Amplicon-Based Detection and Sequencing of SARS-CoV-2 in Nasopharyngeal Swabs from Patients With COVID-19 and Identification of Deletions in the Viral Genome That Encode Proteins Involved in Interferon Antagonism

 , , , , , , , , , , , , , , ,
, , , , , , , , , , , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics and Clinical Information
2.2. RNA Extraction and Preparation
2.3. Primer Design
2.4. RT and PCR
2.5. Library Preparation for MinION Sequencing
2.6. Informatics Analysis
3. Results
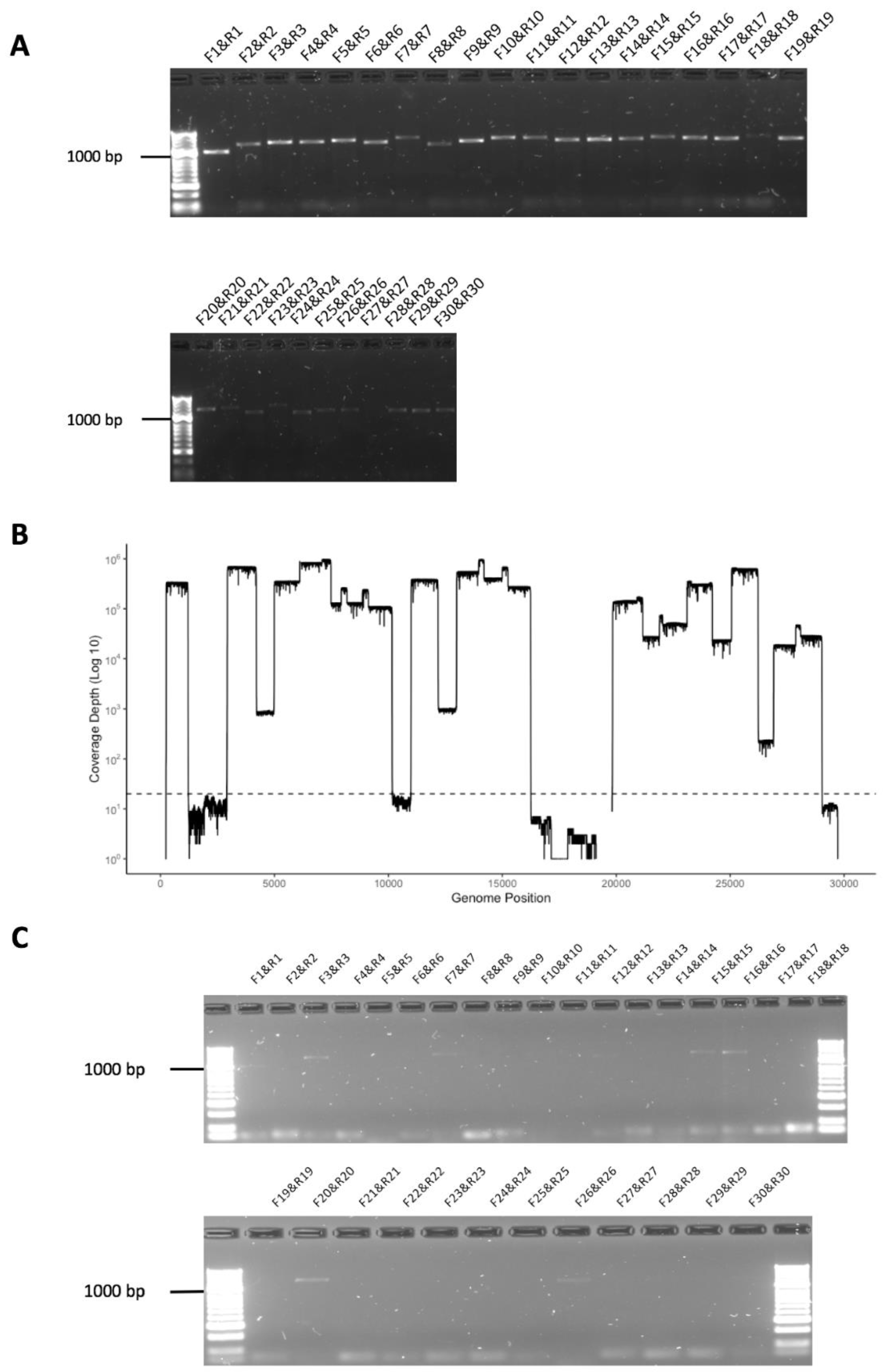
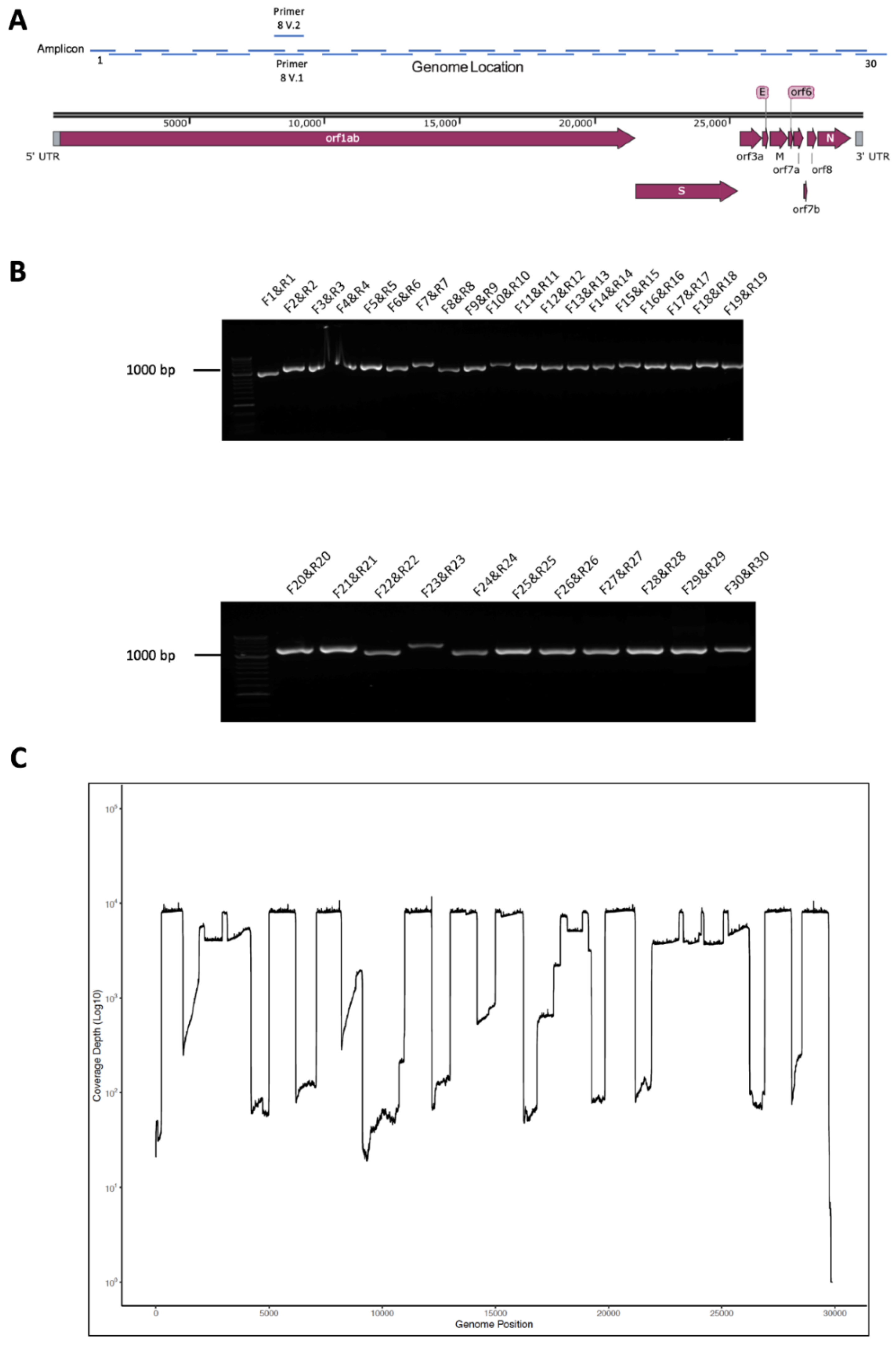
3.1. Primer and Amplicon Design
3.2. Validation of Amplicon Generation and Oxford Nanopore Sequencing Using RNA from Cells Infected with SARS-CoV-2
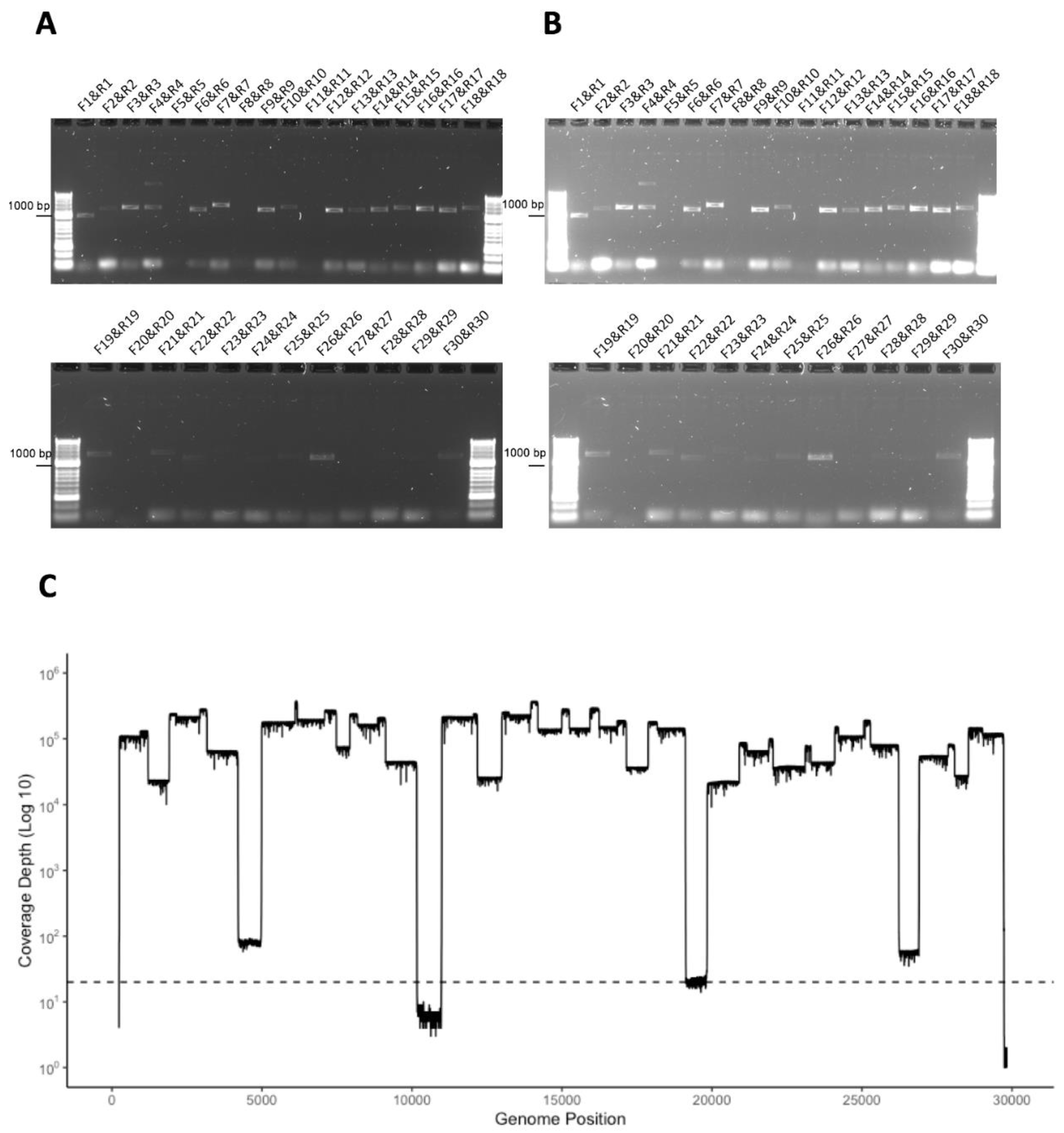
3.3. Initial Evaluation of the Amplicon Approach to Detect SARS-CoV-2 on RNA Purified from Nasopharyngeal Swabs Collected from Patients with COVID-19 Correctly Identified a Positive but also a False Negative Sample
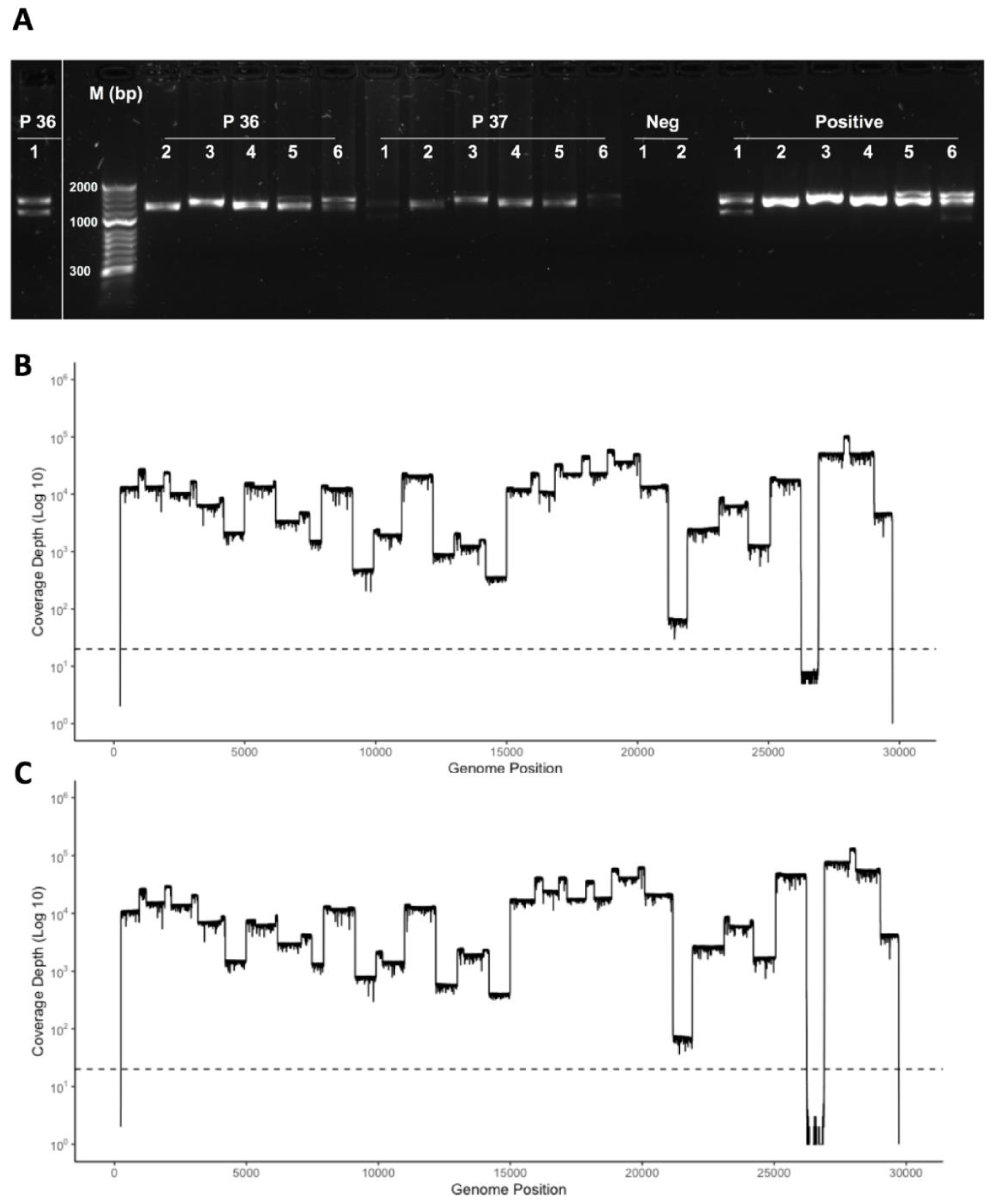
3.4. Development of a Multiplex Method to Generate Amplicons
3.5. Identification of Deletions in SARS-CoV-2 from Patients with COVID-19
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in SARS-CoV-2, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Docherty, A.B.; Harrison, E.M.; Green, C.A.; Hardwick, H.E.; Pius, R.; Norman, L.; Holden, K.A.; Read, J.M.; Dondelinger, F.; Carson, G.; et al. Features of 20 133 UK patients in hospital with covid-19 using the ISARIC WHO Clinical Characterisation Protocol: Prospective observational cohort study. BMJ 2020, 369, m1985. [Google Scholar] [CrossRef] [PubMed]
