
Atomic Interactions and Order–Disorder Transition in FCC-Type FeCoNiAl1−xTix High-Entropy Alloys



1
College of Mechanical and Vehicle Engineering, Hunan University, Changsha 410082, China
2
College of Artificial Intelligence and Big Data for Medical Sciences, Shandong First Medical University & Shandong Academy of Medical Sciences, Jinan 250117, China
3
State Key Laboratory of Advanced Design and Manufacturing for Vehicle Body, Hunan University, Changsha 410082, China
4
School of Materials Science and Engineering, Xiangtan University, Xiangtan 411105, China
*
Author to whom correspondence should be addressed.
Materials 2022, 15(11), 3992; https://0-doi-org.brum.beds.ac.uk/10.3390/ma15113992
Submission received: 2 May 2022
/
Revised: 20 May 2022
/
Accepted: 27 May 2022
/
Published: 3 June 2022
(This article belongs to the Special Issue Advances in High Entropy Materials)
Abstract
:Single-phase high-entropy alloys with compositionally disordered elemental arrangements have excellent strength, but show a serious embrittlement effect with increasing strength. Precipitation-hardened high-entropy alloys, such as those strengthened by L12-type ordered intermetallics, possess a superior synergy of strength and ductility. In this work, we employ first-principles calculations and thermodynamic simulations to explore the atomic interactions and order–disorder transitions in FeCoNiAl1−xTix high-entropy alloys. Our calculated results indicate that the atomic interactions depend on the atomic size of the alloy components. The thermodynamic stability behaviors of L12 binary intermetallics are quite diverse, while their atomic arrangements are short-range in FeCoNiAl1−xTix high-entropy alloys. Moreover, the order–disorder transition temperatures decrease with increasing Ti content in FeCoNiAl1−xTix high-entropy alloys, the characteristics of order–disorder transition from first-principles calculations are in line with experimental observations and CALPHAD simulations. The results of this work provide a technique strategy for proper control of the order–disorder transitions that can be used for further optimizing the microstructure characteristics as well as the mechanical properties of FeCoNiAl1−xTixhigh-entropy alloys.
1. Introduction
High-entropy alloys (HEAs) are usually defined as those alloys having at least five major metallic elements and atomic concentrations in the range between 5 and 35 at.%. The premise on which these alloys have been conceptualized is high configurational entropy, due to a possible random mixing of constituent alloying elements in these multi-principal-element alloy systems, which reaches a maximum when the alloy is equiatomic [1,2,3,4]. Based on the entropy concept, the so-called HEAs are also generally defined as alloys having a high configurational entropy of more than 1.5 R (R being the gas constant) [5]. Due to the excellent strength and ductility of the single-phase HEAs at cryogenic temperatures, one of the main focus areas of HEAs studied earlier was to design a single-phase solid solution phase by adjusting the configurational entropy, however, they usually possess low ductility when the high strength is achieved, which makes them inappropriate for engineering applications. Therefore, extensive efforts have been turned toward dual-phase or multi-phase HEAs for achieving a combination of strength and ductility [6,7,8,9].
In recent work, the mechanical properties of HEAs containing ordered L12 precipitate have been investigated comprehensively. It was supposed that the formation of precipitate phases could contribute to achieving a balance between ductility and strength [10,11,12,13,14,15]. Lu et al. found that the balanced tensile properties were achieved in an fcc FeCoNiCr HEA due to the formation of a unique microstructure of the L12–Ni3(Ti,Al) nano-sized coherent reinforcing phase in the matrix [10]. The same was true in the investigation by Kai et al. for CoCrNi medium-entropy alloy containing nanoscale L12-(Ni, Co, Cr)3(Ti, Al)-type particles, compared to the single-phase CoCrNi medium-entropy alloy, the yield strength and the tensile strength of the precipitation-strengthened CoCrNi medium-entropy alloy were increased by ~70% to ~750 MPa and ~44% to ~1.3 GPa, respectively, whereas a good ductility, ~45%, was still achieved [11]. In an experimental study of the effect of coherent L12 nano-precipitates on the tensile behavior of a novel-designed Al0.2CrFeCoNi2Cu0.2 [12], Wang et al. found that the presence of L12 nano-precipitates resulted in increases of 259 MPa and 316 MPa in the yield strength and ultimate tensile strength, respectively, accompanied by the maintenance of a high elongation (30.4%). Moreover, it was reported by Gwalani et al. that the L12 phase could be stabilized by Cu addition in the Al0.3CrCuFeNi2 HEA [13]. A similar mechanical response was also observed when modulating the Al and Ti contents in the FeCoNiAlxTiy HEAs [16,17], while the Ti/Al ratios ranging from 0.7 to 2 would prefer the formation of the L12 phase in the CoCrFeNi-based HEAs [14]. This was also confirmed by Joseph et al. in the Al–Co–Cr–Fe–Ni–Ti HESAs, where the authors found that the phase equilibrium and yield strength of the Al–Co–Cr–Fe–Ni–Ti HEAs could be affected by the (Al/Ti)-ratio [18]. Moreover, the chemical instability of the supersaturation and migration of grain boundaries could cause the formation of cellular precipitate, which in turn affects the tensile properties of the precipitation-strengthened HEAs [15].
The fundamental understanding of the thermodynamic and mechanical stability of precipitates in HEAs is of major importance for the design of new alloys and for an accurate thermodynamic description of the materials containing such precipitates using atomistic modeling and thermodynamic simulation, i.e., first-principles calculations and the calculation of phase diagrams (CALPHAD) method [19]. Using first-principles calculations, Widom et al. found that the configurational entropy could stabilize a single body-centered cubic (BCC) phase from 1700 K up to melting, while a complex intermetallic was favored at lower temperatures [20]. Hu et al. reported that the HfNbTaTiZr HEA decomposed most favorably to BCC NbTa-rich and HfZr-rich phases below the critical temperature of 1298 K [21]. Lai et al. calculated the formation enthalpy and cohesive energy of a list of potential inter-metallic compounds in the FeTiCoNiVCrMnCuAl HEA and showed that FeTi, Fe2Ti, AlCrFe2, Co2Ti, AlMn2V and Mn2Ti phases were preferential in the formation process of the FeTiCoNiVCrMnCuAl HEA [22]. Using density functional theory (DFT) calculations, the ordering-induced elastic anomaly in the FeCoNiAl1−xTix were investigated, it was found that the Ti addition could be attributed to the presence of an ordered L12 phase [23]. Sluiter et al. investigated the order–disorder transition in NbMoTaW HEA, they found that the B2(Mo;Ta) and B32(Nb;W) were stable from an energetic point of view in the ground state and revealed a B2(Mo,W;Ta,Nb) ordering at ambient temperatures [24]. Using CALPHAD methods, the content of L12–Ni3(Ti, Al) phase contents in NiCoAlTiFeCr HEA [25], the grain size as well as the volume fraction of L12 precipitates in FeCoNiAlTi HEA [26], and the phase stability of L12-(Ni,Co,Cr)3(Ti,Al) in CoCrNi-based medium-entropy alloys were investigated [11]. The results indicated that the CALPHAD method could be used as a practical tool for designing microstructure and mechanical properties of HEAs.
