Hepatitis C Virus-Induced FUT8 Causes 5-FU Drug Resistance in Human Hepatoma Huh7.5.1 Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, Viral Infection and Reagents
2.2. Small Interfering RNA (SiRNA) Transfection
2.3. Plasmid Construction and Transfection
2.4. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.5. Lactate Dehydrogenase Release Assay
2.6. Cell Proliferation Assay
2.7. Western Blot Analysis
2.8. Flow Cytometry Analysis
2.9. Statistical Analysis
3. Results
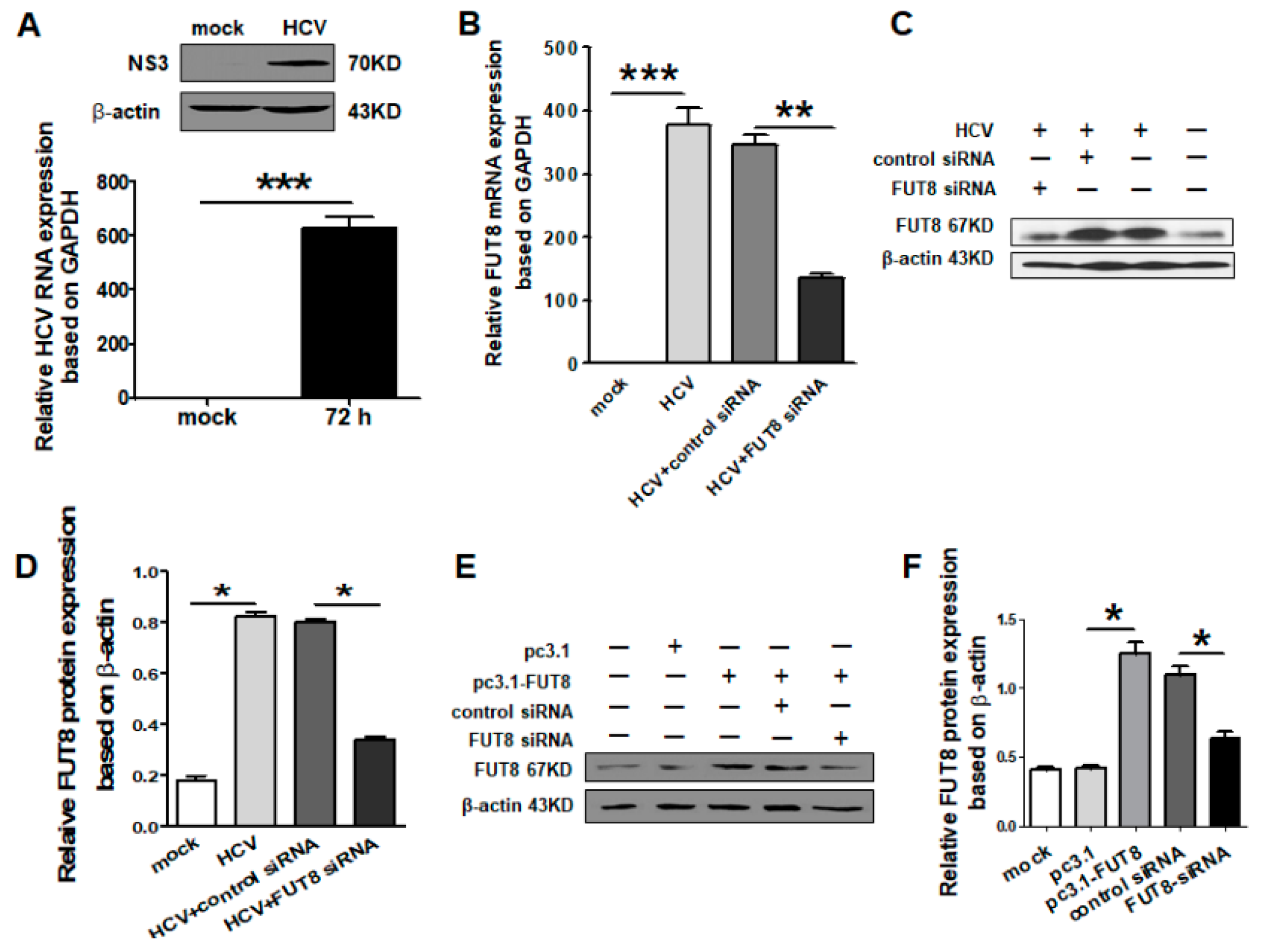
3.1. HCV Infection Induces FUT8 Expression in Huh7.5.1 Cells
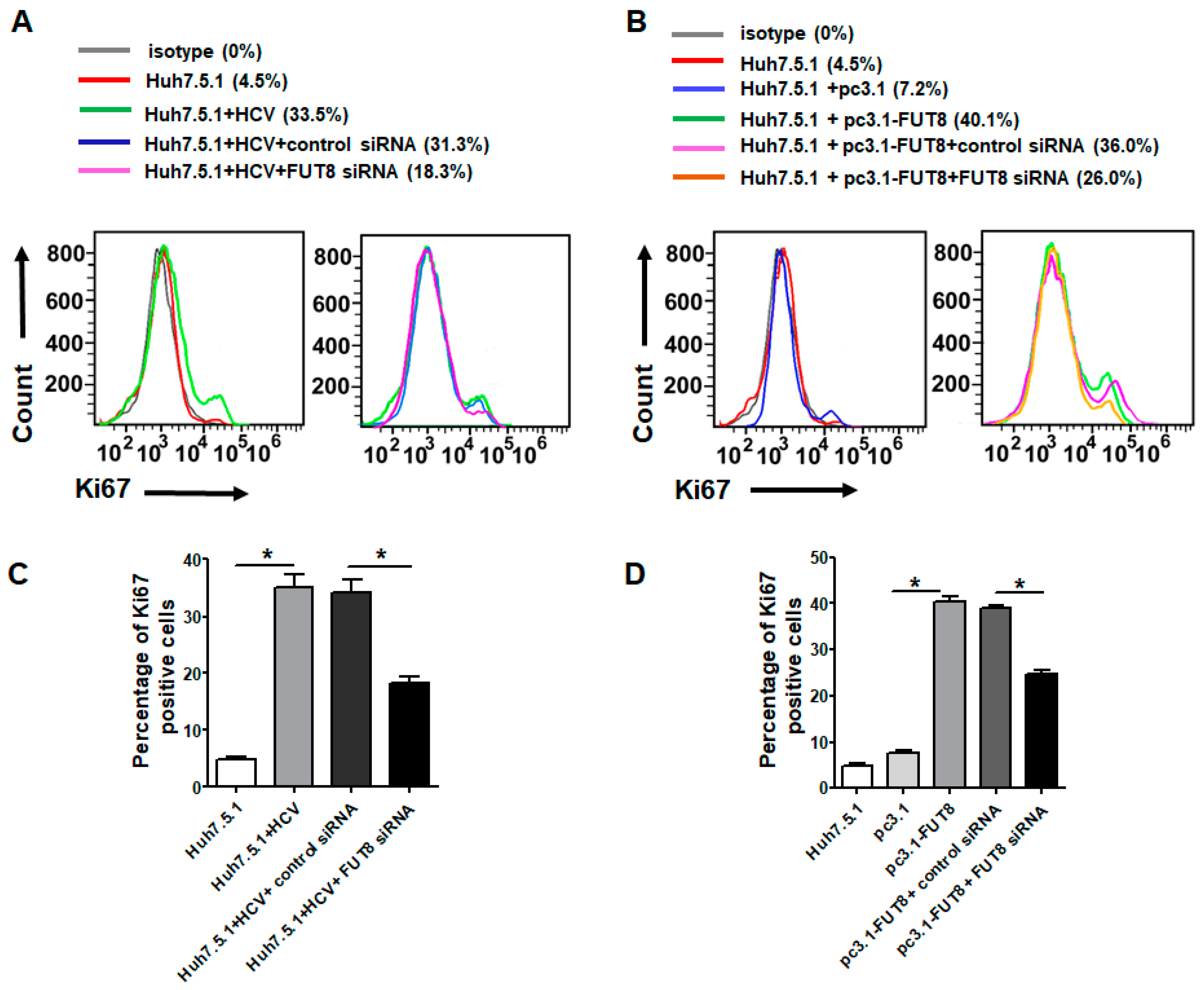
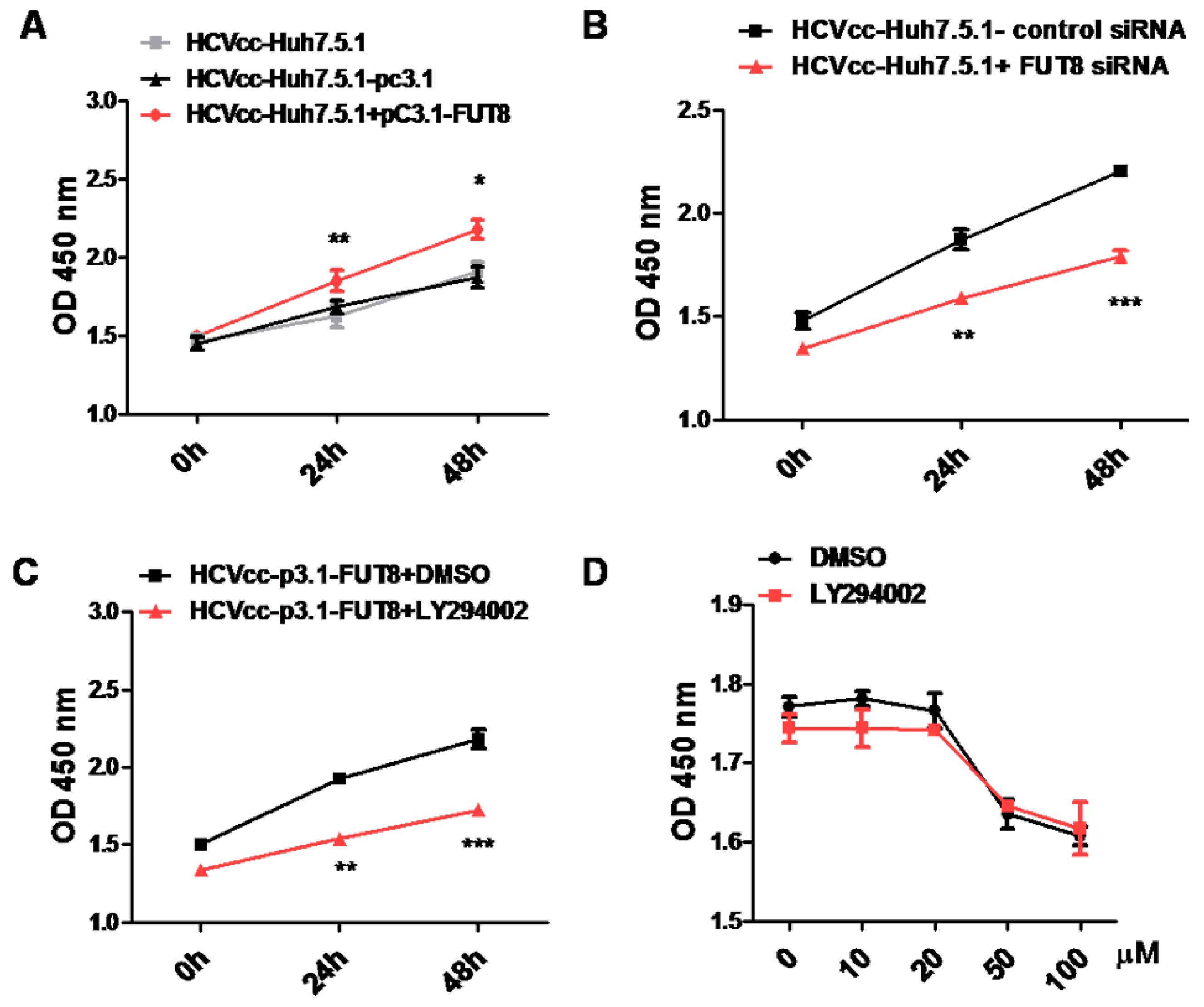
3.2. Both HCV Infection and Overexpression of FUT8 Enhanced Proliferation of Huh7.5.1 Cells
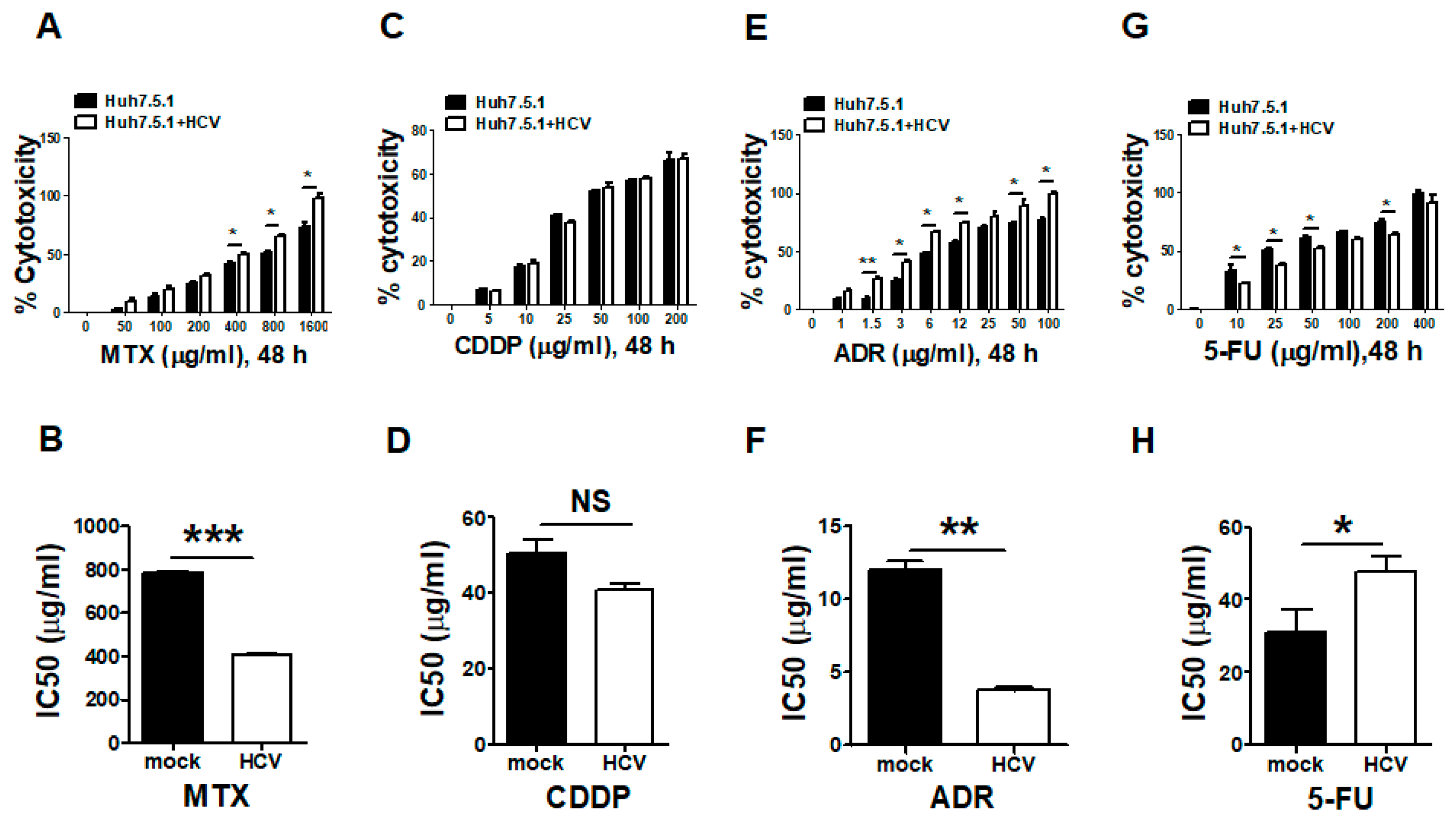
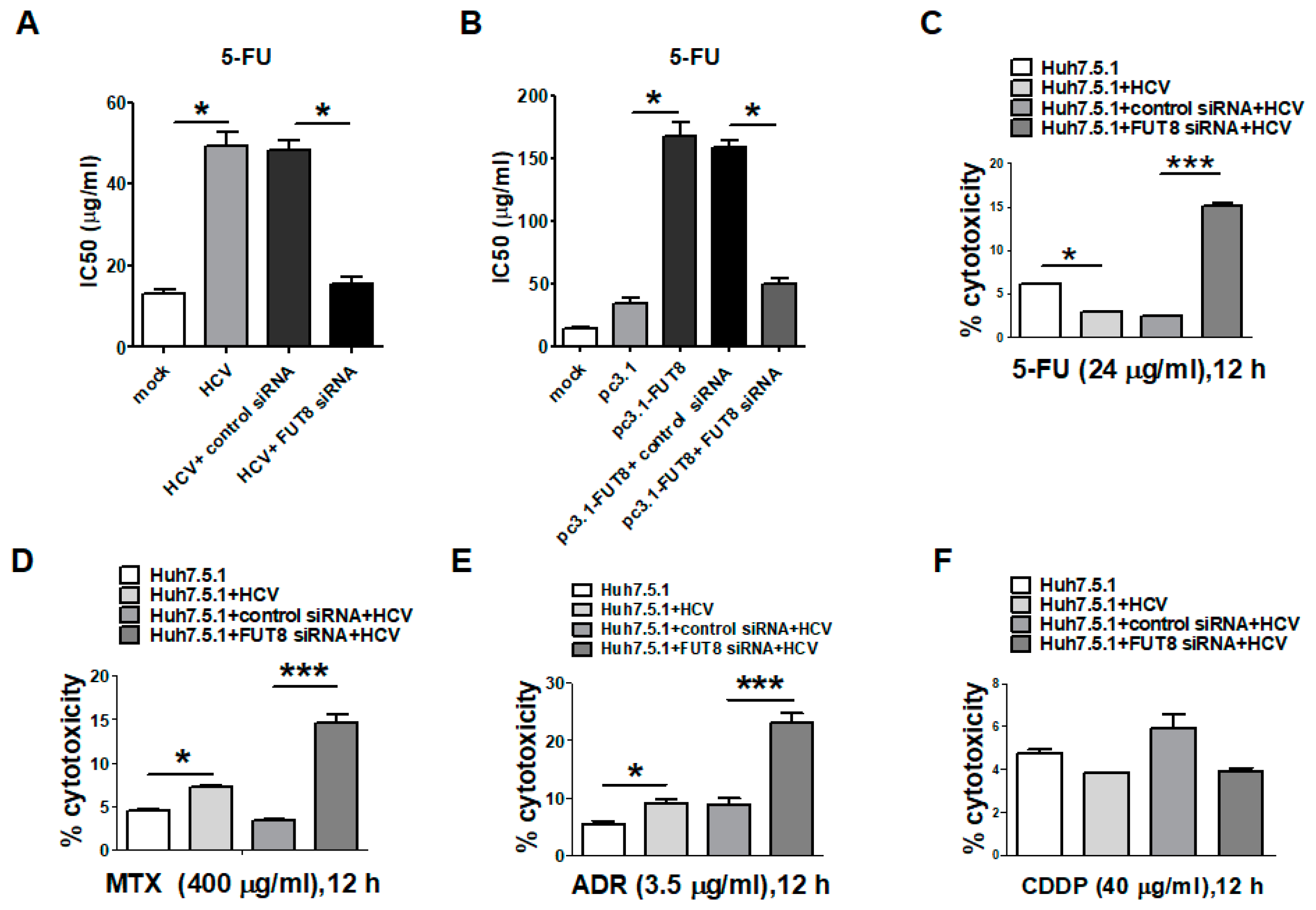
3.3. Silencing the FUT8 Gene Increases the 5-FU Drug Sensitivity of HCV-Infected and FUT8-Overexpressing Huh7.5.1 Cells
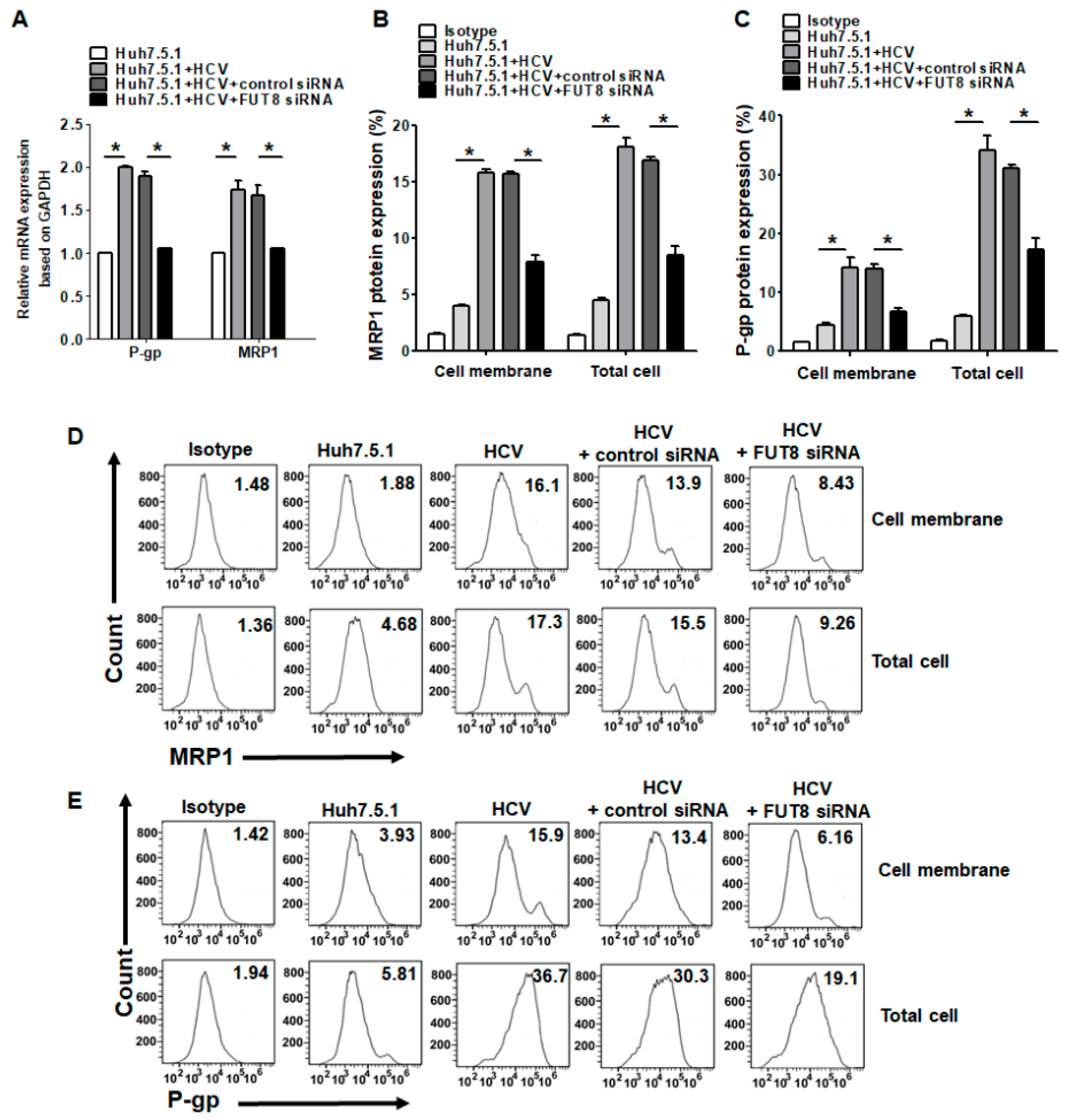
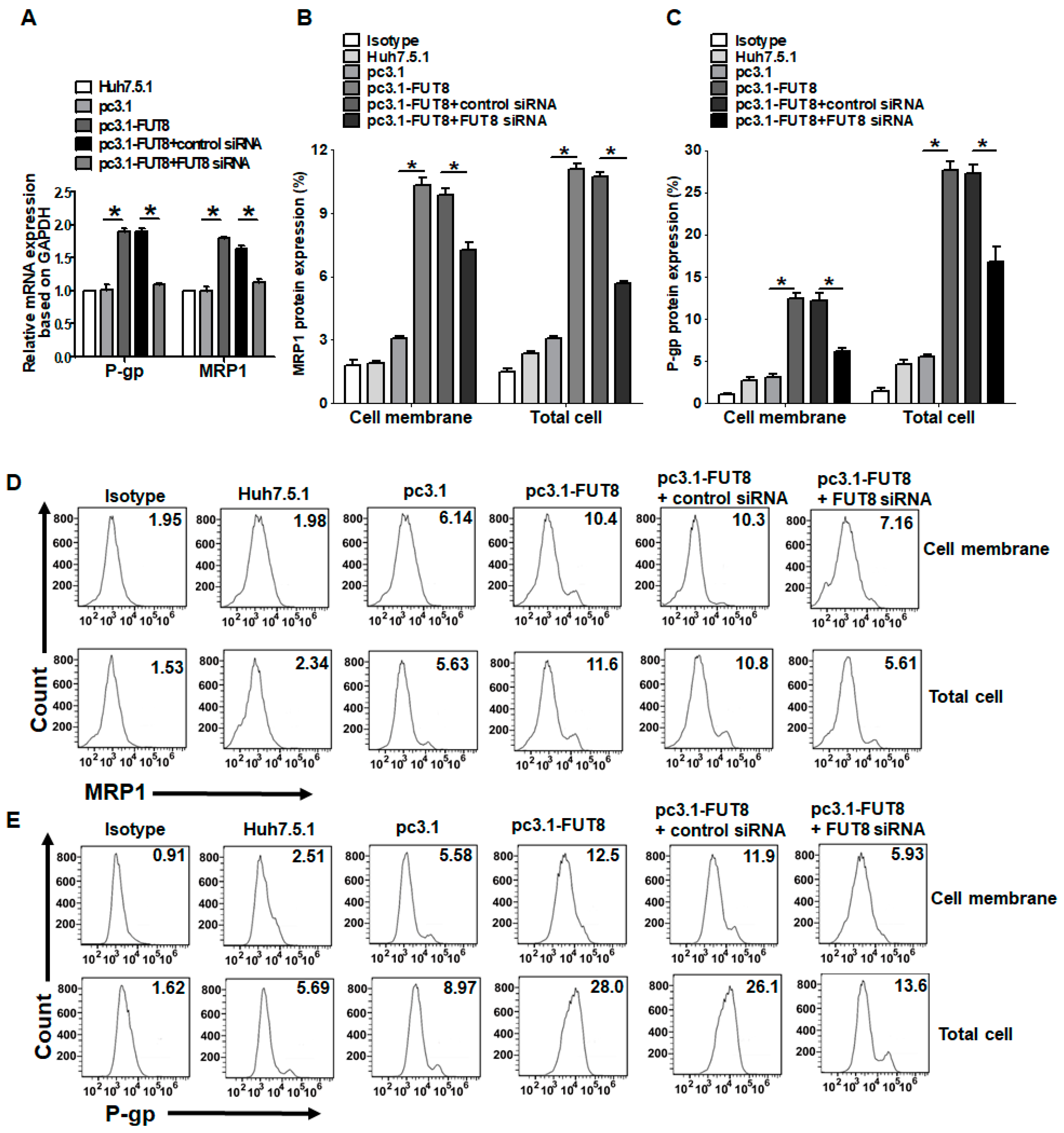
3.4. Upregulation of the Drug Resistance Genes P-gp and MRP1 after HCV Infection and FUT8 Overexpression
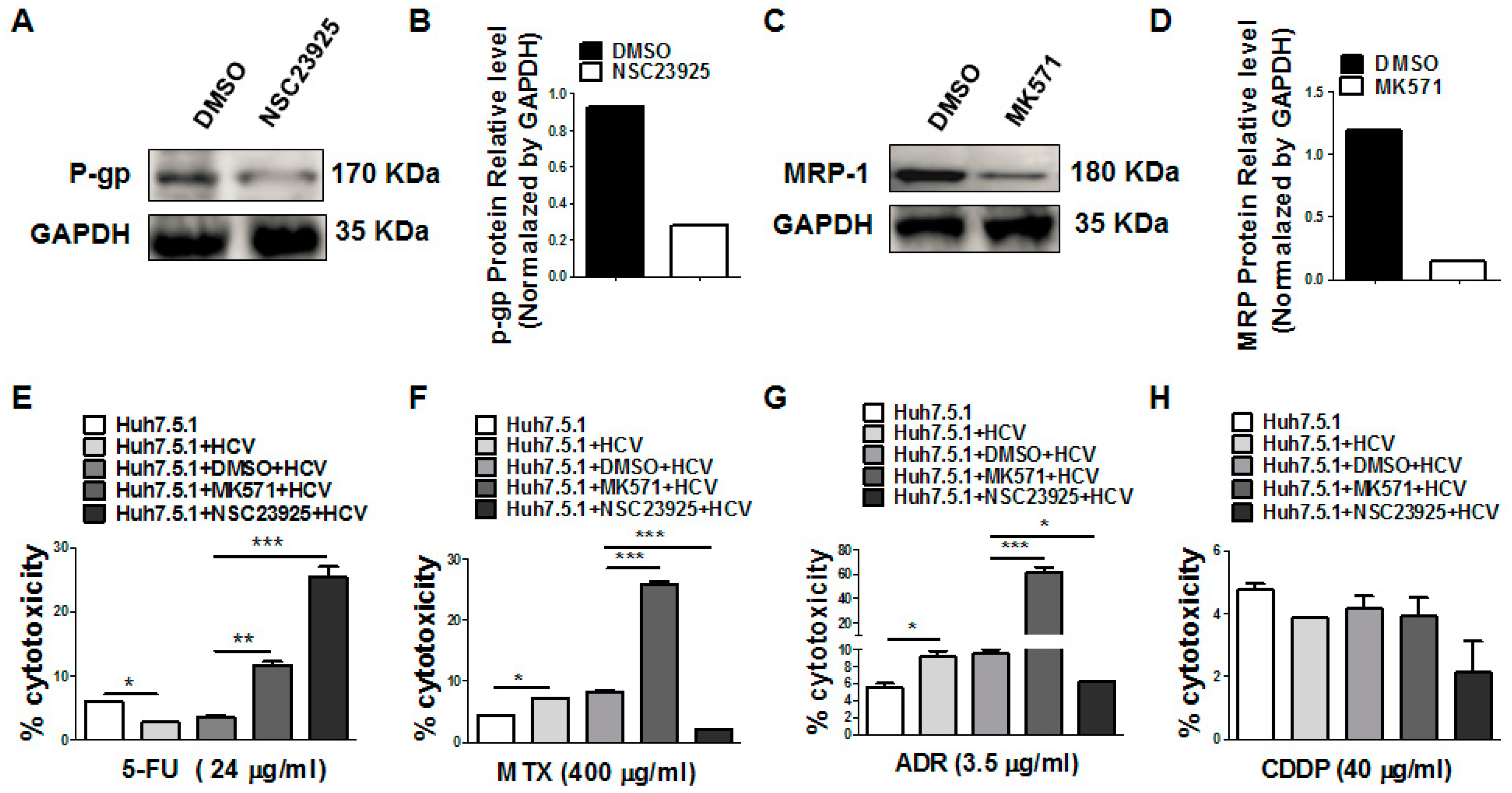
3.5. Inhibition of P-gp and MRP1 Increases the 5-FU Drug Sensitivity after HCV Infection
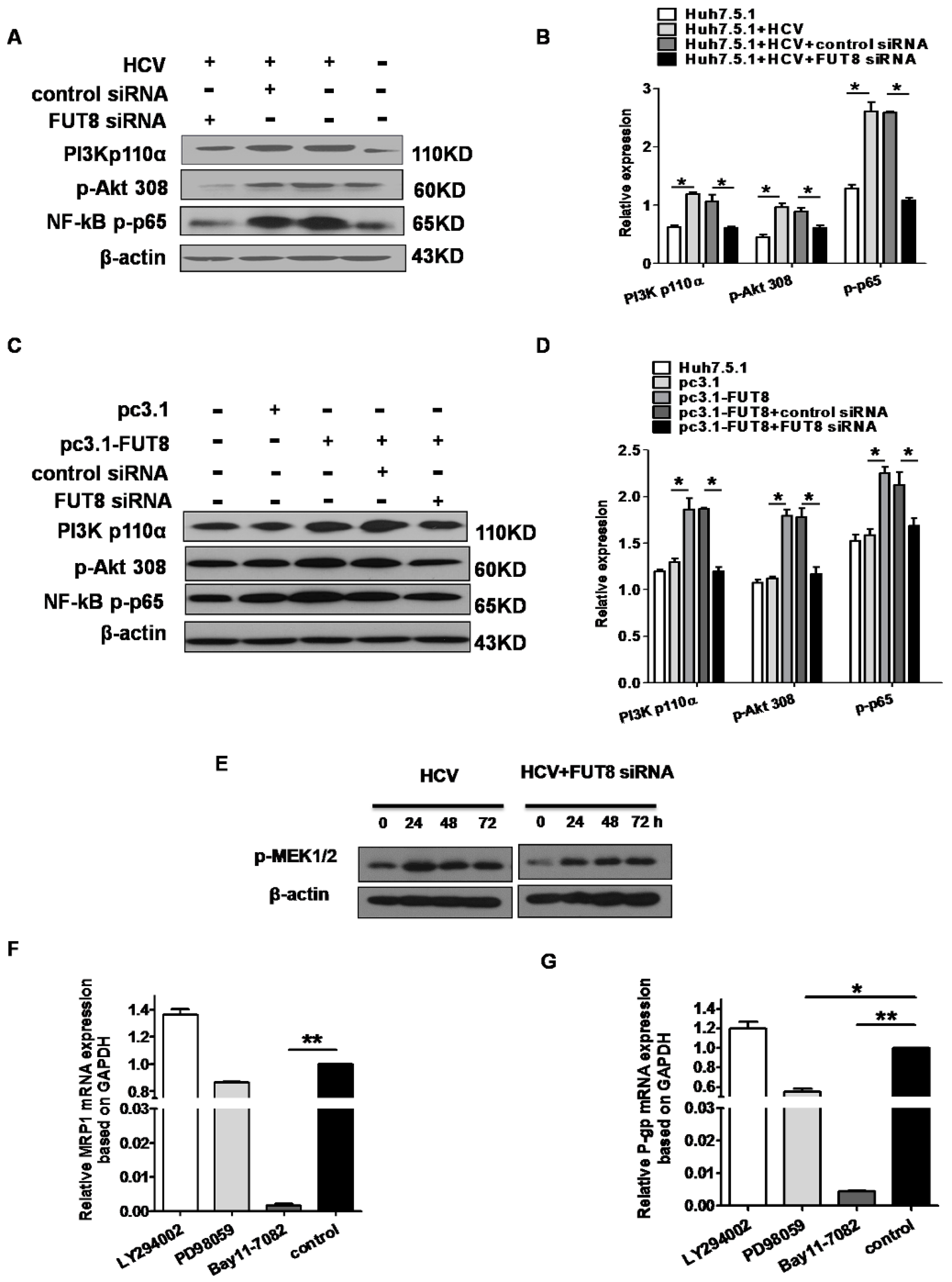
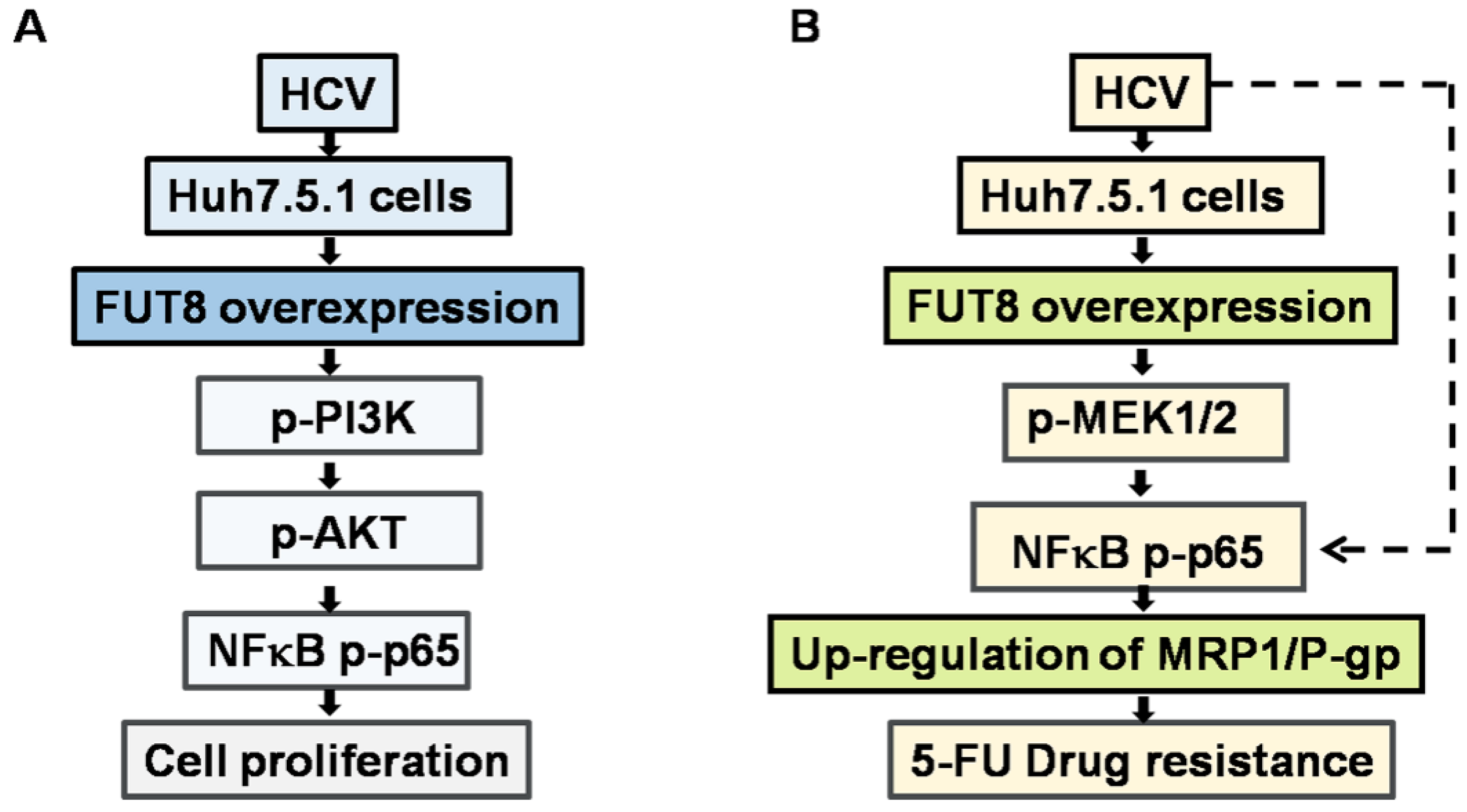
3.6. NF-κB is Critical for FUT8-Mediated 5-FU Resistance in Huh7.5.1 Cells
3.7. FUT8 Promotes the Proliferation of HCVcc-Infected Huh7.5.1 Cells through Activating the PI3K Signaling Pathway
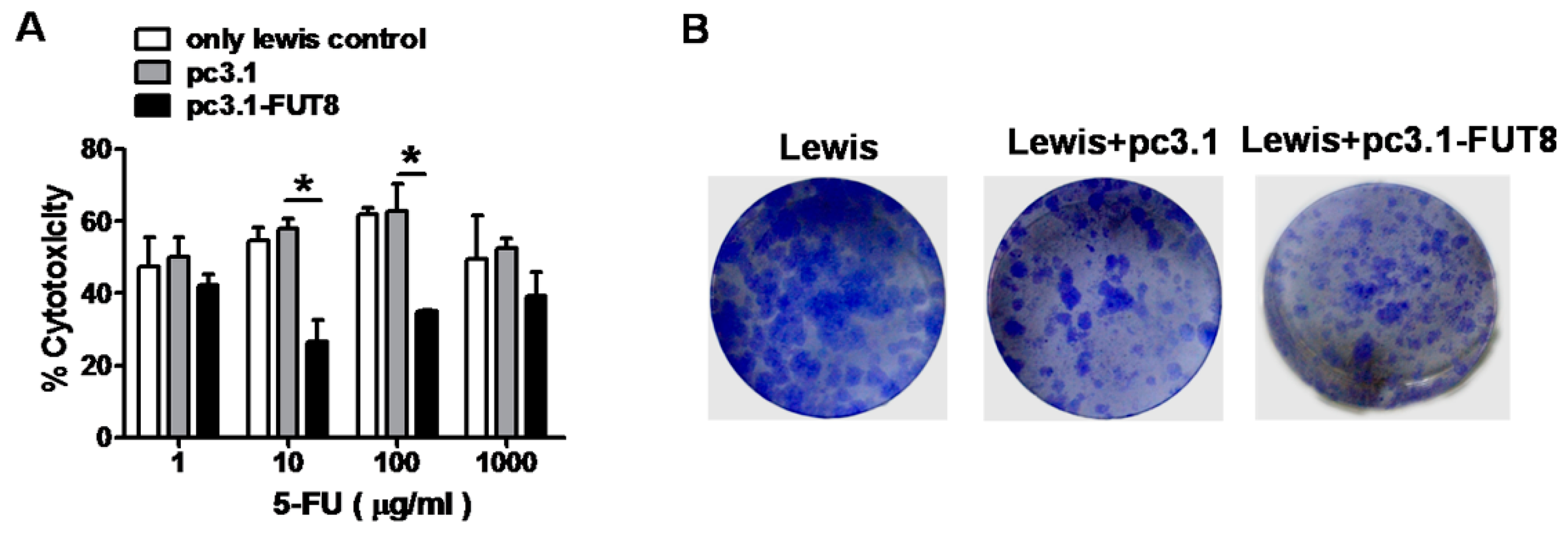
3.8. FUT8 is Highly Related to 5-FU Resistance in the Lewis Cell Model