- Knight, S.R.; Ho, A.; Pius, R.; Buchan, I.; Carson, G.; Drake, T.M.; Dunning, J.; Fairfield, C.J.; Gamble, C.; Green, C.A.; et al. Risk stratification of patients admitted to hospital with covid-19 using the ISARIC WHO Clinical Characterisation Protocol: Development and validation of the 4C Mortality Score. BMJ 2020, 370, m3339. [Google Scholar] [CrossRef]
- Kottier, S.A.; Cavanagh, D.; Britton, P. Experimental evidence of recombination in coronavirus infectious bronchitis virus. Virology 1995, 213, 569–580. [Google Scholar] [CrossRef]
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 2020, 176, 104742. [Google Scholar] [CrossRef]
- Davidson, A.D.; Williamson, M.K.; Lewis, S.; Shoemark, D.; Carroll, M.W.; Heesom, K.J.; Zambon, M.; Ellis, J.; Lewis, P.A.; Hiscox, J.A.; et al. Characterisation of the transcriptome and proteome of SARS-CoV-2 reveals a cell passage induced in-frame deletion of the furin-like cleavage site from the spike glycoprotein. Genome Med. 2020, 12, 68. [Google Scholar] [CrossRef]
- Cavanagh, D. Severe acute respiratory syndrome vaccine development: Experiences of vaccination against avian infectious bronchitis coronavirus. Avian Pathol. 2003, 32, 567–582. [Google Scholar] [CrossRef] [Green Version]
