 99 | ||
| 8 | Standard | 4 | 48 h | 10 | 90,000 | 318 | ||
| 9 |  | P3HT | Reverse | 2.3 | 24 h | 75 | 150,000 | 904 |
| 10 | Standard | 2.3 | 24 h | 26 | 81,000 | 488 | ||
| 11 | Standard | 4 | 24 h | 25 | 65,000 | 395 | ||
| 12 | Standard | 4 | 48 h | 72 | 65,000 | 395 |
| Reverse Addition | Standard Addition | |
|---|---|---|
| Solvent Composition | Pure chlorobenzene | Binary mixture of chlorobenzene and acetonitrile |
| [FeCl2] in Solution | Low | High |
| Relative Oxidant/Monomer Ratio | Begins high and decreases to theoretical value | Begins low and increases to theoretical value |
| Oxidation Potential of Solution | High | Low |
| Polymerization Mechanism | More step growth-like | More chain growth-like |
| Xw | Low | High |
| Yield | High | Low |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hebert, D.D.; Naley, M.A.; Cunningham, C.C.; Sharp, D.J.; Murphy, E.E.; Stanton, V.; Irvin, J.A. Enabling Conducting Polymer Applications: Methods for Achieving High Molecular Weight in Chemical Oxidative Polymerization in Alkyl- and Ether-Substituted Thiophenes. Materials 2021, 14, 6146. https://0-doi-org.brum.beds.ac.uk/10.3390/ma14206146
Hebert DD, Naley MA, Cunningham CC, Sharp DJ, Murphy EE, Stanton V, Irvin JA. Enabling Conducting Polymer Applications: Methods for Achieving High Molecular Weight in Chemical Oxidative Polymerization in Alkyl- and Ether-Substituted Thiophenes. Materials. 2021; 14(20):6146. https://0-doi-org.brum.beds.ac.uk/10.3390/ma14206146
Chicago/Turabian StyleHebert, David D., Michael A. Naley, Carter C. Cunningham, David J. Sharp, Emma E. Murphy, Venus Stanton, and Jennifer A. Irvin. 2021. "Enabling Conducting Polymer Applications: Methods for Achieving High Molecular Weight in Chemical Oxidative Polymerization in Alkyl- and Ether-Substituted Thiophenes" Materials 14, no. 20: 6146. https://0-doi-org.brum.beds.ac.uk/10.3390/ma14206146