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Waziry, R.; Hajarizadeh, B.; Grebely, J.; Amin, J.; Law, M.; Danta, M.; George, J.; Dore, G.J. Hepatocellular carcinoma risk following direct-acting antiviral HCV therapy: A systematic review, meta-analyses, and meta-regression. J. Hepatol. 2017, 67, 1204–1212. [Google Scholar] [CrossRef]
- Pradat, P.; Virlogeux, V.; Trepo, E. Epidemiology and elimination of HCV-related liver disease. Viruses 2018, 10, 545. [Google Scholar] [CrossRef]
- Magalhaes, A.; Duarte, H.O.; Reis, C.A. Aberrant glycosylation in cancer: A novel molecular mechanism controlling metastasis. Cancer Cell 2017, 31, 733–735. [Google Scholar] [CrossRef]
- Gomes, J.; Mereiter, S.; Magalhaes, A.; Reis, C.A. Early galnac O-glycosylation: Pushing the tumor boundaries. Cancer Cell 2017, 32, 544–545. [Google Scholar] [CrossRef]
- Lucena, M.C.; Carvalho-Cruz, P.; Donadio, J.L.; Oliveira, I.A.; de Queiroz, R.M.; Marinho-Carvalho, M.M.; Sola-Penna, M.; de Paula, I.F.; Gondim, K.C.; McComb, M.E.; et al. Epithelial mesenchymal transition induces aberrant glycosylation through hexosamine biosynthetic pathway activation. J. Biol. Chem. 2016, 291, 12917–12929. [Google Scholar] [CrossRef]
- Iwamori, M.; Tanaka, K.; Kubushiro, K.; Lin, B.; Kiguchi, K.; Ishiwata, I.; Tsukazaki, K.; Nozawa, S. Alterations in the glycolipid composition and cellular properties of ovarian carcinoma-derived rmg-1 cells on transfection of the alpha1,2-fucosyltransferase gene. Cancer Sci. 2005, 96, 26–30. [Google Scholar] [CrossRef]
- Feng, X.; Zhao, L.; Gao, S.; Song, X.; Dong, W.; Zhao, Y.; Zhou, H.; Cheng, L.; Miao, X.; Jia, L. Increased fucosylation has a pivotal role in multidrug resistance of breast cancer cells through mir-224-3p targeting fut4. Gene 2016, 578, 232–241. [Google Scholar] [CrossRef]