- Carroll, M.W.; Matthews, D.A.; Hiscox, J.A.; Elmore, M.J.; Pollakis, G.; Rambaut, A.; Hewson, R.; Garcia-Dorival, I.; Bore, J.A.; Koundouno, R.; et al. Temporal and spatial analysis of the 2014–2015 Ebola virus outbreak in West Africa. Nature 2015, 524, 97–101. [Google Scholar] [CrossRef] [Green Version]
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 2016, 530, 228–232. [Google Scholar] [CrossRef]
- Pini, A.; Zomahoun, D.; Duraffour, S.; Derrough, T.; Charles, M.; Quick, J.; Loman, N.; Cowley, L.; Leno, M.; Ouedraogo, N.; et al. Field investigation with real-time virus genetic characterisation support of a cluster of Ebola virus disease cases in Dubreka, Guinea, April to June 2015. Eurosurveillance 2018, 23, 17-00140. [Google Scholar] [CrossRef]
- Kato, H.; Shimizu, H.; Shibue, Y.; Hosoda, T.; Iwabuchi, K.; Nagamine, K.; Saito, H.; Sawada, R.; Oishi, T.; Tsukiji, J.; et al. Clinical course of 2019 novel coronavirus disease (COVID-19) in individuals present during the outbreak on the Diamond Princess cruise ship. J. Infect. Chemother. 2020, 26, 865–869. [Google Scholar] [CrossRef]
- Russell, T.W.; Hellewell, J.; Jarvis, C.I.; van Zandvoort, K.; Abbott, S.; Ratnayake, R.; CMMID COVID-19 working group; Flasche, S.; Eggo, R.M.; Edmunds, W.J.; et al. Estimating the infection and case fatality ratio for coronavirus disease (COVID-19) using age-adjusted data from the outbreak on the Diamond Princess cruise ship, February 2020. Eurosurveillance 2020, 25, 2000256. [Google Scholar] [CrossRef] [Green Version]