It is known that the phase transformation in HEAs involves the movement of atoms through the metals to rearrange themselves to form the new phases or precipitates, which is also linked to solute mobility and interactions at finite temperatures. Unfortunately, little is known about the atomic interactions in HEAs, even that the atomic interactions of components in HEAs play a critical role in the order–disorder transition in the alloys [24], however, no complete phase diagram is currently available to directly assist in designing FeCoNiAlTi HEAs with desirable micro- and nano-structures. Moreover, it is already known that the Ti addition enhanced the mechanical strengthening of FeCoNiAl1−xTix HEAs [23], but the phase stability and order–disorder transition was not clear in this case. Therefore, the effect of Ti content on the phase stability and order–disorder transition needs to be investigated further. Such theoretical investigations can be used as a strategy for designing the chemical composition and microstructure of FeCoNiAl1−xTix HEAs.
In this work, the phase stability of FeCoNiAl1−xTix HEA was investigated based on the consideration of the interatomic interactions, atomic ordering, and phase transformation. Specifically, the pair interactions of component species in an fcc single-phase lattice were investigated to reveal how these elements interact with each other and how the interactions contribute to the chemical ordering and phase transformation of the FeCoNiAl1−xTix HEAs. Furthermore, Monte Carlo statistical thermodynamic simulations of elevated temperature ordering in FeCoNiAl1−xTix HEAs were conducted and compared with the solidification process simulation using the CALPHAD method. Such an analysis can be directly linked to the precipitation hardening phenomena and used in a consistent approach to identify the influence of each element as well as precipitates in the thermodynamic and mechanical properties of FeCoNiAl1−xTix HEAs, the results of this work will provide solutions for composition and structure optimization to improve the mechanical properties of the FeCoNiAl1−xTix HEAs.
2. Computational Method
2.1. Atomic Interactions
In this study, considering atomic interactions in face-centered cubic (fcc) Ni matrix [27], we obtain the atomic interactions from a set of the total energies of supercells where positions in a given cluster are occupied by atoms in different configurations. These were calculated from two representative cases:
- (i)
- In the binary system of Ni–X, the X–X solute pair interaction at the pth coordination shell is calculated by [27]:where is the total energy of a supercell with two X atoms at the pth coordination shell, is the total energy of a supercell with one X atom, and is the total energy with no impurity;
- (ii)
- In the ternary system of Ni–X–Y, the X–Y solute pair interaction in the pth coordination shell is calculated by:where is the total energy of a supercell with two Y atoms at the pth coordination shell, and is the total energy of the supercell with one X atom and one Y atom at the pth coordination shell. In this work, a negative interaction energy indicates the attraction between solutes.
Although the size mismatches of atoms in FeCoNiAl1−xTix HEAs are rather moderate, due to the equal or nearly equal atomic ratio in HEAs, local lattice relaxations play a critical role in alloy energetics [28,29]. Therefore, the differences between chemical and total interactions are calculated and defined as the strain-induced (SI) interaction:
where is calculated by relaxing the atoms in the supercells, while for the , the atom positions in the supercells are fixed. In both cases, the shape of the supercells is kept at the equilibrium lattice constant [30].
2.2. Thermodynamic Monte Carlo Method
In the statistical thermodynamic Monte Carlo (MC) simulations, the atomic interactions obtained from DFT calculations are used to capture the chemical degree of freedom of alloys. The configurational ordering energy of the FeCoNiAl1−xTix is mapped onto an Ising Hamiltonian employing atomic interactions [24]:
where the second sum runs over all pairs p, while are the atomic interactions between atomic species and in FeCoNiAl1−xTix HEAs . The first sum runs over the species , and , and denotes the concentration variables at lattice sites n and m. The independent concentration fluctuations are given as and = with concentration . The is the site-occupation variable which is equal to 1 (0) if the atom at site i is (is not) occupied by species . In most cases, higher-order terms are found to be quite small and can be neglected [27].
2.3. Formation Enthalpy of L12-A3B Precipitate
The formation enthalpy () of a four-atom L12-A3B intermetallic compound is calculated by using the following equation [31,32]:
where is the total energy of the L12-A3B intermetallic compound, (A) and (B) are the zero-Kelvin total energies of the pure elements at their reference states, namely, fcc, bcc, hcp, fcc, and hcp, are taken as the ground state crystal structures for Ni, Fe, Co, Al, and Ti, respectively. The total energies of the L12-A3B precipitates, as well as Ni, Fe, and Co, are calculated in the ferromagnetic state.
2.4. First-Principles Method
Density functional theory (DFT) total-energy calculations for FeCoNiAl1−xTix HEAs within DFT were performed using the projector augmented-wave (PAW) method [33] as implemented in the Vienna ab initio simulation package (VASP) [34,35,36]. All the calculations were performed using the Perdew–Burke–Ernzerhof (PBE) form for the generalized gradient approximation (GGA) [37,38]. The atomic interactions are determined in the dilute limit using a 3 × 3 × 3 repetition of the initial fcc 4-atom supercell in the ferromagnetic state of Ni. In order to keep the cubic symmetry of the underlying fcc lattice, which should be preserved on average in real alloys, the translation vectors of supercells are fixed. The integration over the Brillouin zone is performed using the 4 × 4 × 4 Monkhorst-Pack grid [39] for the supercell. In the calculations for the atomic interactions for FeCoNiAl1−xTix HEAs, atomic positions are relaxed while the shape of the supercells is fixed. The k-point density is equivalent to the 12 × 12 × 12 k-mesh grids for a four-atom fcc unit cell scale, namely, a 4 × 4 × 4 k-mesh density, is used for a 3 × 3 × 3 conventional fcc supercell. It is helpful for avoiding systematic errors [27,32,40].
Other details for all the VASP-PAW calculations are as follows. The convergence criteria are 10−5 eV per cell. All the atomic positions inside the supercells are relaxed until the forces acting on the atoms are less than 9 × 10−3 eV/Å. The lattice constant is chosen as 3.52 Å, being between the derived T = 0 K value from experimental [41] and theoretical values [30,42]. Plane waves are included up to 500 eV, which is found to be sufficient for the accurate convergence of total energies. The atomic interactions are calculated using a 3 × 3 × 3 conventional fcc supercell [42,43].
2.5. CALPHAD Method
The precipitation temperatures of the L12 phase were calculated using Thermo-Calc 2021b [44] with TCHEA5 databases [45], with the aim of comparing them with those predicted in the Monte Carlo calculations. In the present study, the Sheil–Gulliver model was adopted to simulate the solidification process and the phase precipitation sequence of the FeCoNiAl1−xTix HEAs with different chemical compositions. The assumption of the Sheil model is that the speed of diffusion in the liquid phase is rapid enough to achieve an even composition, and only local equilibrium is reached at liquid/solid interfaces. The calculated results for different alloys are shown in Section 3.5.