- Cheng, L.; Luo, S.; Jin, C.; Ma, H.; Zhou, H.; Jia, L. Fut family mediates the multidrug resistance of human hepatocellular carcinoma via the pi3k/akt signaling pathway. Cell Death Dis. 2013, 4, e923. [Google Scholar] [CrossRef]
- Agrawal, P.; Fontanals-Cirera, B.; Sokolova, E.; Jacob, S.; Vaiana, C.A.; Argibay, D.; Davalos, V.; McDermott, M.; Nayak, S.; Darvishian, F.; et al. A systems biology approach identifies fut8 as a driver of melanoma metastasis. Cancer Cell 2017, 31, 804–819 e807. [Google Scholar] [CrossRef]
- Subramaniam, A.; Shanmugam, M.K.; Perumal, E.; Li, F.; Nachiyappan, A.; Dai, X.; Swamy, S.N.; Ahn, K.S.; Kumar, A.P.; Tan, B.K.; et al. Potential role of signal transducer and activator of transcription (stat)3 signaling pathway in inflammation, survival, proliferation and invasion of hepatocellular carcinoma. Biochim. Biophys. Acta. 2013, 1835, 46–60. [Google Scholar] [CrossRef]
- Wang, C.; Li, X.; Zhang, J.; Ge, Z.; Chen, H.; Hu, J. Ezh2 contributes to 5-fu resistance in gastric cancer by epigenetically suppressing fbxo32 expression. Onco. Targets Ther. 2018, 11, 7853–7864. [Google Scholar] [CrossRef]
- Wang, W.; Guo, W.; Li, L.; Fu, Z.; Liu, W.; Gao, J.; Shu, Y.; Xu, Q.; Sun, Y.; Gu, Y. Andrographolide reversed 5-fu resistance in human colorectal cancer by elevating bax expression. Biochem. Pharmacol. 2016, 121, 8–17. [Google Scholar] [CrossRef]