- Sabir, J.S.; Lam, T.T.; Ahmed, M.M.; Li, L.; Shen, Y.; Abo-Aba, S.E.; Qureshi, M.I.; Abu-Zeid, M.; Zhang, Y.; Khiyami, M.A.; et al. Co-circulation of three camel coronavirus species and recombination of MERS-CoVs in Saudi Arabia. Science 2016, 351, 81–84. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.I.; Kim, Y.J.; Lemey, P.; Lee, I.; Park, S.; Bae, J.Y.; Kim, D.; Kim, H.; Jang, S.I.; Yang, J.S.; et al. The recent ancestry of Middle East respiratory syndrome coronavirus in Korea has been shaped by recombination. Sci. Rep. 2016, 6, 18825. [Google Scholar] [CrossRef]
- Nasir, J.A.; Kozak, R.A.; Aftanas, P.; Raphenya, A.R.; Smith, K.M.; Maguire, F.; Maan, H.; Alruwaili, M.; Banerjee, A.; Mbareche, H.; et al. A Comparison of Whole Genome Sequencing of SARS-CoV-2 Using Amplicon-Based Sequencing, Random Hexamers, and Bait Capture. Viruses 2020, 12, 895. [Google Scholar] [CrossRef]
- Kafetzopoulou, L.E.; Efthymiadis, K.; Lewandowski, K.; Crook, A.; Carter, D.; Osborne, J.; Aarons, E.; Hewson, R.; Hiscox, J.A.; Carroll, M.W.; et al. Assessment of metagenomic Nanopore and Illumina sequencing for recovering whole genome sequences of chikungunya and dengue viruses directly from clinical samples. Eurosurveillance 2018, 23, 1800228. [Google Scholar] [CrossRef] [Green Version]