3. Results and Discussion
3.1. Atomic Interactions of Solute Pairs in FeCoNiAl1−xTix HEAs
In order to investigate the solute pair interactions in fcc single-matrix FeCoNiAl1−xTix HEAs, the atomic interactions of solute pairs in the fcc Ni matrix were taken into account and calculated using supercell models. The first four nearest neighboring atoms (labeled by Arabic numerals 1, 2, 3, and 4) from a reference site (labeled by Arabic numeral 0) are shown in a conventional fcc cell in Figure 1a, which has been proved to be sufficient for reproducing the atomic environment of FeCoNiAl1−xTix HEAs [46]. The atomic configuration of L12-A3B is also shown in Figure 1b, where one can see the ordering at the second and fourth coordination shells.
Using Equations (1) and (2), the atomic interactions for all solute pairs were calculated and listed in Table 1. One can see there exists positive atomic interaction energies for all nearest neighboring solute pairs, meaning that the nearest neighboring solute pairs were repulsive in the fcc Ni matrix. These repulsive interactions would contribute to the atomic ordering as well as the formation of precipitates [27,47,48]. At the time time, it is worth noting that the Co–Co first nearest neighboring atomic interaction was not as large as other solute pairs, which indicates that the Co atoms should have no ordering tendency in the FeCoNiAl1−xTix HEAs. Moreover, it is known that the atomic radii of Al, Co, Fe, Ni, and Ti are 1.25 Å, 1.35 Å, 1.40 Å, 1.35 Å, and 1.40 Å [49], respectively. Among the aforementioned species, Co and Ni have the similar atomic radius, which may be the reason why Co–Co solute pairs showed weak atomic interactions not only at the first nearest neighboring but also at the second, third, and fourth nearest neighboring. As expected, there was a clear correlation between the sizes of the atoms and the local lattice strains, which would affect the atomic interactions and the atomic binding of the atoms.
Moreover, to understand the effect of atom size mismatch on the interactions between atoms in FeCoNiAl1−xTix HEAs, we also show the strain-induced interactions obtained from Equation (3) in Table 1. One notices that the strain-induced interactions of Al–X were quite large compared to those of other solute pairs due to the smallest atomic size of Al (1.25 Å) among the componential species in FeCoNiAl1−xTix HEAs. This also holds true for Co–Co strain-induced interactions: the Co–Co solute pair interactions were negligible for both total atomic interactions and strain-induced interactions due to the similar atomic size between Co and Ni atoms. Moreover, it is not surprising that the strong strain-induced interactions of Al–X in the Ni matrix due to atomic radius differences between Al and other atoms (Co, Fe, and Ti) played an important role in the atomic ordering in the FeCoNiAl1−xTix HEAs. With the local lattice relaxation as obtained in first-principles calculations of atomic interactions, the atomic configurations and order–disorder transitions in FeCoNiAl1−xTix HEAs were investigated using Monte Carlo simulations, which is shown in later sections.
3.2. The Order–Disorder Transition in Equal-Atomic AlCoFeNi HEAs
We now turn to the prediction of finite-temperature atomic configurations of AlCoFeNi HEAs using Monte Carlo simulations. The Monte Carlo simulations were performed for a box containing 62,500 atoms ([25 × 25 × 25] × 4 conventional unit cell) with periodic boundary conditions from 2300 K down to 300 K, including the first four nearest neighboring pair interactions as listed in Table 1. At each temperature, the system was first equilibrated for 4000 Monte Carlo steps/atom. After that, the statistical data were obtained by averaging over additional 4000 Monte Carlo steps/atom [27].
With the DFT calculated pair interactions, the heat capacity of the AlCoFeNi HEAs was obtained using Monte Carlo calculations. The configurational contribution to the specific heat, evaluated via the energy fluctuations, is presented in Figure 2. It is obvious that there exists a peak value for specific heat at 1650 K, indicating that there should be a configurational energy change at this temperature. A phase separation at this temperature in AlCoFeNi HEAs can be observed in AlCoFeNi HEAs, and the evolution of the L12 precipitate was also confirmed in the study of AlxCo1.5CrFeNi1.5Tiy HEAs [50]. However, the mechanism of such a phase evolution has not been discussed in detail. Therefore, the atomic configuration in AlCoFeNiTi HEAs will be shown later in terms of the atom arrangement and short-range order (SRO) parameter of solute pairs, which provides information about the arrangement of atoms in the immediate neighborhood of particular atoms of the lattice [27,46].
3.3. The Microstructure of AlCoFeNi at Elevated and Ambient Temperatures
In Figure 3, we illustrate the atomic configuration of AlCoFeNi HEAs at an elevated temperature of 2300 K, which is 650 K higher than the order–disorder temperature of AlCoFeNi HEAs. One can see that the elementary elements of AlCoFeNi HEAs are randomly distributed in the cubic box, indicating a disordered atomic configuration in the AlCoFeNi HEAs at the elevated temperature above the order–disorder transition point.
In Figure 4, the atomic configuration of AlCoFeNi HEA at 300 K is illustrated with spheres of different colors, where the L12 ordered structure can be seen in the snapshot. We also depict the [100] and [010] crystal planes of three-dimension AlCoFeNi HEAs at the bottom of the figure. Al, Co, Fe, and Ni atoms are depicted here with blue, orange, purple, and green spheres, respectively. It is interesting that two types of L12 precipitates can be observed in this snapshot at 300 K, i.e., Ni3Al and Fe3Al. However, one can also see that some of the A and B sites are occupied by solute except Ni, Al, and Fe atoms, for example, one can see that Co atoms replace Al in Ni3Al precipitates and Al sites in Fe3Al precipitates in the snapshot of Figure 3. Such site preference behaviors of solute in intermetallic compounds transcend the framework of the current paper, it will be a subject of ongoing work [32] and future publications. Moreover, it is obvious that few Co atoms were soluble in the precipitates, however, most Co atoms tended to accumulate and segregate at the interfaces of the precipitates. This is due to the weak atomic interactions between Co atoms, as illustrated in Table 1.
We can therefore conclude that the AlCoFeNi HEAs retain an fcc solid solution down to low temperatures, but they are short-range. According to the Monte Carlo simulation results, the sizes of the aforementioned precipitates are not greater than 35 nm, while the experimental finding indicates that the L12 nano-particles with an average size of 30 nm are embedded in the fcc matrix of Fe–Co–Ni–Al–Ti-based HEA [9]. Our simulated results of precipitate size are in good agreement with the experimental observations.
Figure 5 shows the atomic short-range order (SRO) parameters [27] obtained from the Monte Carlo simulations. It clearly indicates that curves of , , and were very similar, with only small quantitative differences in the whole temperature range. However, was larger and more pronounced than and . At the same time, one notices from Table 1 that the Co–Co and Co–Fe pair interactions were negligible (the strongest interaction energy at the first four coordination shells was found to be less than 0.03 eV), which means that the negligible change in SRO parameters was determined by the weak atomic interactions of the solutes. This result agrees with previous findings in [51], where the authors found that the Co–Ni interactions were below 0.01 eV. In the case of Al–Co, Al–Fe, and Al–Al solute pairs, , , and decreased at 1650 K, which is the order–disorder transition temperature of AlFeCoNi HEA.