- Mata, J.F.; Garcia-Manteiga, J.M.; Lostao, M.P.; Fernandez-Veledo, S.; Guillen-Gomez, E.; Larrayoz, I.M.; Lloberas, J.; Casado, F.J.; Pastor-Anglada, M. Role of the human concentrative nucleoside transporter (hcnt1) in the cytotoxic action of 5[prime]-deoxy-5-fluorouridine, an active intermediate metabolite of capecitabine, a novel oral anticancer drug. Mol. Pharmacol. 2001, 59, 1542–1548. [Google Scholar] [CrossRef]
- Liu, W.; Fang, Y.; Wang, X.T.; Liu, J.; Dan, X.; Sun, L.L. Overcoming 5-fu resistance of colon cells through inhibition of glut1 by the specific inhibitor wzb117. Asian Pac. J. Cancer Prev. 2014, 15, 7037–7041. [Google Scholar] [CrossRef]
- Tyler, A.; Johansson, A.; Karlsson, T.; Gudey, S.K.; Brannstrom, T.; Grankvist, K.; Behnam-Motlagh, P. Targeting glucosylceramide synthase induction of cell surface globotriaosylceramide (gb3) in acquired cisplatin-resistance of lung cancer and malignant pleural mesothelioma cells. Exp. Cell Res. 2015, 336, 23–32. [Google Scholar] [CrossRef]
- Xiang, T.; Yang, G.; Liu, X.; Zhou, Y.; Fu, Z.; Lu, F.; Gu, J.; Taniguchi, N.; Tan, Z.; Chen, X.; et al. Alteration of n-glycan expression profile and glycan pattern of glycoproteins in human hepatoma cells after hcv infection. Biochim. Biophys. Acta. 2017, 1861, 1036–1045. [Google Scholar] [CrossRef]
- Zhao, Y.; Ren, Y.; Zhang, X.; Zhao, P.; Tao, W.; Zhong, J.; Li, Q.; Zhang, X.L. Ficolin-2 inhibits hepatitis c virus infection, whereas apolipoprotein e3 mediates viral immune escape. J. Immunol. 2014, 193, 783–796. [Google Scholar] [CrossRef]
- Hu, Y.L.; Luo, F.L.; Fu, J.L.; Chen, T.L.; Wu, S.M.; Zhou, Y.D.; Zhang, X.L. Early increased ficolin-2 concentrations are associated with severity of liver inflammation and efficacy of anti-viral therapy in chronic hepatitis c patients. Scand. J. Immunol. 2013, 77, 144–150. [Google Scholar] [CrossRef]