- Dudas, G.; Carvalho, L.M.; Bedford, T.; Tatem, A.J.; Baele, G.; Faria, N.R.; Park, D.J.; Ladner, J.T.; Arias, A.; Asogun, D.; et al. Virus genomes reveal factors that spread and sustained the Ebola epidemic. Nature 2017, 544, 309–315. [Google Scholar] [CrossRef] [Green Version]
- So, R.T.Y.; Chu, D.K.W.; Miguel, E.; Perera, R.; Oladipo, J.O.; Fassi-Fihri, O.; Aylet, G.; Ko, R.L.W.; Zhou, Z.; Cheng, M.S.; et al. Diversity of Dromedary Camel Coronavirus HKU23 in African Camels Revealed Multiple Recombination Events among Closely Related Betacoronaviruses of the Subgenus Embecovirus. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef] [Green Version]
- Siu, K.L.; Yuen, K.S.; Castano-Rodriguez, C.; Ye, Z.W.; Yeung, M.L.; Fung, S.Y.; Yuan, S.; Chan, C.P.; Yuen, K.Y.; Enjuanes, L.; et al. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019, 33, 8865–8877. [Google Scholar] [CrossRef] [PubMed]
- Kanzawa, N.; Nishigaki, K.; Hayashi, T.; Ishii, Y.; Furukawa, S.; Niiro, A.; Yasui, F.; Kohara, M.; Morita, K.; Matsushima, K.; et al. Augmentation of chemokine production by severe acute respiratory syndrome coronavirus 3a/X1 and 7a/X4 proteins through NF-kappaB activation. FEBS Lett. 2006, 580, 6807–6812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karjee, S.; Minhas, A.; Sood, V.; Ponia, S.S.; Banerjea, A.C.; Chow, V.T.; Mukherjee, S.K.; Lal, S.K. The 7a accessory protein of severe acute respiratory syndrome coronavirus acts as an RNA silencing suppressor. J. Virol. 