Implications of HIV-1 Nef for “Shock and Kill” Strategies to Eliminate Latent Viral Reservoirs


1
Department of Molecular Biology and Biochemistry, Simon Fraser University, Burnaby, BC V5A 1S6, Canada
2
Faculty of Health Sciences, Simon Fraser University, Burnaby, BC V5A 1S6, Canada
3
British Columbia Centre for Excellence in HIV/AIDS, Vancouver, BC V6Z 1Y6, Canada
*
Author to whom correspondence should be addressed.
Viruses 2018, 10(12), 677; https://0-doi-org.brum.beds.ac.uk/10.3390/v10120677
Submission received: 7 November 2018
/
Revised: 26 November 2018
/
Accepted: 28 November 2018
/
Published: 30 November 2018
(This article belongs to the Special Issue CSV2018: The 2nd symposium of the Canadian Society for Virology (CSV))
{kind=link}
Abstract
:Finding a cure for HIV is challenging because the virus is able to integrate itself into the host cell genome and establish a silent state, called latency, allowing it to evade antiviral drugs and the immune system. Various “shock and kill” strategies are being explored in attempts to eliminate latent HIV reservoirs. The goal of these approaches is to reactivate latent viruses (“shock”), thereby exposing them to clearance by viral cytopathic effects or immune-mediated responses (“kill”). To date, there has been limited clinical success using these methods. In this review, we highlight various functions of the HIV accessory protein Nef and discuss their double-edged effects that may contribute to the limited effectiveness of current “shock and kill” methods to eradicate latent HIV reservoirs in treated individuals.
1. Introduction
The presence of long-lived latent HIV reservoirs is the major hurdle to achieving combination antiretroviral therapy (cART)-free viral remission and a potential cure. To date, the only case of an apparently successful HIV cure is the “Berlin patient”, who received two hematopoietic stem cell transplants from separate CCR5∆32 homozygous donors to treat his leukemia [1,2]. He displays no evidence of HIV infection despite remaining off therapy since 2007. Such transplants are exceptionally high-risk procedures and are thus not applicable to the global population of approximately 37 million HIV-infected individuals [3]. Furthermore, subsequent attempts to use similar transplantation strategies in HIV-infected individuals who were also undergoing cancer therapy have been unsuccessful, with viral rebound observed within weeks to months following cART discontinuation [4]. Therefore, the development of safer and more effective methods to reduce or eliminate latent HIV reservoirs in cART-treated individuals is a high priority for researchers and the community.
Different potentially curative approaches for HIV are currently under development, ranging from pharmacological approaches to immune-based and genetic therapies. Of these, the most intensively investigated strategies are the “shock and kill” methods to reduce or eliminate replication-competent latent HIV reservoirs in cART-treated individuals [5]. However, this strategy requires the induction of viral protein expression, including the regulatory and accessory proteins Tat, Rev, Nef, Vif, Vpr and Vpu, which could interfere with this process. In this article, we introduce the “shock and kill” method, describe the multi-functional viral accessory protein Nef, and consider how Nef may alter the efficiency of HIV cure approaches by modulating the viral reactivation from latency or the subsequent elimination by host immune mechanisms.
2. “Shock and Kill” Method
An illustration of the “shock and kill” method to eliminate latent HIV-infected cells in cART-suppressed individuals is shown in Figure 1A. Using latency-reversing agents (LRAs) that modulate cellular chromatin structure or otherwise stimulate the HIV 5’ LTR promoter, viral gene transcription is reactivated (“shock”) in latent HIV-infected cells. The subsequent viral protein expression, followed by the proteasomal processing and presentation of viral antigens on the cell surface in complex with human leukocyte antigen class I (HLA-I) molecules is then expected to result in the elimination (“kill”) of these cells by cytotoxic T lymphocytes (CTL). Alternatively, reactivated cells may undergo apoptosis due to the accumulation of viral cytopathic effects (CPE). By maintaining individuals on cART treatment during this process, viral replication and seeding of new HIV reservoirs is avoided.
2.1. Inefficient Viral Reactivation Using LRAs
Different classes of LRAs have been identified and tested for their ability to “shock” the latent HIV reservoir. In particular, pan-histone deacetylase inhibitors (HDACi), such as vorinostat [6], romidepsin [7], and panobinostat [8], are currently among the most promising classes of LRAs. Through the inhibition of multiple HDAC enzymes, HDACi increases the overall level of acetylation on histone molecules. This ultimately reduces chromatin condensation and promotes nonspecific increases in both host and viral gene expression. Many HDACi are FDA-approved for cancer treatment, and their pharmacological and toxicological profiles are known. Hence, HDACi have advanced quickly to human clinical trials in the context of HIV cure strategies, where they have demonstrated a range of abilities to induce latent viral reservoirs that broadly reflect their potency [9,10]. Several other classes of LRAs have also been tested in clinical studies. For example, disulfiram modestly reverses HIV latency by depleting PTEN (phosphatase and tensin homolog), which subsequently results in the activation of the PI3K/Akt pathway [11]. Protein kinase C (PKC) activators, such as prostratin and bryostatin, potently initiate HIV transcription in ex vivo experiments [12,13]; however, treatment with tolerable doses of bryostatin showed minimal ability to reactivate latent HIV in vivo in human studies [14]. Additional LRAs such as Toll-like receptor (TLR) agonists [15] and cytokines (i.e., interleukin-7 and -15) [16] are also being examined. Overall, none of these clinically relevant LRAs has been shown to reverse HIV latency potently in infected individuals. In fact, one ex vivo study indicated that many latent virus-infected cells remained uninduced despite strong T cell stimulation using phytohemagglutinin (PHA) or phorbol 12-myristate 13-acetate (PMA) plus ionomycin [17], suggesting that repeated induction using more potent LRAs may be necessary to achieve a clinically beneficial outcome.
2.2. Ineffective Clearance of Reactivated Cells
Despite some success with inducing latent HIV gene expression in cART-treated individuals, no significant reductions in viral reservoir size have been observed in vivo. This suggests that immune-mediated clearance of reactivated cells and/or viral CPE is inefficient. While it is often assumed that the production of HIV proteins such as Vif and Vpr could cause cell death due to viral CPE [18], Shan et al. demonstrated that the presence of viral protein expression was not associated with a spontaneous reduction of latent HIV-infected cells following reactivation using vorinostat [19]. In addition to the limited impact of viral CPE, the same study showed that CTL isolated from most cART-treated individuals were unable to eliminate latent cells reactivated ex vivo with HDACi efficiently without pre-stimulation using HIV antigens [19]. Nevertheless, a more recent study using Nef- and Gag-stimulated CTL was unsuccessful in eliminating reactivated cells and reducing the size of latent reservoirs [20]. The lack of CTL-mediated killing is potentially attributed to impaired CTL functionality and/or limited viral peptide presentation by reactivated cells. While there has been controversy regarding LRA-associated CTL impairment, results from clinical studies showed no evidence of CTL dysfunction in patients who were treated with HDACi [7,21]. Nonetheless, increasing evidence from in vitro studies are reporting associations between treatment with selected LRAs and CTL dysfunction. In particular, romidepsin, panobinostat, and vorinostat appeared to reduce the production of cytokines interferon-γ, tumor necrosis factor-α (TNF- α) and interleukin-2 [20,22]. Correspondingly, these HDACi-treated CTL displayed an impaired ability to eliminate HIV-infected cells [22]. On the other hand, limited studies have investigated HIV peptide presentation by reactivating cells. Clutton et al. observed impaired antigen presentation in reactivating cells due to inadvertent reduction in HLA class I expression following HDACi stimulation [23].