If we look at the SRO parameter in the second coordination shell, which governs the L1 ordering in the fcc matrix (the nearest A–A sites in L12 A3B are located at the second nearest neighboring of the fcc lattice), one can see the dramatic changes in and at the order–disorder transition temperature, i.e., 1650 K. Clearly, the ordering of Al–Al and Al–Fe solute pairs in the second coordination shell was needed for the formation of L12 Ni3Al and Fe3Al. The thermodynamic stability of some L12 Ni3X intermetallic compounds have been investigated previously [32] and a more comprehensive investigation on the stability of L12-A3B intermetallic compounds is shown in later sections.
3.4. Alloying Effect of Ti on The Microstructure Evolution of FeCoNiAl1−xTix HEAs
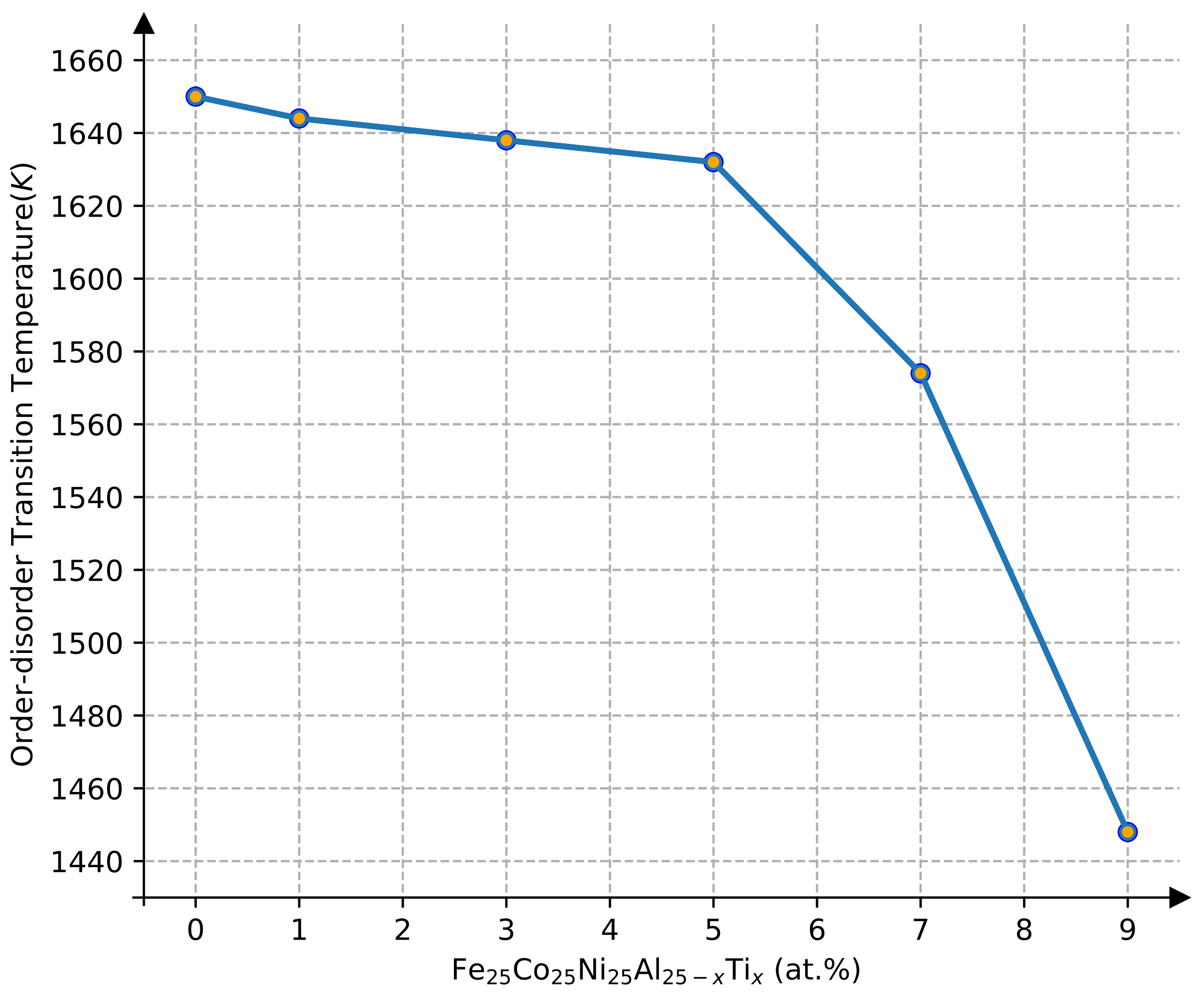
The effect of Ti content on the transition temperature was investigated in the range of 1–9 Ti at.% using Monte Carlo simulations, while the contents of Ti and Al were kept balanced in the FeCoNiAl1−xTix HEAs. In the following discussion, the FeCoNiAl 1−xTix was also written as Fe25Co25Ni25Al25−xTix for quasi-quaternary-type equiatomic HEAs. The concentration dependence of the order–disorder phase transition temperature is shown in Figure 6. It was found that the order–disorder transition temperature decreased smoothly with the concentration of Ti in the Fe25Co25Ni25Al25−xTix HEAs in the range of 0–5 Ti at.%, while it declined dramatically with the concentration of Ti when the Ti content was higher than 5 Ti at.%. One should note that L12Ni3Ti (−0.429 eV) has significantly lower formation enthalpy compared to the Ni3Al (−0.475 eV), indicating that Ni3Ti is much more stable than Ni3Al [32]. This means that the L12 of the Fe25Co25Ni25Al25−xTix HEAs can be stabilized by adding or increasing Ti. The calculated results are subsequently in agreement with the experimental observations, in which the as-heat-treated Al01Ti04 and Al02Ti03 alloys exhibited stable L12 precipitates [5].
The snapshots of the Monte Carlo simulation box of Fe25Co25Ni25Al20Ti5 HEA at 300 K are shown in Figure 7. One notices that there was a phase separation transition where the L12 ordered structure precipitated in Fe25Co25Ni25Al20Ti5 HEA. The main chemical compositions of the ordered structure were indeed Ni3Al, Ni3Ti, and Fe3Al precipitates, as framed in Figure 7. Moreover, it is the same as AlCoFeNi HEAs, one can also see the accumulation and segregation of Co atoms at the interfaces of the precipitates in Fe25Co25Ni25Al20Ti5 HEA due to the weak atomic interactions between Co atoms.