2010, 84, 10395–10401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, B.E.; Fong, S.W.; Chan, Y.H.; Mak, T.M.; Ang, L.W.; Anderson, D.E.; Lee, C.Y.; Amrun, S.N.; Lee, B.; Goh, Y.S.; et al. Effects of a major deletion in the SARS-CoV-2 genome on the severity of infection and the inflammatory response: An observational cohort study. Lancet 2020, 396, 603–611. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, J.; Xiao, Y.; Zhang, J.; Wang, Y.; Chen, L.; Paranhos-Baccala, G.; Ren, L.; Wang, J. Genotype shift in human coronavirus OC43 and emergence of a novel genotype by natural recombination. J. Infect. 2015, 70, 641–650. [Google Scholar] [CrossRef] [Green Version]
- Hewson, K.A.; Noormohammadi, A.H.; Devlin, J.M.; Browning, G.F.; Schultz, B.K.; Ignjatovic, J. Evaluation of a novel strain of infectious bronchitis virus emerged as a result of spike gene recombination between two highly diverged parent strains. Avian Pathol. 2014, 43, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Terada, Y.; Matsui, N.; Noguchi, K.; Kuwata, R.; Shimoda, H.; Soma, T.; Mochizuki, M.; Maeda, K. Emergence of pathogenic coronaviruses in cats by homologous recombination between feline and canine coronaviruses. PLoS ONE 2014, 9, e106534. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence 5′–3′ | Location | Size (bp) | Multiplex Pool | |
|---|---|---|---|---|---|
| Start | End | ||||
| SARS-CoV-2_1_F | GTGTGACCGAAAGGTAAGATGG | 248 | 269 | 956 | 1 |