In summary, clinical studies have not reported a successful reduction of the latent viral reservoir in vivo [6,7,10,21]. The major hurdles encountered by these strategies include the inefficient induction of viral protein expression and the ineffective clearance of reactivated cells by the immune system.
3. Modulation of HIV-Infected Cells by Nef
HIV-1 Nef is a ~27 kDa myristoylated protein. It is encoded by the highly variable nef gene, which is located near the 3’ end of the viral genome. Nef is one of the earliest and most abundant viral proteins expressed by cells following infection [24,25,26,27], and presumably, following viral reactivation. Although Nef is often not required for HIV replication in vitro, it has been shown to be crucial for viral pathogenesis in vivo. Nef does not display any enzymatic activity; rather, it serves as a multi-functional adaptor protein that interacts with host proteins to interfere with a variety of processes in infected cells [28,29].
Nef downregulates CD4 expression on the surface of virus-infected cells [30] through clathrin-mediated endocytosis [31,32] and the increased endosomal retention [33,34] of CD4 molecules. Because CD4 is the primary receptor for HIV attachment and the entry into target cells, reduced CD4 expression allows a more efficient release of newly formed HIV particles [35,36], enhances virion infectivity [37] and inhibits superinfection [38]. Perhaps more important in the context of viral reactivation from latency, the interaction between CD4 and Env glycoproteins on the same cell has been shown to alter the conformation of Env to expose epitopes that are recognized by antibodies with potent antibody-dependent cellular cytotoxicity (ADCC) activity [39,40,41]. Hence, the efficient downregulation of CD4 by Nef can also protect infected cells from elimination by ADCC [42].
Nef is also well-known for its ability to evade the host immune response by selectively downregulating two HLA-I molecules, HLA-A and HLA-B [43,44,45]. This activity of Nef is genetically separable and mechanistically distinct from that of CD4 downregulation [46,47]. HLA-restricted CTL responses are associated with better control of viremia during primary HIV infection [48,49] and differential rates of clinical disease progression [50,51]. Thus, the reduced expression of HLA-A and HLA-B molecules on the surface of infected cells can protect them from CTL recognition and elimination [52]. In addition, the retention of HLA-C and HLA-E can inhibit the cytolytic activity of natural killer (NK) cells [44,45], preventing virus-infected cells from being eliminated through this innate immune mechanism.
A novel strategy to explain how Nef enhances viral infectivity was elucidated by two groups of researchers in 2015, who demonstrated that Nef can antagonize host restriction factors serine incorporator 3 and 5 (SERINC3/5) [53,54]. While understanding the precise mechanisms responsible for SERINC-mediated antiviral activity is currently an area of active investigation [55,56], the incorporation of SERINC3 or 5 into the membrane of newly formed virions significantly reduces their ability to form fusion pores with target cells, resulting in lower HIV infectivity [57]. To counteract these host restriction factors, Nef can downregulate SERINC3/5 from the surface of infected cells, which ultimately leads to the production of progeny virions that display higher infectivity [58].
Another critical role of Nef during HIV infection is its ability to modulate T cell signaling events. By downregulating CD4 and CD28 molecules on the surface of virus-infected T cells, Nef reduces the efficiency of T cell activation mediated through the T cell receptor (TCR) [30,59]. To further suppress the antigen-mediated stimulation of infected T cells, Nef binds Lck and redirects it to the trans-Golgi network (TGN), away from the plasma membrane where it can no longer participate in proximal TCR signal amplification events [60,61,62]. Together, the reduced availability of CD4, CD28 and Lck signaling molecules prevents the formation of an immunological synapse at the plasma membrane [60,61,63]. Paradoxically, while the altered trafficking of Lck interrupts TCR-mediated signaling at the plasma membrane, it permits the activation of Ras and downstream mitogen-activated protein kinase/extracellular signal-regulated kinases (MAPK/ERK) signaling events at the intracellular TGN compartment by forming a large complex that has been referred to as the Nef “signalosome” [62]. Alternatively, Nef can induce Ras activity via the formation of a Nef-associated kinase complex (NAKC), which is comprised of Nef, Lck, linker of activated T cells (LAT) and Ras proteins [62,64]. In synergy with activated Ras signaling, interaction between Nef and the endoplasmic reticulum-resident inositol triphosphate receptor (IP3R) can trigger calcium flux into the cytosol and induce TCR-independent activation of nuclear factor of activated T cells (NFAT) [65,66]. Together, Nef’s uncoupled effects on T cell activation pathways can simultaneously suppress activation-induced cell death (AICD) triggered by extracellular antigen recognition and also increase viral gene transcription.
Current evidence indicates that Nef may protect virus-infected cells from apoptosis, while simultaneously eliciting the death of bystander immune cells, which may enhance pathogenesis. To prevent infected cells from undergoing programmed cell death, Nef inhibits the activities of apoptosis signal-regulating kinase 1 (ASK1) [67], tumor suppressor p53 [68] and the pro-apoptotic protein Bcl-2-associated death promoter (BAD) [69]. In contrast, secreted Nef can upregulate Fas ligand induced apoptosis of uninfected bystander CD4+ T cells and CTL [70,71,72], thereby dampening the local immune response against HIV-infected cells. Transgenic mice expressing Nef display AIDS-like pathologies [73], raising the possibility that the induction of Nef by “shock and kill” methods may lead to toxicity, particularly in localized tissues that harbor latent viral reservoirs, such as lymph nodes or the central nervous system [74,75].
Finally, by manipulating cytoskeletal dynamics, Nef may promote a more permissive cellular environment to support viral replication or spread. Nef associates with the serine/threonine kinase p21 activated kinase 2 (PAK2) in a multiprotein complex and redirects its phosphorylation to a novel target, the actin depolymerization factor cofilin [76,77], which results in reduced F-actin turnover and actin cytoskeleton remodeling [78,79]. Consequently, this prevents F-actin accumulation at the immunological synapses upon TCR engagement [61], thereby contributing to the inhibition of AICD and prolonging the survival of infected cells [80].
4. The Double-Edged Effect of HIV-1 Nef
4.1. How Nef Might Enhance “Shock and Kill” Strategies
Many factors that promote HIV latency are likely to contribute to the inducibility of viral reservoirs upon treatment with an LRA. Even though Nef’s role in the context of latency is not fully characterized, several studies have highlighted its ability to induce viral reactivation. For example, Fujinaga et al. demonstrated that exogenous Nef activated virus production in latent cell lines (i.e., MOLT-20-2 and U1) as well as in peripheral blood mononuclear cells (PBMC) isolated from asymptomatic HIV-infected individuals [81]. Follow-up studies by the same group suggested that this effect was driven by Nef’s ability to induce Ras-mediated MAPK/ERK signaling [82]. The effect of Nef on latency reversal was confirmed in a separate study using U1 cells [83]. More recently, treatment using exogenous Nef alone was also found to be sufficient to activate the PI3K/Akt pathway and to increase HIV reactivation in the Jurkat-derived 1G5 latent T cell line [84].