The atomic interactions of any two of these components are shown in Table 1, which indicates that the atomic interactions govern the ordering evolution and play a critical role in the formation of L12-type precipitates. To be more quantitative, the thermodynamic properties of L12 intermetallic compounds formed by any two of the components in AlFeCoNiTi HEAs are also calculated and shown in Figure 8 as a form of color map. It may be seen from the formation energy color map that the formation enthalpies of Ni3Al and Ni3Ti were found to be −0.44 eV and −0.47 eV, respectively. Both of the formation enthalpies were the most stable chemical compositions in L12 in AlFeCoNiTi HEAs, which is in agreement with the snapshots of the atomic results in Figure 4 and Figure 7, where L12 Ni3Al and Ni3Ti are confirmed. The formation enthalpy of Fe3Al was −0.20 eV, which was found in the AlCoFeNi HEA at low temperature. Moreover, one can find that the intermetallic compounds containing Al and Ti have lower formation enthalpies, which means that they should be stable at low temperatures in AlCoFeNiTi HEAs.
In [32], it is reported that Ni3Al could be the most stable chemical composition in the L12–Ni3X-type intermetallic compounds selected in the list (X = Al, Ti, Mo, Cr, Mo, Fe). Specifically, the formation energy of L12–Ni3Al at 0 K was −0.43 eV/atom, which indicates a strong formation tendency of the L12 structure in Ni–Al alloys as well as other alloys containing Ni and Al. In this study, it was also found that the L12–Ni3Al precipitates formed at 300 K in AlCoFeNi HEAs, as shown in the [010] crystal plane of Figure 4. However, the sub-lattices of Ni and Al in L12–Ni3Al precipitates can also be replaced by other elements, for instance, Fe, Co, or Ti, which are also discussed in [32]. In [52], the formation energies of Fe–Al intermetallic compounds are investigated using first-principles calculations and CALPHAD simulations. The formation energy of L12–Fe3Al at 0 K was found to be −0.19 eV/atom, before dropping down to −0.20 eV/atom at 298 K. This means that the L12 structure exhibits a high stability in Fe–Al alloys as well as other alloys containing Fe and Al. Moreover, L12–Fe3Al had a stronger formation tendency at the higher temperature (298 K).
One can still check the SRO parameters again in AlFeCoNiTi HEAs, as they are shown in Figure 9. The SRO parameters for the first coordination shell are similar above and below the transition temperature, i.e., 1632 K. The dramatic change in Al–Al, Al–Ti, Al–Fe, and Al–Co at the second coordination shell can be seen, indicating an atomic ordering for Al, Ti, Fe, and Co in AlFeCoNiTi HEAs, as shown in Figure 7, where Ni3Al, Ni3Ti, and small volumes of Al3Fe and Fe3Co were found to be stable at ambient temperature.
3.5. The Verification from CALPHAD
As shown in Figure 10, the Sheil solidification curve was calculated to predict the L12 precipitation temperature of alloy AlCoFeNi. Our calculated results indicate that the phase precipitation sequence of this alloy is Liquid–Bcc–L12, while the predicted precipitation temperature of L12 is 1640 K. This result is in good agreement with the temperature (1650 K) calculated from the Monte Carlo simulation. Due to the limitation of current Monte Carlo simulations of fcc-based phase transformation, some disagreements between the CALPHAD simulated results based on the current databases and Monte Carlo as well as experimental results are still present [5].
Figure 11 shows the alloying effect of the Ti element on the precipitation temperature of the L12 phase. Four alloys with different compositions were obtained by replacing Al with Ti: Co25Fe25Ni25Al24Ti1, Co25Fe25Ni25Al22Ti3, Co25Fe25Ni25Al20Ti5, and Co25Fe25Ni25Al18Ti7. The calculated precipitation temperatures of the L12 phase were 1600 K, 1540 K, 1490 K, and 1465 K, respectively. The precipitation temperature decreased with increasing contents of Ti. The order–disorder transition temperatures for the four aforementioned HEAs in the Monte Carlo simulation were 1644 K, 1638 K, 1632 K, and 1574 K, respectively. It is worth noting that the results from the CALPHAD method were much higher than the results calculated using the Monte Carlo simulation, as shown in Figure 6, when the Ti content was less than 5%. One reason could be that the severe lattice distortion caused by Ti alloying significantly changed the atomic configuration, which is a sensitive parameter for Monte Carlo simulations in comparison to CALPHAD calculations.
Another interesting finding from CALPHAD modeling was that the Laves phase began to precipitate at about 1300 K when the Ti content was large enough. Laves are phases with high hardness but poor toughness, they could thus form a disadvantageous precipitate for structural applications. Therefore, the modulation of the Ti content would be an effective way to affect the microstructure and mechanical properties of the HEAs. However, although an investigation into the higher Ti content in Co25Fe25Ni25Al25−xTix transcends the scope of the present paper, it would be an interesting subject for future investigations.
4. Conclusions
The atomic interactions and order-disorder transition in the FeCoNiAl25−xTix HEAs have been investigated by using first-principles calculations and thermodynamic simulations comprehensively. The following conclusions may be drawn:
- (1)
- The chemical ordering in fcc-type FeCoNiAl1−xTix HEAs is short-range, while strain-induced interactions play an important role in the atomic ordering. Co atoms were found to accumulate at the interfaces between precipitates and matrix due to their weak interaction in FeCoNiAl1−xTix, the strong interactions of Al–Al and Ti–Ti atom pairs caused Al and Ti to act as the main components for forming the L12 precipitates.
- (2)
- The presence of the L12 phase and the chemical composition of L12 have been investigated comprehensively for the FeCoNiAl1−xTix high-entropy alloys. The site occupation in the L12 phase was found to be complex, and it requires further study. The order–disorder transition temperature from first-principles-based Monte Carlo simulations was 1650 K in FeCoNiAl HEAs, which is also consistent with the CALPHAD results. Moreover, the transition temperature decreased with increasing Ti contents in FeCoNiAl1−xTix HEAs.
- (3)
- The order–disorder transition results are in line with existing experimental observations, which means that a selected computer-aided design of the chemical composition can be used for further optimization of the microstructure characteristics in the FeCoNiAl1−xTix HEAs.
However, the order–disorder transition in FeCoNiAl1−xTix HEAs is complicated. Beyond what was performed in the current investigation, it also involves element site occupation, element segregation at interfaces, and phase competition. All of these require systematic study in further research.
Author Contributions
Conceptualization, Z.L. and S.H.; methodology, Z.L. and S.H.; software, Z.L. and S.H.; validation, Y.W., Z.L. and S.H.; formal analysis, Y.W.; investigation, Y.W.; resources, Y.W.; data curation, Y.W.; writing—original draft preparation, Y.W.; writing—review and editing, Y.W., Z.L., H.F. and S.H.; visualization, Y.W.; supervision, H.F.; project administration, Y.W.; funding acquisition, Y.W., Z.L. and S.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China, grant numbers 52101152 and 52001098, the Key Laboratory of Cloud Computing of Gansu Province, grant number HNU20210107.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors acknowledge Wei Wang (Northeast Electric Power University) for helpful advice and inspiring discussions. The numerical calculations in this work were performed at the National SuperComputer Center (NSCC) in Changsha and Tianjin.
Conflicts of Interest
The authors declare that there are no competing interests in their work.