| SARS-CoV-2_1_R | TTGCATTCATTTGGTGACGC | 1203 | 1184 | ||
| SARS-CoV-2_2_F | GGTGTATACTGCTGCCGTGA | 944 | 963 | 1213 | 2 |
| SARS-CoV-2_2_R | GCCAATCAAGGACGGGTTTG | 2156 | 2137 | ||
| SARS-CoV-2_3_F | CCGCACTCTTGAAACTGCTC | 1912 | 1931 | 1254 | 3 |
| SARS-CoV-2_3_R | GCAGAAGTGGCACCAAATTC | 3165 | 3146 | ||
| SARS-CoV-2_4_F | ACACCACTGGGCATTGATTTAG | 2936 | 2957 | 1264 | 4 |
| SARS-CoV-2_4_R | TTTCAGTAGTGCCACCAGCC | 4199 | 4180 | ||
| SARS-CoV-2_5_F | CTTCATCCAGATTCTGCCAC | 4052 | 4071 | 1296 | 5 |
| SARS-CoV-2_5_R | AGCAGGTGGATTAAACTTCAACTC | 5347 | 5324 | ||
| SARS-CoV-2_6_F | CAACATTAACCTCCACACGC | 4990 | 5009 | 1189 | 6 |
| SARS-CoV-2_6_R | ATCAATAGCCACCACATCACC | 6178 | 6158 | ||
| SARS-CoV-2_7_F | AGAAACCTGCTTCAAGAGAGC | 6108 | 6128 | 1373 | 1 |
| SARS-CoV-2_7_R | ATTACAACCGTCTACAACATGCAC | 7480 | 7457 | ||
| SARS-CoV-2_8_F | GTCACTATTGCAACCTACTGTAC | 7091 | 7113 | 1093 | 2 |
| SARS-CoV-2_8_R | CTTGCCGAGCTGCTGAAATA | 8183 | 8164 | ||
| SARS-CoV-2_9_F | AATCAGCGTCTGTTTACTACAGTC | 7929 | 7952 | 1192 | 3 |
| SARS-CoV-2_9_R | GTGTCAGGGCGTAAACTTTC | 9120 | 9101 | ||
| SARS-CoV-2_10_F | TTGTCGTGCCTGGTTTGC | 8856 | 8873 | 1303 | 4 |
| SARS-CoV-2_10_R | ACGTCATCAAGCCAAAGACC | 10158 | 10139 | ||
| SARS-CoV-2_11_F | AGTGGAGCAATGGATACAAC | 9917 | 9936 | 1239 | 5 |
| SARS-CoV-2_11_R | AGCTACAGTGGCAAGAGAAG | 11209 | 11190 | ||
| SARS-CoV-2_12_F | AGGGTACACACCACTGGTTG | 10995 | 11014 | 1185 | 6 |