- Chen, Y.L.; Yang, T.Y.; Chen, K.C.; Wu, C.L.; Hsu, S.L.; Hsueh, C.M. Hypoxia can impair doxorubicin resistance of non-small cell lung cancer cells by inhibiting mrp1 and p-gp expression and boosting the chemosensitizing effects of mrp1 and p-gp blockers. Cell. Oncol. 2016, 39, 411–433. [Google Scholar] [CrossRef]
- Bai, J.; Yong, H.M.; Chen, F.F.; Mei, P.J.; Liu, H.; Li, C.; Pan, Z.Q.; Wu, Y.P.; Zheng, J.N. Cullin1 is a novel marker of poor prognosis and a potential therapeutic target in human breast cancer. Ann. Oncol. 2013, 24, 2016–2022. [Google Scholar] [CrossRef] [Green Version]
- Zhong, J.; Gastaminza, P.; Cheng, G.; Kapadia, S.; Kato, T.; Burton, D.R.; Wieland, S.F.; Uprichard, S.L.; Wakita, T.; Chisari, F.V. Robust hepatitis c virus infection in vitro. Proc. Natl. Acad. Sci. USA 2005, 102, 9294–9299. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Wang, L.X. Mammalian alpha-1,6-fucosyltransferase (fut8) is the sole enzyme responsible for the n-acetylglucosaminyltransferase i-independent core fucosylation of high-mannose n-glycans. J. Biol. Chem. 2016, 291, 11064–11071. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, H.; Yang, L.; Wu, Q.; Liu, W.; Fu, Q.; Zhang, W.; Zhang, H.; Xu, J.; Gu, J. Loss of n-acetylgalactosaminyltransferase-4 orchestrates oncogenic microrna-9 in hepatocellular carcinoma. J. Biol. Chem. 2017, 292, 3186–3200. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.E.; Diepeveen, L.A.; Stubbs, K.A.; Yeoh, G.C. Glycosylation-related diagnostic and therapeutic drug target markers in hepatocellular carcinoma. J. Gastrointestin. Liver Dis. 2015, 24, 349–357. [Google Scholar] [PubMed]