In addition to Ras and Akt, Nef can also regulate the cellular activation status by interacting with other host proteins. Hence, it is not entirely surprising that Nef could activate latent HIV-infected cell lines. For instance, the presence of Nef can trigger the formation of NAKC and induce downstream Ras/MAPK activity [62,64]. Through its interaction with IP3R, Nef can trigger calcium flux into the cytosol and induce NFAT activation [65,66]. In both cases, early production of Nef during viral reactivation might enhance latent T cell activation. Moreover, previous studies reported that Nef can be released into the extracellular space either in soluble form [85,86] or within exosomes [87,88]. Both soluble and exosome-associated Nef have been shown to induce HIV reactivation in latently infected cells [81,89], but their proposed molecular mechanisms are distinct. In particular, soluble Nef may bind non-specifically to the surface of latent HIV-infected cells and be internalized via endocytosis [90,91]. After entering the cell, Nef can induce Ras/MAPK [82] and PI3K/Akt [84] signaling pathways that ultimately activate viral gene transcription. On the other hand, Nef increases the production of exosomes containing activated ADAM17 (a disintegrin and metalloprotease domain 17) [92], an enzyme that converts pro-TNF-α into its active form. The uptake of ADAM17-containing exosomes by target cells can induce the release of TNF-α [93], which subsequently binds to TNF receptor type 1 and activates NF-κB and c-Jun N-terminal kinase (JNK) pathways [94]. Additionally, Nef has been shown to increase exosome release, which presumably enhances the transfer of Nef-associated signaling activities to nearby cells [95]. Nef-mediated effects on cellular signaling are complex and their potential impacts on viral reactivation are not mutually exclusive. In fact, based on these previous findings, we speculate that Nef’s ability to enhance viral reactivation may be attributed to a positive feedback loop of cellular activation. Specifically, upon stimulation with LRAs, early Nef expression may increase viral gene expression. Subsequent secretion of soluble Nef and Nef/ADAM17-contaning exosomes could further increase the activation of latent cells through direct effects of Nef or TNF-mediated signaling pathways.
4.2. How Nef Might Impair “Shock and Kill” Strategies
Recent results by Huang et al. suggested that replication-competent latent proviruses may display resistance to elimination by HIV-specific CTL [96]. Hence, apart from LRA-associated impairments in CTL functions, the expression of Nef immediately following viral reactivation may further reduce the ability of CTL to recognize and eliminate latent reservoirs. Specifically, the ability of Nef to selectively downregulate surface HLA-I molecules [43,44,45] may allow reactivated cells to evade immune surveillance. In support of this theory, Mujib et al. used small molecules designed to inhibit Nef, which partially reversed HLA downregulation and promoted the elimination of reactivating cells by HIV-specific CTL [97]. While the ability of Nef to downregulate CD4 can prevent the ADCC-mediated elimination of productive virus-infected cells [42], no studies have examined this question in the context of latent viral reservoirs.
As the leading class of LRAs, HDACi triggers various apoptotic pathways to induce tumor cell death (reviewed in Reference [98]). While this strongly suggests that the use of certain LRAs could inadvertently induce apoptosis of latent reservoirs upon viral reactivation, the mechanism(s) involved have not been explored. Nonetheless, the ability of Nef to counteract multiple apoptotic pathways and promote cell survival could further hinder the clearance of reactivating reservoirs. First, Nef can bind directly to ASK-1 [67], an importance intermediate of Fas- and TNF-α-induced death signaling cascades [99,100], thereby preventing its dissociation from negative regulator thioredoxin [101]. Consequently, this inhibits the ASK-1-mediated activation of the downstream JNK signaling pathway to induce apoptosis [102]. Second, Nef can protect cells from undergoing p53-mediated apoptosis by binding and destabilizing p53, causing an overall reduction of this protein [68]. Third, the ability of Nef to associate with PI3K can induce downstream PAK-mediated phosphorylation of pro-apoptotic protein BAD [69]. Since phosphorylated BAD is incapable of forming heterodimers with anti-apoptotic proteins Bcl-2 and Bcl-XL, these proteins remain active for the promotion of cell survival [103].
Furthermore, broad reactivation of HIV proteins using LRAs may lead to AICD among the proportion of reservoir cells that is HIV-specific [104]. In this case, Nef’s ability to downregulate CD4 expression, modulate T cell signaling and cytoskeleton rearrangement may protect these cells from undergoing AICD. Taken together, early Nef expression following LRA-induced viral reactivation could inhibit CTL-mediated killing, apoptosis and AICD of latent reservoir, which may contribute to the lack of success seen using current “shock and kill” methods.
5. Conclusions
The efficiency of “shock and kill” strategies is determined by the degree to which latent HIV reservoirs are reactivated and subsequently eliminated in the host. We hypothesize that Nef might play a “dual” role in modulating both of these important factors (as illustrated in Figure 1B). While studies have demonstrated the use of exogeneous Nef to induce viral reactivation, Nef’s ability to mediate immune evasion and to enhance cell survival through the inhibition of apoptosis are also well documented. Nef leads to the downregulation of HLA-I molecules on the cell surface [43,44,45], which reduces the presentation of viral peptide antigens and impairs CTL-mediated recognition and cytolytic activity against reactivating reservoirs [52]. Additionally, Nef’s ability to modulate apoptotic pathways may prevent reactivated cells from dying due to viral cytopathic effects [67,69]. In contrast, latent cells that lack functional Nef may be unable to produce viral proteins efficiently. As a result, the presentation of viral peptides may be limited despite retaining high levels of HLA-I expression on the cell surface. Hence, the diverse roles played by Nef may create double-edged effects in the setting of a “shock and kill” strategy. Further studies to explore the possible impact of Nef and other viral accessory proteins, such as Vpr and Vpu, during HIV reactivation from latency may lead to enhanced clinical interventions.
Funding
This research was funded by the Canadian Institutes for Health Research (CIHR) (PJT-148621, HIG-133050) and the National Institutes of Health, USA (UM1-AI126617). The article processing charges were funded by CIHR. X.T.K. received Frederick Banting and Charles Best Canada Graduate Scholarship Doctoral Award from CIHR. M.A.B. holds a Canada Research Chair, Tier 2, in Viral Pathogenesis and Immunity.