Abbreviations
The following abbreviations are used in this manuscript:
| HEAs | High-entropy alloys |
| CALPHAD | Calculation of phase diagrams |
| VASP | Vienna ab initio simulation package |
| PAW | Projector augmented-wave |
| GGA | Gerneralized gradient approximation |
| PBE | Perdew–Burke–Ernzerhof |
| DFT | Density functional theory |
| MC | Monte Carlo |
| SRO | Short-range order |
| BCC | Body-centered cubic |
| FCC | Face-centered cubic |
| HCP | Hexagonal close-packed |
References
- Yeh, J.W.; Chen, S.K.; Lin, S.J.; Gan, J.Y.; Chin, T.S.; Shun, T.T.; Tsau, C.H.; Chang, S.Y. Nanostructured High-Entropy Alloys with Multiple Principal Elements: Novel Alloy Design Concepts and Outcomes. Adv. Eng. Mater. 2004, 6, 299–303. [Google Scholar] [CrossRef]
- Cantor, B.; Chang, I.; Knight, P.; Vincent, A. Microstructural development in equiatomic multicomponent alloys. Mater. Sci. Eng. A 2004, 375–377, 213–218. [Google Scholar] [CrossRef]
- Otto, F.; Dlouhý, A.; Somsen, C.; Bei, H.; Eggeler, G.; George, E. The influences of temperature and microstructure on the tensile properties of a CoCrFeMnNi high-entropy alloy. Acta Mater. 2013, 61, 5743–5755. [Google Scholar] [CrossRef] [Green Version]
- Srivatsan, T.; Gupta, M. High Entropy Alloys:Innovations, Advances, and Applications, 1st ed.; CRC Press: Boca Raton, FL, USA, 2020. [Google Scholar] [CrossRef]
- Yang, T.; Zhao, Y.; Liu, W.; Kai, J.; Liu, C. L12-strengthened high-entropy alloys for advanced structural applications. J. Mater. Res. 2018, 33, 2983–2997. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Bei, H.; Pharr, G.; George, E. Temperature dependence of the mechanical properties of equiatomic solid solution alloys with face-centered cubic crystal structures. Acta Mater. 2014, 81, 428–441. [Google Scholar] [CrossRef]
- Zhang, Y.; Zuo, T.T.; Tang, Z.; Gao, M.C.; Dahmen, K.A.; Liaw, P.K.; Lu, Z.P. Microstructures and properties of high-entropy alloys. Prog. Mater. Sci. 2014, 61, 1–93. [Google Scholar] [CrossRef]
- Liu, W.; Lu, Z.; He, J.; Luan, J.; Wang, Z.; Liu, B.; Liu, Y.; Chen, M.; Liu, C. Ductile CoCrFeNiMox high entropy alloys strengthened by hard intermetallic phases. Acta Mater. 2016, 116, 332–342. [Google Scholar] [CrossRef]
- Liu, W.; Yang, T.; Liu, C. Precipitation hardening in CoCrFeNi-based high entropy alloys. Mater. Chem. Phys. 2018, 210, 2–11. [Google Scholar] [CrossRef]
- He, J.; Wang, H.; Huang, H.; Xu, X.; Chen, M.; Wu, Y.; Liu, X.; Nieh, T.; An, K.; Lu, Z. A precipitation-hardened high-entropy alloy with outstanding tensile properties. Acta Mater. 2016, 102, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Yang, T.; Tong, Y.; Wang, J.; Luan, J.; Jiao, Z.; Chen, D.; Yang, Y.; Hu, A.; Liu, C.; et al. Heterogeneous precipitation behavior and stacking-fault-mediated deformation in a CoCrNi-based medium-entropy alloy. Acta Mater. 2017, 138, 72–82. [Google Scholar] [CrossRef]
- Wang, Z.; Zhou, W.; Fu, L.; Wang, J.; Luo, R.; Han, X.; Chen, B.; Wang, X. Effect of coherent L12 nanoprecipitates on the tensile behavior of a fcc-based high-entropy alloy. Mater. Sci. Eng. A 2017, 696, 503–510. [Google Scholar] [CrossRef]
- Gwalani, B.; Choudhuri, D.; Soni, V.; Ren, Y.; Styles, M.; Hwang, J.; Nam, S.; Ryu, H.; Hong, S.; Banerjee, R. Cu assisted stabilization and nucleation of L12 precipitates in Al0.3CuFeCrNi2 fcc-based high entropy alloy. Acta Mater. 2017, 129, 170–182. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; He, F.; Han, B.; Wu, Q.; Tong, Y.; Zhao, Y.; Wang, Z.; Wang, J.; Kai, J. Synergistic effect of Ti and Al on L12-phase design in CoCrFeNi-based high entropy alloys. Intermetallics 2019, 110, 106476. [Google Scholar] [CrossRef]
- Chang, Y.J.; Yeh, A.C. The formation of cellular precipitate and its effect on the tensile properties of a precipitation strengthened high entropy alloy. Mater. Chem. Phys. 2018, 210, 111–119. [Google Scholar] [CrossRef]
- Fu, Z.; Chen, W.; Chen, Z.; Wen, H.; Lavernia, E.J. Influence of Ti addition and sintering method on microstructure and mechanical behavior of a medium-entropy Al0.6CoNiFe alloy. Mater. Sci. Eng. A 2014, 619, 137–145. [Google Scholar] [CrossRef]
- Fu, Z.; Chen, W.; Wen, H.; Morgan, S.; Chen, F.; Zheng, B.; Zhou, Y.; Zhang, L.; Lavernia, E.J. Microstructure and mechanical behavior of a novel Co20Ni20Fe20Al20Ti20 alloy fabricated by mechanical alloying and spark plasma sintering. Mater. Sci. Eng. A 2015, 644, 10–16. [Google Scholar] [CrossRef]
- Joseph, J.; Annasamy, M.; Kada, S.; Hodgson, P.; Barnett, M.; Fabijanic, D. Optimising the Al and Ti compositional window for the design of γ′ L12-strengthened Al-Co-Cr-Fe-Ni-Ti high entropy alloys. Mater. Sci. Eng. A 2022, 835, 142620. [Google Scholar] [CrossRef]
- Ferrari, A.; Dutta, B.; Gubaev, K.; Ikeda, Y.; Srinivasan, P.; Grabowski, B.; Körmann, F. Frontiers in atomistic simulations of high entropy alloys. J. Appl. Phys. 2020, 128, 150901. [Google Scholar] [CrossRef]
- Feng, R.; Liaw, P.K.; Gao, M.C.; Widom, M. First-Principles prediction of high-entropy-alloy stability. Npj Comput. Mater. 2017, 3, 50. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.M.; Ma, Z.J.; Qiu, S.; Zhang, L.J.; Zhang, S.Z.; Yang, R.; Hu, Q.M. Phase decomposition and strengthening in HfNbTaTiZr high entropy alloy from first-principles calculations. Acta Mater. 2022, 225, 117582. [Google Scholar] [CrossRef]