- Tu, C.F.; Wu, M.Y.; Lin, Y.C.; Kannagi, R.; Yang, R.B. Fut8 promotes breast cancer cell invasiveness by remodeling tgf-beta receptor core fucosylation. Breast Cancer Res. 2017, 19, 111. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Cui, X.; Wang, H.; Liu, J.; Qin, H.; Liu, S.; Yan, Q. Fut8 drives the proliferation and invasion of trophoblastic cells via igf-1/igf-1r signaling pathway. Placenta 2019, 75, 45–53. [Google Scholar] [CrossRef]
- Holohan, C.; van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Roussel, B.; Johnson-Farley, N.; Kerrigan, J.E.; Scotto, K.W.; Banerjee, D.; Felczak, K.; Pankiewicz, K.W.; Gounder, M.; Lin, H.; Abali, E.E.; et al. A second target of benzamide riboside: Dihydrofolate reductase. Cancer Biol. Ther. 2012, 13, 1290–1298. [Google Scholar] [CrossRef]
- Zhou, Z.; Hu, Y.; Shan, X.; Li, W.; Bai, X.; Wang, P.; Lu, X. Revealing three stages of DNA-cisplatin reaction by a solid-state nanopore. Sci. Rep. 2015, 5, 11868. [Google Scholar] [CrossRef]
- Chou, A.J.; Gorlick, R. Chemotherapy resistance in osteosarcoma: Current challenges and future directions. Expert Rev. Anticancer Ther. 2006, 6, 1075–1085. [Google Scholar] [CrossRef]
- Momparler, R.L.; Karon, M.; Siegel, S.E.; Avila, F. Effect of adriamycin on DNA, RNA, and protein synthesis in cell-free systems and intact cells. Cancer Res. 1976, 36, 2891–2895. [Google Scholar]
- Kamiyama, T.; Yokoo, H.; Furukawa, J.; Kurogochi, M.; Togashi, T.; Miura, N.; Nakanishi, K.; Kamachi, H.; Kakisaka, T.; Tsuruga, Y.; et al. Identification of novel serum biomarkers of hepatocellular carcinoma using glycomic analysis. Hepatology 2013, 57, 2314–2325. [Google Scholar] [CrossRef] [Green Version]