Acknowledgments
The authors are grateful to the Canadian HIV Cure Enterprise (CanCURE) and the BELIEVE Martin Delaney Collaboratory for research and training support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hutter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Mussig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kucherer, C.; Blau, O.; et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Yukl, S.A.; Boritz, E.; Busch, M.; Bentsen, C.; Chun, T.W.; Douek, D.; Eisele, E.; Haase, A.; Ho, Y.C.; Hutter, G.; et al. Challenges in detecting HIV persistence during potentially curative interventions: A study of the Berlin patient. PLoS Pathog. 2013, 9, e1003347. [Google Scholar] [CrossRef] [PubMed]
- UNAIDS/WHO. Global HIV & AIDS Statistics—2018 Fact Sheet; UNAIDS/WHO: Geneva, Switzerland, 2018. [Google Scholar]
- Henrich, T.J.; Hanhauser, E.; Marty, F.M.; Sirignano, M.N.; Keating, S.; Lee, T.H.; Robles, Y.P.; Davis, B.T.; Li, J.Z.; Heisey, A.; et al. Antiretroviral-free HIV-1 remission and viral rebound after allogeneic stem cell transplantation: Report of 2 cases. Ann. Intern. Med. 2014, 161, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G. HIV: Shock and kill. Nature 2012, 487, 439–440. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.; Choudhary, S.K.; Kuruc, J.D.; Crooks, A.M.; Parker, D.C.; Anderson, E.M.; Kearney, M.F.; Strain, M.C.; et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sogaard, O.S.; Graversen, M.E.; Leth, S.; Olesen, R.; Brinkmann, C.R.; Nissen, S.K.; Kjaer, A.S.; Schleimann, M.H.; Denton, P.W.; Hey-Cunningham, W.J.; et al. The Depsipeptide Romidepsin Reverses HIV-1 Latency In Vivo. PLoS Pathog. 2015, 11, e1005142. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.A.; Tolstrup, M.; Brinkmann, C.R.; Olesen, R.; Erikstrup, C.; Solomon, A.; Winckelmann, A.; Palmer, S.; Dinarello, C.; Buzon, M.; et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: A phase 1/2, single group, clinical trial. Lancet HIV 2014, 1, e13–e21. [Google Scholar] [CrossRef]
- Delagreverie, H.M.; Delaugerre, C.; Lewin, S.R.; Deeks, S.G.; Li, J.Z. Ongoing Clinical Trials of Human Immunodeficiency Virus Latency-Reversing and Immunomodulatory Agents. Open Forum Infect. Dis. 2016, 3, ofw189. [Google Scholar] [CrossRef] [PubMed]
- Thorlund, K.; Horwitz, M.S.; Fife, B.T.; Lester, R.; Cameron, D.W. Landscape review of current HIV ‘kick and kill’ cure research—Some kicking, not enough killing. BMC Infect. Dis. 2017, 17, 595. [Google Scholar] [CrossRef] [PubMed]
- Doyon, G.; Zerbato, J.; Mellors, J.W.; Sluis-Cremer, N. Disulfiram reactivates latent HIV-1 expression through depletion of the phosphatase and tensin homolog. AIDS 2013, 27, F7–F11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korin, Y.D.; Brooks, D.G.; Brown, S.; Korotzer, A.; Zack, J.A. Effects of prostratin on T-cell activation and human immunodeficiency virus latency. J. Virol. 2002, 76, 8118–8123. [Google Scholar] [CrossRef] [PubMed]
- Mehla, R.; Bivalkar-Mehla, S.; Zhang, R.; Handy, I.; Albrecht, H.; Giri, S.; Nagarkatti, P.; Nagarkatti, M.; Chauhan, A. Bryostatin modulates latent HIV-1 infection via PKC and AMPK signaling but inhibits acute infection in a receptor independent manner. PLoS ONE 2010, 5, e11160. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, C.; Serrano-Villar, S.; Madrid-Elena, N.; Perez-Elias, M.J.; Martin, M.E.; Barbas, C.; Ruiperez, J.; Munoz, E.; Munoz-Fernandez, M.A.; Castor, T.; et al. Bryostatin-1 for latent virus reactivation in HIV-infected patients on antiretroviral therapy. AIDS 2016, 30, 1385–1392. [Google Scholar] [CrossRef] [PubMed]
- Vibholm, L.; Schleimann, M.H.; Hojen, J.F.; Benfield, T.; Offersen, R.; Rasmussen, K.; Olesen, R.; Dige, A.; Agnholt, J.; Grau, J.; et al. Short-Course Toll-Like Receptor 9 Agonist Treatment Impacts Innate Immunity and Plasma Viremia in Individuals with Human Immunodeficiency Virus Infection. Clin. Infect. Dis. 2017, 64, 1686–1695. [Google Scholar] [CrossRef] [PubMed]
- Sereti, I.; Dunham, R.M.; Spritzler, J.; Aga, E.; Proschan, M.A.; Medvik, K.; Battaglia, C.A.; Landay, A.L.; Pahwa, S.; Fischl, M.A.; et al. IL-7 administration drives T cell-cycle entry and expansion in HIV-1 infection. Blood 2009, 113, 6304–6314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, Y.C.; Shan, L.; Hosmane, N.N.; Wang, J.; Laskey, S.B.; Rosenbloom, D.I.; Lai, J.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 2013, 155, 540–551. [Google Scholar] [CrossRef] [PubMed]
- Stellbrink, H.J.; van Lunzen, J.; Westby, M.; O’Sullivan, E.; Schneider, C.; Adam, A.; Weitner, L.; Kuhlmann, B.; Hoffmann, C.; Fenske, S.; et al. Effects of interleukin-2 plus highly active antiretroviral therapy on HIV-1 replication and proviral DNA (COSMIC trial). AIDS 2002, 16, 1479–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, L.; Deng, K.; Shroff, N.S.; Durand, C.M.; Rabi, S.A.; Yang, H.C.; Zhang, H.; Margolick, J.B.; Blankson, J.N.; Siliciano, R.F. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity 2012, 36, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Walker-Sperling, V.E.; Pohlmeyer, C.W.; Tarwater, P.M.; Blankson, J.N. The Effect of Latency Reversal Agents on Primary CD8+ T Cells: Implications for Shock and Kill Strategies for Human Immunodeficiency Virus Eradication. EBioMedicine 2016, 8, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.H.; Wightman, F.; Solomon, A.; Ghneim, K.; Ahlers, J.; Cameron, M.J.; Smith, M.Z.; Spelman, T.; McMahon, J.; Velayudham, P.; et al. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog. 2014, 10, e1004473. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.B.; O’Connor, R.; Mueller, S.; Foley, M.; Szeto, G.L.; Karel, D.; Lichterfeld, M.; Kovacs, C.; Ostrowski, M.A.; Trocha, A.; et al. Histone deacetylase inhibitors impair the elimination of HIV-infected cells by cytotoxic T-lymphocytes. PLoS Pathog. 2014, 10, e1004287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clutton, G.; Xu, Y.; Baldoni, P.L.; Mollan, K.R.; Kirchherr, J.; Newhard, W.; Cox, K.; Kuruc, J.D.; Kashuba, A.; Barnard, R.; et al. The differential short- and long-term effects of HIV-1 latency-reversing agents on T cell function. Sci. Rep. 2016, 6, 30749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.Y.; Byrn, R.; Groopman, J.; Baltimore, D. Temporal aspects of DNA and RNA synthesis during human immunodeficiency virus infection: Evidence for differential gene expression. J. Virol. 1989, 63, 3708–3713. [Google Scholar] [PubMed]