- Nonh, Z.-S.; Zhu, J.-C.; Yu, H.-L.; Lai, Z.-H. First principles calculation of intermetallic compounds in FeTiCoNiVCrMnCuAl system high entropy alloy. Trans. Nonfer. Met. Soc. China 2012, 22, 1437–1444. [Google Scholar] [CrossRef]
- Huang, S. The chemical ordering and elasticity in FeCoNiAl1−xTix high-entropy alloys. Scr. Mater. 2019, 168, 5–9. [Google Scholar] [CrossRef]
- Körmann, F.; Ruban, A.V.; Sluiter, M.H. Long-ranged interactions in bcc NbMoTaW high-entropy alloys. Mater. Res. Lett. 2017, 5, 35–40. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Hu, L.; Li, L.; Gao, J.; Zhang, Q. On the correlation between L12 nanoparticles and mechanical properties of (NiCo)52+2x(AlTi)4+2xFe29−4xCr15 (x = 0–4) high-entropy alloys. J. Alloys Compd. 2020, 817, 152750. [Google Scholar] [CrossRef]
- Guo, L.; Gu, J.; Gong, X.; Ni, S.; Song, M. CALPHAD aided design of high entropy alloy to achieve high strength via precipitate strengthening. Sci. China Mater. 2020, 63, 288–299. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Peng, P.; Gorbatov, O.I.; Ruban, A.V. Effective interactions and atomic ordering in Ni-rich Ni-Re alloys. Phys. Rev. B 2016, 94, 024111. [Google Scholar] [CrossRef] [Green Version]
- Drexler, A.; He, S.; Razumovskiy, V.; Romaner, L.; Ecker, W.; Pippan, R. Verification of the generalised chemical potential for stress-driven hydrogen diffusion in nickel. Philos. Mag. Lett. 2020, 100, 513–523. [Google Scholar] [CrossRef]
- Drexler, A.; He, S.; Pippan, R.; Romaner, L.; Razumovskiy, V.; Ecker, W. Hydrogen segregation near a crack tip in nickel. Scr. Mater. 2021, 194, 113697. [Google Scholar] [CrossRef]
- He, S.; Popov, M.N.; Ecker, W.; Pippan, R.; Razumovskiy, V. A theoretical insight into hydrogen clustering at defects in Ni. Philos. Mag. Lett. 2021, 101, 68–78. [Google Scholar] [CrossRef]
- He, S.; Peng, P.; Peng, L.; Chen, Y.; Wei, H.; Hu, Z. An interplay of sulfur and phosphorus at the γ-Ni/γ′-Ni3Al interface. J. Alloys Compd. 2014, 597, 243–248. [Google Scholar] [CrossRef]
- Kulo, N.; He, S.; Ecker, W.; Pippan, R.; Antretter, T.; Razumovskiy, V.I. Thermodynamic and mechanical stability of Ni3X-type intermetallic compounds. Intermetallics 2019, 114, 106604. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Open-Shell Transition Metals. Phys. Rev. B 1993, 48, 13115–13118. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- He, S.; Scheiber, D.; Jechtl, T.; Moitzi, F.; Peil, O.; Romaner, L.; Zamberger, S.; Povoden-Karadeniz, E.; Razumovskiy, V.; Ruban, A.V. Solubility and segregation of B in paramagnetic fcc Fe. Phys. Rev. Mater. 2022, 6, 023604. [Google Scholar] [CrossRef]
- Bandyopadhyay, J.; Gupta, K. Low temperature lattice parameter of nickel and some nickel-cobalt alloys and Grüneisen parameter of nickel. Cryogenics 1977, 17, 345–347. [Google Scholar] [CrossRef]
- He, S.; Ecker, W.; Pippan, R.; Razumovskiy, V.I. Hydrogen-enhanced decohesion mechanism of the special Σ5(012)[100] grain boundary in Ni with Mo and C solutes. Comput. Mater. Sci. 2019, 167, 100–110. [Google Scholar] [CrossRef]
- He, S.; Ecker, W.; Peil, O.E.; Pippan, R.; Razumovskiy, V.I. The effect of solute atoms on the bulk and grain boundary cohesion in Ni: Implications for hydrogen embrittlement. Materialia 2022, 21, 101293. [Google Scholar] [CrossRef]
- Andersson, J.O.; Helander, T.; Hoglund, L.; Shi, P.; Sundman, B. Thermo-Calc and DICTRA, computational tools for materials science. Calphad 2002, 26, 273–312. [Google Scholar] [CrossRef]
- Mao, H.H.; Chen, H.L.; Chen, Q. TCHEA1 A Thermodynamic Database Not Limited for High Entropy Alloys. J. Phase Equilib. Diffus. 2017, 38, 353–368. [Google Scholar] [CrossRef] [Green Version]
- Schönfeld, B.; Sax, C.R.; Zemp, J.; Engelke, M.; Boesecke, P.; Kresse, T.; Boll, T.; Al-Kassab, T.; Peil, O.E.; Ruban, A.V. Local order in Cr-Fe-Co-Ni: Experiment and electronic structure calculations. Phys. Rev. B 2019, 99, 014206. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; He, S.; Yi, Z.; Peng, P. Impact of correlative defects induced by double Re-addition on the ideal shear strength of γ′-Ni3Al phases. Comput. Mater. Sci. 2018, 152, 408–416. [Google Scholar] [CrossRef]
- Chen, Y.; He, S.; Yi, Z.; Peng, P. A synergistic reinforcement of Re and W for ideal shear strengths of γ′-Ni3Al phases. J. Phys. Chem. Solids 2019, 131, 34–43. [Google Scholar] [CrossRef]
- Slater, J.C. Atomic Radii in Crystals. J. Chem. Phys. 1964, 41, 3199–3204. [Google Scholar] [CrossRef]
- Chang, Y.J.; Yeh, A.C. The evolution of microstructures and high temperature properties of AlxCo1.5CrFeNi1.5Tiy high entropy alloys. J. Alloys Compd. 2015, 653, 379–385. [Google Scholar] [CrossRef]
- Kostiuchenko, T.; Ruban, A.V.; Neugebauer, J.; Shapeev, A.; Körmann, F. Short-range order in face-centered cubic VCoNi alloys. Phys. Rev. Mater. 2020, 4, 113802. [Google Scholar] [CrossRef]
- Zheng, W.; He, S.; Selleby, M.; He, Y.; Li, L.; Lu, X.-G.; Ågren, J. Thermodynamic assessment of the Al-C-Fe system. Calphad 2017, 58, 34–49. [Google Scholar] [CrossRef]
Figure 1.
First four coordination shell numbers in fcc lattice (a) and the atomic schematics of the L12-A3B intermetallic compound (b). The Arabic numerals 1, 2, 3, and 4 on the sphere of (a) present the coordination shell indexes 110, 200, 211, and 220, respectively, while “0” is the reference site. The positions of B atoms are indicated with golden spheres, while the positions of A atoms are indicated with blue spheres in (b).
Figure 1.