- Pascal, J.; Ashley, C.E.; Wang, Z.; Brocato, T.A.; Butner, J.D.; Carnes, E.C.; Koay, E.J.; Brinker, C.J.; Cristini, V. Mechanistic modeling identifies drug-uptake history as predictor of tumor drug resistance and nano-carrier-mediated response. ACS Nano 2013, 7, 11174–11182. [Google Scholar] [CrossRef]
- Hayes, C.N.; Zhang, P.; Zhang, Y.; Chayama, K. Molecular mechanisms of hepatocarcinogenesis following sustained virological response in patients with chronic hepatitis c virus infection. Viruses 2018, 10, 531. [Google Scholar] [CrossRef] [PubMed]
- Nanbo, A.; Inoue, K.; Adachi-Takasawa, K.; Takada, K. Epstein-barr virus rna confers resistance to interferon-alpha-induced apoptosis in burkitt’s lymphoma. EMBO J. 2002, 21, 954–965. [Google Scholar] [CrossRef]
- Lee, S.; Jang, J.; Jeon, H.; Lee, J.; Yoo, S.M.; Park, J.; Lee, M.S. Latent kaposi’s sarcoma-associated herpesvirus infection in bladder cancer cells promotes drug resistance by reducing reactive oxygen species. J. Microbiol. 2016, 54, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.F.; Pokharel, D.; Bebawy, M. Mrp1 and its role in anticancer drug resistance. Drug Metab. Rev. 2015, 47, 406–419. [Google Scholar] [CrossRef]
- Johnson, Z.L.; Chen, J. Structural basis of substrate recognition by the multidrug resistance protein mrp1. Cell 2017, 168, 1075–1085 e1079. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.Y.; Lv, Y.P.; Yan, D.F.; Gao, F.L. Knockdown of mdr1 increases the sensitivity to adriamycin in drug resistant gastric cancer cells. Asian Pac. J. Cancer Prev. 2013, 14, 6757–6760. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Huan, M.L.; Liu, M.; Cheng, Y.; Sun, Y.; Cui, H.; Liu, D.Z.; Mei, Q.B.; Zhou, S.Y. Doxorubicin and resveratrol co-delivery nanoparticle to overcome doxorubicin resistance. Sci. Rep. 2016, 6, 35267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakata, S.; Fujiwara, M.; Ohtsuka, K.; Kamma, H.; Nagane, M.; Sakamoto, A.; Fujioka, Y. Atp-binding cassette transporters in primary central nervous system lymphoma: Decreased expression of mdr1 p-glycoprotein and breast cancer resistance protein in tumor capillary endothelial cells. Oncol. Rep. 2011, 25, 333–339. [Google Scholar]
- Liu, Y.; Lou, G.; Wu, W.; Shi, Y.; Zheng, M.; Chen, Z. Interferon-alpha sensitizes hbx-expressing hepatocarcinoma cells to chemotherapeutic drugs through inhibition of hbx-mediated NF-κb activation. Virol. J. 2013, 10, 168. [Google Scholar] [CrossRef]











© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Liu, X.-Y.; Pan, Q.; Wu, J.; Liu, Z.-H.; Wang, Y.; Liu, M.; Zhang, X.-L. Hepatitis C Virus-Induced FUT8 Causes 5-FU Drug Resistance in Human Hepatoma Huh7.5.1 Cells. Viruses 2019, 11, 378. https://0-doi-org.brum.beds.ac.uk/10.3390/v11040378
Li S, Liu X-Y, Pan Q, Wu J, Liu Z-H, Wang Y, Liu M, Zhang X-L. Hepatitis C Virus-Induced FUT8 Causes 5-FU Drug Resistance in Human Hepatoma Huh7.5.1 Cells. Viruses. 2019; 11(4):378. https://0-doi-org.brum.beds.ac.uk/10.3390/v11040378
Chicago/Turabian StyleLi, Shu, Xiao-Yu Liu, Qiu Pan, Jian Wu, Zhi-Hao Liu, Yong Wang, Min Liu, and Xiao-Lian Zhang. 2019. "Hepatitis C Virus-Induced FUT8 Causes 5-FU Drug Resistance in Human Hepatoma Huh7.5.1 Cells" Viruses 11, no. 4: 378. https://0-doi-org.brum.beds.ac.uk/10.3390/v11040378