- Robert-Guroff, M.; Popovic, M.; Gartner, S.; Markham, P.; Gallo, R.C.; Reitz, M.S. Structure and expression of tat-, rev-, and nef-specific transcripts of human immunodeficiency virus type 1 in infected lymphocytes and macrophages. J. Virol. 1990, 64, 3391–3398. [Google Scholar] [PubMed]
- Klotman, M.E.; Kim, S.; Buchbinder, A.; DeRossi, A.; Baltimore, D.; Wong-Staal, F. Kinetics of expression of multiply spliced RNA in early human immunodeficiency virus type 1 infection of lymphocytes and monocytes. Proc. Natl. Acad. Sci. USA 1991, 88, 5011–5015. [Google Scholar] [CrossRef] [PubMed]
- Zaunders, J.J.; Geczy, A.F.; Dyer, W.B.; McIntyre, L.B.; Cooley, M.A.; Ashton, L.J.; Raynes-Greenow, C.H.; Learmont, J.; Cooper, D.A.; Sullivan, J.S. Effect of long-term infection with nef-defective attenuated HIV type 1 on CD4+ and CD8+ T lymphocytes: Increased CD45RO+CD4+ T lymphocytes and limited activation of CD8+ T lymphocytes. AIDS Res. Hum. Retroviruses 1999, 15, 1519–1527. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, E.N.; Dikeakos, J.D. HIV-1 Nef: A master manipulator of the membrane trafficking machinery mediating immune evasion. Biochim. Biophys. Acta 2015, 1850, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Faust, T.B.; Binning, J.M.; Gross, J.D.; Frankel, A.D. Making Sense of Multifunctional Proteins: Human Immunodeficiency Virus Type 1 Accessory and Regulatory Proteins and Connections to Transcription. Annu. Rev. Virol. 2017, 4, 241–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, J.V.; Miller, A.D. Serine phosphorylation-independent downregulation of cell-surface CD4 by nef. Nature 1991, 350, 508–511. [Google Scholar] [CrossRef] [PubMed]
- Aiken, C.; Konner, J.; Landau, N.R.; Lenburg, M.E.; Trono, D. Nef induces CD4 endocytosis: Requirement for a critical dileucine motif in the membrane-proximal CD4 cytoplasmic domain. Cell 1994, 76, 853–864. [Google Scholar] [CrossRef]
- Laguette, N.; Bregnard, C.; Bouchet, J.; Benmerah, A.; Benichou, S.; Basmaciogullari, S. Nef-induced CD4 endocytosis in human immunodeficiency virus type 1 host cells: Role of p56lck kinase. J. Virol. 2009, 83, 7117–7128. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Chang, S.H.; Kwon, J.H.; Rhee, S.S. HIV-1 Nef plays an essential role in two independent processes in CD4 down-regulation: Dissociation of the CD4-p56(lck) complex and targeting of CD4 to lysosomes. Virology 1999, 257, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Rose, J.J.; Janvier, K.; Chandrasekhar, S.; Sekaly, R.P.; Bonifacino, J.S.; Venkatesan, S. CD4 down-regulation by HIV-1 and simian immunodeficiency virus (SIV) Nef proteins involves both internalization and intracellular retention mechanisms. J. Biol. Chem. 2005, 280, 7413–7426. [Google Scholar] [CrossRef] [PubMed]
- Ross, T.M.; Oran, A.E.; Cullen, B.R. Inhibition of HIV-1 progeny virion release by cell-surface CD4 is relieved by expression of the viral Nef protein. Curr. Biol. CB 1999, 9, 613–621. [Google Scholar] [CrossRef]
- Lama, J.; Mangasarian, A.; Trono, D. Cell-surface expression of CD4 reduces HIV-1 infectivity by blocking Env incorporation in a Nef- and Vpu-inhibitable manner. Curr. Biol. 1999, 9, 622–631. [Google Scholar] [CrossRef]
- Arganaraz, E.R.; Schindler, M.; Kirchhoff, F.; Cortes, M.J.; Lama, J. Enhanced CD4 down-modulation by late stage HIV-1 nef alleles is associated with increased Env incorporation and viral replication. J. Biol. Chem. 2003, 278, 33912–33919. [Google Scholar] [CrossRef] [PubMed]
- Wildum, S.; Schindler, M.; Munch, J.; Kirchhoff, F. Contribution of Vpu, Env, and Nef to CD4 down-modulation and resistance of human immunodeficiency virus type 1-infected T cells to superinfection. J. Virol. 2006, 80, 8047–8059. [Google Scholar] [CrossRef] [PubMed]
- Veillette, M.; Desormeaux, A.; Medjahed, H.; Gharsallah, N.E.; Coutu, M.; Baalwa, J.; Guan, Y.; Lewis, G.; Ferrari, G.; Hahn, B.H.; et al. Interaction with cellular CD4 exposes HIV-1 envelope epitopes targeted by antibody-dependent cell-mediated cytotoxicity. J. Virol. 2014, 88, 2633–2644. [Google Scholar] [CrossRef] [PubMed]
- Veillette, M.; Coutu, M.; Richard, J.; Batraville, L.A.; Dagher, O.; Bernard, N.; Tremblay, C.; Kaufmann, D.E.; Roger, M.; Finzi, A. The HIV-1 gp120 CD4-bound conformation is preferentially targeted by antibody-dependent cellular cytotoxicity-mediating antibodies in sera from HIV-1-infected individuals. J. Virol. 2015, 89, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Prevost, J.; Richard, J.; Medjahed, H.; Alexander, A.; Jones, J.; Kappes, J.C.; Ochsenbauer, C.; Finzi, A. Incomplete Downregulation of CD4 Expression Affects HIV-1 Env Conformation and Antibody-Dependent Cellular Cytotoxicity Responses. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Alsahafi, N.; Ding, S.; Richard, J.; Markle, T.; Brassard, N.; Walker, B.; Lewis, G.K.; Kaufmann, D.E.; Brockman, M.A.; Finzi, A. Nef Proteins from HIV-1 Elite Controllers Are Inefficient at Preventing Antibody-Dependent Cellular Cytotoxicity. J. Virol. 2015, 90, 2993–3002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, O.; Marechal, V.; Le Gall, S.; Lemonnier, F.; Heard, J.M. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat. Med. 1996, 2, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, S.; Erdtmann, L.; Benichou, S.; Berlioz-Torrent, C.; Liu, L.; Benarous, R.; Heard, J.M.; Schwartz, O. Nef interacts with the mu subunit of clathrin adaptor complexes and reveals a cryptic sorting signal in MHC I molecules. Immunity 1998, 8, 483–495. [Google Scholar] [CrossRef]
- Cohen, G.B.; Gandhi, R.T.; Davis, D.M.; Mandelboim, O.; Chen, B.K.; Strominger, J.L.; Baltimore, D. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 1999, 10, 661–671. [Google Scholar] [CrossRef]