First four coordination shell numbers in fcc lattice (a) and the atomic schematics of the L12-A3B intermetallic compound (b). The Arabic numerals 1, 2, 3, and 4 on the sphere of (a) present the coordination shell indexes 110, 200, 211, and 220, respectively, while “0” is the reference site. The positions of B atoms are indicated with golden spheres, while the positions of A atoms are indicated with blue spheres in (b).

Figure 2.
Specific heat capacity of AlCoFeNi HEAs.

Figure 3.
Atomic configuration of AlCoFeNi at 2300 K. Al, Co, Fe, and Ni atoms are indicated with blue, orange, purple, and green spheres, respectively.
Figure 3.
Atomic configuration of AlCoFeNi at 2300 K. Al, Co, Fe, and Ni atoms are indicated with blue, orange, purple, and green spheres, respectively.

Figure 4.
Atomic configuration of AlCoFeNi HEAs at 300 K. The [100] and [010] crystal planes of the three-dimension AlCoFeNi HEAs are also shown at the bottom. Here Al, Co, Fe, and Ni atoms are indicated with blue, orange, purple, and green spheres, respectively. The L12-type precipitates are also framed in the snapshot.
Figure 4.
Atomic configuration of AlCoFeNi HEAs at 300 K. The [100] and [010] crystal planes of the three-dimension AlCoFeNi HEAs are also shown at the bottom. Here Al, Co, Fe, and Ni atoms are indicated with blue, orange, purple, and green spheres, respectively. The L12-type precipitates are also framed in the snapshot.

Figure 5.
Atomic SRO parameters of AlCoFeNi HEA at the first and second coordination shell from Monte Carlo simulations with the solute interactions from DFT calculations. The dashed line shows the order–disorder transition temperature.
Figure 5.
Atomic SRO parameters of AlCoFeNi HEA at the first and second coordination shell from Monte Carlo simulations with the solute interactions from DFT calculations. The dashed line shows the order–disorder transition temperature.

Figure 6.
Order–disorder transition temperature as a function of Ti contents in FeCoNiAl25−xTix HEAs.
Figure 6.
Order–disorder transition temperature as a function of Ti contents in FeCoNiAl25−xTix HEAs.

Figure 7.
Atomic configuration of AlCoFeNiTi HEAs at 300 K. The [100] and [010] crystal planes of the three-dimension AlCoFeNiTi HEAs are also shown at the bottom. Here, Al, Co, Fe, Ni, and Ti atoms are indicated with blue, orange, purple, green, and black spheres, respectively. The L12-type precipitates are also framed in the snapshot.
Figure 7.
Atomic configuration of AlCoFeNiTi HEAs at 300 K. The [100] and [010] crystal planes of the three-dimension AlCoFeNiTi HEAs are also shown at the bottom. Here, Al, Co, Fe, Ni, and Ti atoms are indicated with blue, orange, purple, green, and black spheres, respectively. The L12-type precipitates are also framed in the snapshot.

Figure 8.
A color map of the formation enthalpies (eV/atom) of L12-A3B precipitates in AlCoFeNiTi HEAs. The color scale ranges from green to red, with dark green indicating a formation energy value of −0.50 eV and dark red a formation energy value of 0.20 eV.
Figure 8.
A color map of the formation enthalpies (eV/atom) of L12-A3B precipitates in AlCoFeNiTi HEAs. The color scale ranges from green to red, with dark green indicating a formation energy value of −0.50 eV and dark red a formation energy value of 0.20 eV.

Figure 9.
Atomic SRO parameters of Co25Fe25Ni25Al20Ti5 HEA at the first and second coordination shells from Monte Carlo simulations with the solute interactions from DFT calculations. The dashed line shows the order–disorder transition temperature.
Figure 9.
Atomic SRO parameters of Co25Fe25Ni25Al20Ti5 HEA at the first and second coordination shells from Monte Carlo simulations with the solute interactions from DFT calculations. The dashed line shows the order–disorder transition temperature.

Figure 10.
The solidification of an equal-atomic AlCoFeNi alloy.

Figure 11.
The solidification of the equal-atomic Co25Fe25Ni25Al25−xTix alloy. Here, Ti is substituted for the content of a quarter Al in Co25Fe25Ni25Al25−xTix.
Figure 11.
The solidification of the equal-atomic Co25Fe25Ni25Al25−xTix alloy. Here, Ti is substituted for the content of a quarter Al in Co25Fe25Ni25Al25−xTix.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Pair interaction (in eV) of solute pairs in fcc Ni matrix. : Coordination shell index.
| 110 | 200 | 211 | 220 | 110 | 200 | 211 | 220 | |
|---|---|---|---|---|---|---|---|---|
| Al–Al | 0.243 | −0.009 | 0.003 | −0.013 | −0.099 | −0.002 | −0.003 | 0.044 |
| Al–Co | 0.063 | −0.010 | −0.016 | −0.012 | −0.083 | −0.003 | 0.000 | −0.012 |
| Al–Fe | 0.077 | −0.021 | −0.013 | 0.006 | −0.173 | −0.003 | 0.002 | 0.044 |
| Al–Ti | 0.127 | −0.024 | −0.012 | 0.007 | −0.144 | 0.003 | −0.195 | −0.001 |
| Co–Co | 0.003 | −0.002 | 0.001 | 0.010 | 0.000 | −0.001 | 0.001 | −0.001 |
| Co–Fe | 0.029 | 0.001 | 0.004 | −0.002 | −0.018 | 0.001 | −0.001 | 0.001 |
| Co–Ti | 0.203 | −0.082 | −0.022 | −0.020 | −0.011 | −0.005 | −0.007 | −0.012 |
| Fe–Fe | 0.115 | −0.015 | 0.015 | 0.010 | −0.014 | 0.000 | 0.001 | 0.000 |
| Fe–Ti | 0.129 | −0.073 | −0.018 | −0.013 | −0.025 | −0.004 | −0.003 | −0.016 |
| Ti–Ti | 0.367 | −0.129 | −0.052 | −0.054 | 0.087 | −0.053 | −0.059 | 0.087 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wu, Y.; Li, Z.; Feng, H.; He, S. Atomic Interactions and Order–Disorder Transition in FCC-Type FeCoNiAl1−xTix High-Entropy Alloys. Materials 2022, 15, 3992. https://0-doi-org.brum.beds.ac.uk/10.3390/ma15113992
AMA Style
Wu Y, Li Z, Feng H, He S. Atomic Interactions and Order–Disorder Transition in FCC-Type FeCoNiAl1−xTix High-Entropy Alloys. Materials. 2022; 15(11):3992. https://0-doi-org.brum.beds.ac.uk/10.3390/ma15113992
Chicago/Turabian StyleWu, Ying, Zhou Li, Hui Feng, and Shuang He. 2022. "Atomic Interactions and Order–Disorder Transition in FCC-Type FeCoNiAl1−xTix High-Entropy Alloys" Materials 15, no. 11: 3992. https://0-doi-org.brum.beds.ac.uk/10.3390/ma15113992
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.