- Mangasarian, A.; Piguet, V.; Wang, J.K.; Chen, Y.L.; Trono, D. Nef-induced CD4 and major histocompatibility complex class I (MHC-I) down-regulation are governed by distinct determinants: N-terminal alpha helix and proline repeat of Nef selectively regulate MHC-I trafficking. J. Virol. 1999, 73, 1964–1973. [Google Scholar] [PubMed]
- Pereira, E.A.; daSilva, L.L. HIV-1 Nef: Taking Control of Protein Trafficking. Traffic 2016, 17, 976–996. [Google Scholar] [CrossRef] [PubMed]
- Koup, R.A.; Safrit, J.T.; Cao, Y.; Andrews, C.A.; McLeod, G.; Borkowsky, W.; Farthing, C.; Ho, D.D. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J. Virol. 1994, 68, 4650–4655. [Google Scholar] [PubMed]
- Borrow, P.; Lewicki, H.; Wei, X.; Horwitz, M.S.; Peffer, N.; Meyers, H.; Nelson, J.A.; Gairin, J.E.; Hahn, B.H.; Oldstone, M.B.; et al. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat. Med. 1997, 3, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Kiepiela, P.; Leslie, A.J.; Honeyborne, I.; Ramduth, D.; Thobakgale, C.; Chetty, S.; Rathnavalu, P.; Moore, C.; Pfafferott, K.J.; Hilton, L.; et al. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature 2004, 432, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Altfeld, M.; Kalife, E.T.; Qi, Y.; Streeck, H.; Lichterfeld, M.; Johnston, M.N.; Burgett, N.; Swartz, M.E.; Yang, A.; Alter, G.; et al. HLA Alleles Associated with Delayed Progression to AIDS Contribute Strongly to the Initial CD8(+) T Cell Response against HIV-1. PLoS Med. 2006, 3, e403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, K.L.; Chen, B.K.; Kalams, S.A.; Walker, B.D.; Baltimore, D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 1998, 391, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.; Chande, A.; Ziglio, S.; de Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usami, Y.; Wu, Y.; Gottlinger, H.G. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trautz, B.; Wiedemann, H.; Luchtenborg, C.; Pierini, V.; Kranich, J.; Glass, B.; Krausslich, H.G.; Brocker, T.; Pizzato, M.; Ruggieri, A.; et al. The host-cell restriction factor SERINC5 restricts HIV-1 infectivity without altering the lipid composition and organization of viral particles. J. Biol. Chem. 2017, 292, 13702–13713. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhou, T.; Yang, J.; Lin, Y.; Shi, J.; Zhang, X.; Frabutt, D.A.; Zeng, X.; Li, S.; Venta, P.J.; et al. Identification of SERINC5-001 as the Predominant Spliced Isoform for HIV-1 Restriction. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Sood, C.; Marin, M.; Chande, A.; Pizzato, M.; Melikyan, G.B. SERINC5 protein inhibits HIV-1 fusion pore formation by promoting functional inactivation of envelope glycoproteins. J. Biol. Chem. 2017, 292, 6014–6026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trautz, B.; Pierini, V.; Wombacher, R.; Stolp, B.; Chase, A.J.; Pizzato, M.; Fackler, O.T. The Antagonism of HIV-1 Nef to SERINC5 Particle Infectivity Restriction Involves the Counteraction of Virion-Associated Pools of the Restriction Factor. J. Virol. 2016, 90, 10915–10927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swigut, T.; Shohdy, N.; Skowronski, J. Mechanism for down-regulation of CD28 by Nef. EMBO J. 2001, 20, 1593–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoulouze, M.I.; Sol-Foulon, N.; Blanchet, F.; Dautry-Varsat, A.; Schwartz, O.; Alcover, A. Human immunodeficiency virus type-1 infection impairs the formation of the immunological synapse. Immunity 2006, 24, 547–561. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, J.M.; Eickel, N.; Haller, C.; Schindler, M.; Fackler, O.T. Inhibition of T-cell receptor-induced actin remodeling and relocalization of Lck are evolutionarily conserved activities of lentiviral Nef proteins. J. Virol. 2009, 83, 11528–11539. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Rudolph, J.M.; Abraham, L.; Habermann, A.; Haller, C.; Krijnse-Locker, J.; Fackler, O.T. HIV-1 Nef compensates for disorganization of the immunological synapse by inducing trans-Golgi network-associated Lck signaling. Blood 2012, 119, 786–797. [Google Scholar] [CrossRef] [PubMed]
- Haller, C.; Rauch, S.; Fackler, O.T. HIV-1 Nef employs two distinct mechanisms to modulate Lck subcellular localization and TCR induced actin remodeling. PLoS ONE 2007, 2, e1212. [Google Scholar] [CrossRef] [PubMed]
- Witte, V.; Laffert, B.; Gintschel, P.; Krautkramer, E.; Blume, K.; Fackler, O.T.; Baur, A.S. Induction of HIV transcription by Nef involves Lck activation and protein kinase C theta raft recruitment leading to activation of ERK1/2 but not NFκB. J. Immunol. 2008, 181, 8425–8432. [Google Scholar] [CrossRef] [PubMed]
- Manninen, A.; Renkema, G.H.; Saksela, K. Synergistic activation of NFAT by HIV-1 nef and the Ras/MAPK pathway. J. Biol. Chem. 2000, 275, 16513–16517. [Google Scholar] [CrossRef] [PubMed]
- Manninen, A.; Saksela, K. HIV-1 Nef interacts with inositol trisphosphate receptor to activate calcium signaling in T cells. J. Exp. Med. 2002, 195, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Geleziunas, R.; Xu, W.; Takeda, K.; Ichijo, H.; Greene, W.C. HIV-1 Nef inhibits ASK1-dependent death signalling providing a potential mechanism for protecting the infected host cell. Nature 2001, 410, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Greenway, A.L.; McPhee, D.A.; Allen, K.; Johnstone, R.; Holloway, G.; Mills, J.; Azad, A.; Sankovich, S.; Lambert, P. Human immunodeficiency virus type 1 Nef binds to tumor suppressor p53 and protects cells against p53-mediated apoptosis. J. Virol. 2002, 76, 2692–2702. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Witte, V.; Laffert, B.; Blume, K.; Stromer, E.; Trapp, S.; d’Aloja, P.; Schurmann, A.; Baur, A.S. HIV-1 Nef associated PAK and PI3-kinases stimulate Akt-independent Bad-phosphorylation to induce anti-apoptotic signals. Nat. Med. 2001, 7, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.H.; Tudor-Williams, G.; Banda, N.K.; Cotton, M.F.; Curiel, T.; Monks, C.; Baba, T.W.; Ruprecht, R.M.; Kupfer, A. Apoptosis occurs predominantly in bystander cells and not in productively infected cells of HIV- and SIV-infected lymph nodes. Nat. Med. 1995, 1, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.N.; Laffert, B.; Screaton, G.R.; Kraft, M.; Wolf, D.; Kolanus, W.; Mongkolsapay, J.; McMichael, A.J.; Baur, A.S. Induction of Fas ligand expression by HIV involves the interaction of Nef with the T cell receptor zeta chain. J. Exp. Med. 1999, 189, 1489–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muthumani, K.; Choo, A.Y.; Hwang, D.S.; Premkumar, A.; Dayes, N.S.; Harris, C.; Green, D.R.; Wadsworth, S.A.; Siekierka, J.J.; Weiner, D.B. HIV-1 Nef-induced FasL induction and bystander killing requires p38 MAPK activation. Blood 2005, 106, 2059–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, Z.; Weng, X.; Kay, D.G.; Poudrier, J.; Lowell, C.; Jolicoeur, P. The pathogenicity of human immunodeficiency virus (HIV) type 1 Nef in CD4C/HIV transgenic mice is abolished by mutation of its SH3-binding domain, and disease development is delayed in the absence of Hck. J. Virol. 2001, 75, 9378–9392. [Google Scholar] [CrossRef] [PubMed]
- Spector, S.A.; Rappaport, J. HIV cure strategists: Ignore the central nervous system at your patients’ peril. AIDS 2017, 31, 167–168. [Google Scholar] [CrossRef] [PubMed]
- Gama, L.; Abreu, C.; Shirk, E.N.; Queen, S.E.; Beck, S.E.; Metcalf Pate, K.A.; Bullock, B.T.; Zink, M.C.; Mankowski, J.L.; Clements, J.E. SIV Latency in Macrophages in the CNS. Curr. Top. Microbiol. Immunol. 2018. [Google Scholar] [CrossRef]
- Fackler, O.T.; Luo, W.; Geyer, M.; Alberts, A.S.; Peterlin, B.M. Activation of Vav by Nef induces cytoskeletal rearrangements and downstream effector functions. Mol. Cell 1999, 3, 729–739. [Google Scholar] [CrossRef]
- Rauch, S.; Pulkkinen, K.; Saksela, K.; Fackler, O.T. Human immunodeficiency virus type 1 Nef recruits the guanine exchange factor Vav1 via an unexpected interface into plasma membrane microdomains for association with p21-activated kinase 2 activity. J. Virol. 2008, 82, 2918–2929. [Google Scholar] [CrossRef] [PubMed]
- Stolp, B.; Reichman-Fried, M.; Abraham, L.; Pan, X.; Giese, S.I.; Hannemann, S.; Goulimari, P.; Raz, E.; Grosse, R.; Fackler, O.T. HIV-1 Nef interferes with host cell motility by deregulation of Cofilin. Cell Host Microbe 2009, 6, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Stolp, B.; Abraham, L.; Rudolph, J.M.; Fackler, O.T. Lentiviral Nef proteins utilize PAK2-mediated deregulation of cofilin as a general strategy to interfere with actin remodeling. J. Virol. 2010, 84, 3935–3948. [Google Scholar] [CrossRef] [PubMed]
- Fackler, O.T.; Alcover, A.; Schwartz, O. Modulation of the immunological synapse: A key to HIV-1 pathogenesis? Nat. Rev. Immunol. 2007, 7, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Fujinaga, K.; Zhong, Q.; Nakaya, T.; Kameoka, M.; Meguro, T.; Yamada, K.; Ikuta, K. Extracellular Nef protein regulates productive HIV-1 infection from latency. J. Immunol. 1995, 155, 5289–5298. [Google Scholar] [PubMed]
- Tobiume, M.; Fujinaga, K.; Suzuki, S.; Komoto, S.; Mukai, T.; Ikuta, K. Extracellular Nef protein activates signal transduction pathway from Ras to mitogen-activated protein kinase cascades that leads to activation of human immunodeficiency virus from latency. AIDS Res. Hum. Retroviruses 2002, 18, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Varin, A.; Manna, S.K.; Quivy, V.; Decrion, A.Z.; Van Lint, C.; Herbein, G.; Aggarwal, B.B. Exogenous Nef protein activates NF-κB, AP-1, and c-Jun N-terminal kinase and stimulates HIV transcription in promonocytic cells. Role in AIDS pathogenesis. J. Biol. Chem. 2003, 278, 2219–2227. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Abbas, W.; Colin, L.; Khan, K.A.; Bouchat, S.; Varin, A.; Larbi, A.; Gatot, J.S.; Kabeya, K.; Vanhulle, C.; et al. Tuning of AKT-pathway by Nef and its blockade by protease inhibitors results in limited recovery in latently HIV infected T-cell line. Sci. Rep. 2016, 6, 24090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, Y.; Otake, K.; Tashiro, M.; Adachi, A. Soluble Nef antigen of HIV-1 is cytotoxic for human CD4+ T cells. FEBS Lett. 1996, 393, 93–96. [Google Scholar] [CrossRef] [Green Version]
- Federico, M.; Percario, Z.; Olivetta, E.; Fiorucci, G.; Muratori, C.; Micheli, A.; Romeo, G.; Affabris, E. HIV-1 Nef activates STAT1 in human monocytes/macrophages through the release of soluble factors. Blood 2001, 98, 2752–2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenassi, M.; Cagney, G.; Liao, M.; Vaupotic, T.; Bartholomeeusen, K.; Cheng, Y.; Krogan, N.J.; Plemenitas, A.; Peterlin, B.M. HIV Nef is secreted in exosomes and triggers apoptosis in bystander CD4+ T cells. Traffic 2010, 11, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Raymond, A.D.; Campbell-Sims, T.C.; Khan, M.; Lang, M.; Huang, M.B.; Bond, V.C.; Powell, M.D. HIV Type 1 Nef is released from infected cells in CD45(+) microvesicles and is present in the plasma of HIV-infected individuals. AIDS Res. Hum. Retroviruses 2011, 27, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Arenaccio, C.; Anticoli, S.; Manfredi, F.; Chiozzini, C.; Olivetta, E.; Federico, M. Latent HIV-1 is activated by exosomes from cells infected with either replication-competent or defective HIV-1. Retrovirology 2015, 12, 87. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Takei, R.; Tashiro, M. Nef protein of HIV-1 induces apoptotic cytolysis of murine lymphoid cells independently of CD95 (Fas) and its suppression by serine/threonine protein kinase inhibitors. FEBS Lett. 1997, 417, 61–64. [Google Scholar] [CrossRef] [Green Version]
- Alessandrini, L.; Santarcangelo, A.C.; Olivetta, E.; Ferrantelli, F.; d’Aloja, P.; Pugliese, K.; Pelosi, E.; Chelucci, C.; Mattia, G.; Peschle, C.; et al. T-tropic human immunodeficiency virus (HIV) type 1 Nef protein enters human monocyte-macrophages and induces resistance to HIV replication: A possible mechanism of HIV T-tropic emergence in AIDS. J. Gen. Virol. 2000, 81, 2905–2917. [Google Scholar] [CrossRef] [PubMed]
- Gooz, M. ADAM-17: The enzyme that does it all. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 146–169. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Wittki, S.; Brau, T.; Dreyer, F.S.; Kratzel, K.; Dindorf, J.; Johnston, I.C.; Gross, S.; Kremmer, E.; Zeidler, R.; et al. HIV Nef, paxillin, and Pak1/2 regulate activation and secretion of TACE/ADAM10 proteases. Mol. Cell 2013, 49, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Goeddel, D.V. TNF-R1 signaling: A beautiful pathway. Science 2002, 296, 1634–1635. [Google Scholar] [CrossRef] [PubMed]
- Muratori, C.; Cavallin, L.E.; Kratzel, K.; Tinari, A.; De Milito, A.; Fais, S.; D’Aloja, P.; Federico, M.; Vullo, V.; Fomina, A.; et al. Massive secretion by T cells is caused by HIV Nef in infected cells and by Nef transfer to bystander cells. Cell Host Microbe 2009, 6, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.H.; Ren, Y.; Thomas, A.S.; Chan, D.; Mueller, S.; Ward, A.R.; Patel, S.; Bollard, C.M.; Cruz, C.R.; Karandish, S.; et al. Latent HIV reservoirs exhibit inherent resistance to elimination by CD8+ T cells. J. Clin. Investig. 2018, 128, 876–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mujib, S.; Saiyed, A.; Fadel, S.; Bozorgzad, A.; Aidarus, N.; Yue, F.Y.; Benko, E.; Kovacs, C.; Emert-Sedlak, L.A.; Smithgall, T.E.; et al. Pharmacologic HIV-1 Nef blockade promotes CD8 T cell-mediated elimination of latently HIV-1-infected cells in vitro. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhong, Q. Histone deacetylase inhibitors and cell death. Cell. Mol. Life Sci. 2014, 71, 3885–3901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.Y.; Nishitoh, H.; Yang, X.; Ichijo, H.; Baltimore, D. Activation of apoptosis signal-regulating kinase 1 (ASK1) by the adapter protein Daxx. Science 1998, 281, 1860–1863. [Google Scholar] [CrossRef] [PubMed]
- Nishitoh, H.; Saitoh, M.; Mochida, Y.; Takeda, K.; Nakano, H.; Rothe, M.; Miyazono, K.; Ichijo, H. ASK1 is essential for JNK/SAPK activation by TRAF2. Mol. Cell 1998, 2, 389–395. [Google Scholar] [CrossRef]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichijo, H.; Nishida, E.; Irie, K.; ten Dijke, P.; Saitoh, M.; Moriguchi, T.; Takagi, M.; Matsumoto, K.; Miyazono, K.; Gotoh, Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997, 275, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Yu, S.; Eder, A.; Mao, M.; Bast, R.C., Jr.; Boyd, D.; Mills, G.B. Regulation of BAD phosphorylation at serine 112 by the Ras-mitogen-activated protein kinase pathway. Oncogene 1999, 18, 6635–6640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douek, D.C.; Brenchley, J.M.; Betts, M.R.; Ambrozak, D.R.; Hill, B.J.; Okamoto, Y.; Casazza, J.P.; Kuruppu, J.; Kunstman, K.; Wolinsky, S.; et al. HIV preferentially infects HIV-specific CD4+ T cells. Nature 2002, 417, 95–98. [Google Scholar] [CrossRef] [PubMed]
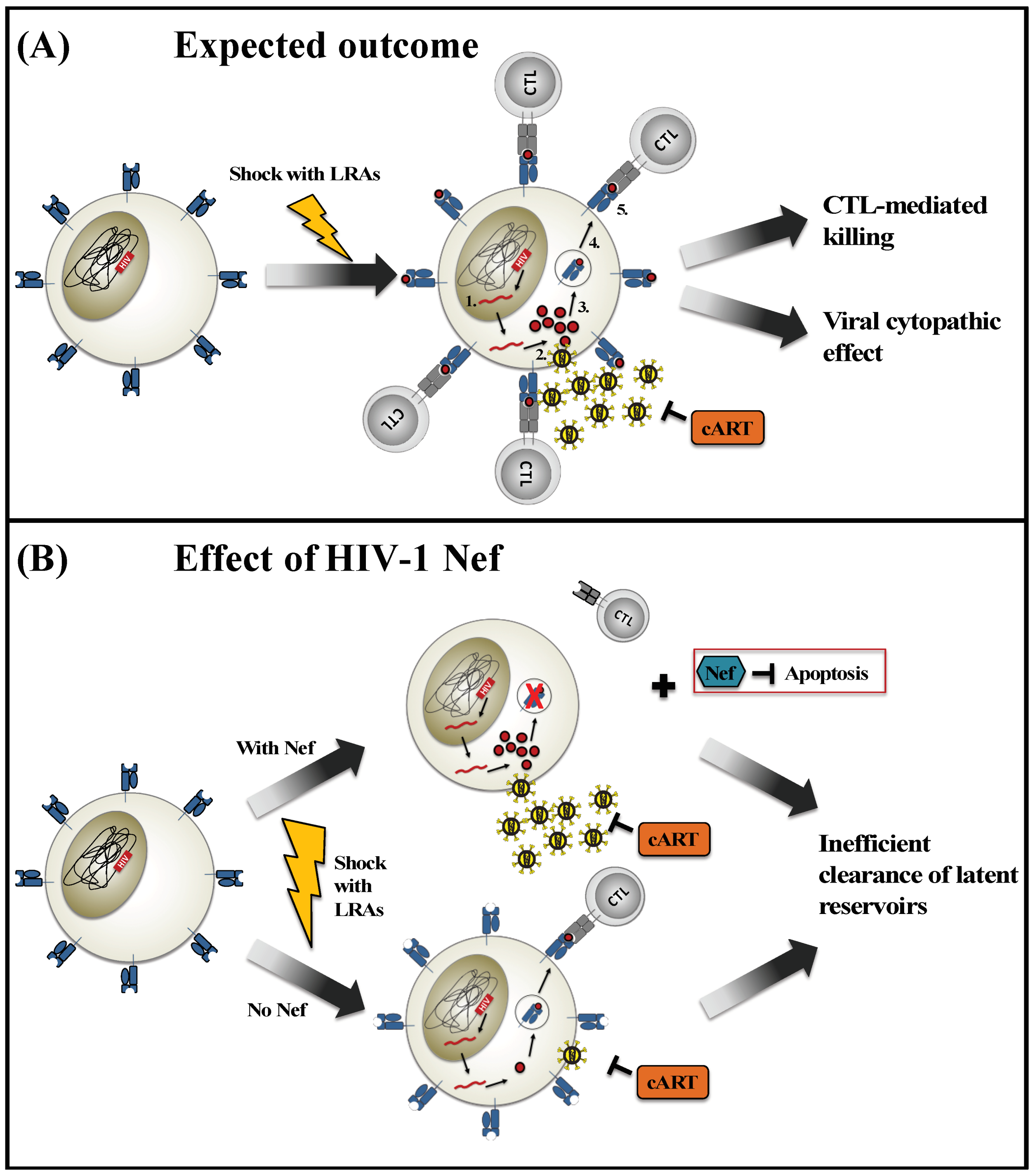
Figure 1.
Impact of Nef on “shock and kill” methods to eradicate HIV reservoirs. (A) This illustration displays the expected outcome of a latent HIV-infected T cell following induction with latency-reversing agents (LRAs) (“shock”) in the presence of combination antiretroviral therapy (cART). The integrated HIV proviral genome is transcribed (1) and translated into viral proteins (2). Some viral proteins are degraded into peptide antigens and loaded onto HLA class I molecules (3) for presentation at the cell surface (4). The recognition of peptide-HLA complexes by cytotoxic T lymphocytes (CTL) (5) induces cytolytic mechanisms that kill the virus-infected cell. Alternatively, the expression of viral proteins may induce viral cytopathic effects that result in the death of the infected cell. (B) This illustration displays the potential contributions of the viral Nef protein to modulate the reactivation and elimination of latent HIV-infected cells by “shock and kill” methods. In the presence of Nef, viral protein expression is robust, but HLA class I molecules are down-regulated from the cell surface and cellular apoptosis is inhibited. In the absence of Nef, viral protein expression is reduced, thus limiting the amount of viral antigen that is available for presentation on HLA class I. In both scenarios, CTL-mediated recognition and elimination of newly reactivated HIV-infected cells may be hindered.
Figure 1.
Impact of Nef on “shock and kill” methods to eradicate HIV reservoirs. (A) This illustration displays the expected outcome of a latent HIV-infected T cell following induction with latency-reversing agents (LRAs) (“shock”) in the presence of combination antiretroviral therapy (cART). The integrated HIV proviral genome is transcribed (1) and translated into viral proteins (2). Some viral proteins are degraded into peptide antigens and loaded onto HLA class I molecules (3) for presentation at the cell surface (4). The recognition of peptide-HLA complexes by cytotoxic T lymphocytes (CTL) (5) induces cytolytic mechanisms that kill the virus-infected cell. Alternatively, the expression of viral proteins may induce viral cytopathic effects that result in the death of the infected cell. (B) This illustration displays the potential contributions of the viral Nef protein to modulate the reactivation and elimination of latent HIV-infected cells by “shock and kill” methods. In the presence of Nef, viral protein expression is robust, but HLA class I molecules are down-regulated from the cell surface and cellular apoptosis is inhibited. In the absence of Nef, viral protein expression is reduced, thus limiting the amount of viral antigen that is available for presentation on HLA class I. In both scenarios, CTL-mediated recognition and elimination of newly reactivated HIV-infected cells may be hindered.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kuang, X.T.; Brockman, M.A. Implications of HIV-1 Nef for “Shock and Kill” Strategies to Eliminate Latent Viral Reservoirs. Viruses 2018, 10, 677. https://0-doi-org.brum.beds.ac.uk/10.3390/v10120677
AMA Style
Kuang XT, Brockman MA. Implications of HIV-1 Nef for “Shock and Kill” Strategies to Eliminate Latent Viral Reservoirs. Viruses. 2018; 10(12):677. https://0-doi-org.brum.beds.ac.uk/10.3390/v10120677
Chicago/Turabian StyleKuang, Xiaomei T., and Mark A. Brockman. 2018. "Implications of HIV-1 Nef for “Shock and Kill” Strategies to Eliminate Latent Viral Reservoirs" Viruses 10, no. 12: 677. https://0-doi-org.brum.beds.ac.uk/10.3390/v10120677
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.