| SARS-CoV-2_12_R | CACCATTAGCAACAGCCTGC | 12179 | 12160 | ||
| SARS-CoV-2_13_F | GTGAAGAAATGCTGGACAACAG | 12057 | 12078 | 1180 | 1 |
| SARS-CoV-2_13_R | GCACCACCAAAGGATTCTTG | 13236 | 13217 | ||
| SARS-CoV-2_14_F | TAGTTTAGCTGCCACAGTACG | 12997 | 13017 | 1200 | 2 |
| SARS-CoV-2_14_R | AGTTAAAGCCCTGGTCAAGG | 14196 | 14177 | ||
| SARS-CoV-2_15_F | ATACGCCAACTTAGGTGAACG | 13962 | 13982 | 1284 | 3 |
| SARS-CoV-2_15_R | AACATGTTGTGCCAACCACC | 15245 | 15226 | ||
| SARS-CoV-2_16_F | TGAGTTATGAGGATCAAGATGCAC | 14996 | 15019 | 1243 | 4 |
| SARS-CoV-2_16_R | GCCTGTAAGACTGTATGCGG | 16238 | 16219 | ||
| SARS-CoV-2_17_F | CCCAGATCCATCAAGAATCCTAG | 15933 | 15955 | 1214 | 5 |
| SARS-CoV-2_17_R | TGCGAGCAGAAGGGTAGTAG | 17146 | 17127 | ||
| SARS-CoV-2_18_F | AAGGTGACTATGGTGATGCTG | 16841 | 16861 | 1336 | 6 |
| SARS-CoV-2_18_R | GGTATGCCAGGTATGTCAACAC | 18176 | 18155 | ||
| SARS-CoV-2_19_F | ACTCAAACCACTGAAACAGCTC | 17875 | 17896 | 1239 | 1 |
| SARS-CoV-2_19_R | GTCACTACAAGGCTGTGCATC | 19113 | 19093 | ||
| SARS-CoV-2_20_F | AGCTAGTTGTGATGCAATCATGAC | 18846 | 18869 | 1235 | 2 |
| SARS-CoV-2_20_R | CTTGTTTGGGACCTACAGATGG | 20098 | 20077 | ||
| SARS-CoV-2_21_F | TTTGGGTGTGGACATTGCTG | 19842 | 19861 | 1323 | 3 |
| SARS-CoV-2_21_R | ATAGCCACGGAACCTCCAAG | 21164 | 21145 | ||
| SARS-CoV-2_22_F | TAAGACAGTGGTTGCCTACG | 20912 | 20931 | 1125 | 4 |
| SARS-CoV-2_22_R | TCTGAACTCACTTTCCATCCAAC | 22036 | 22014 | ||
| SARS-CoV-2_23_F | TTCGAAGACCCAGTCCCTAC | 21895 | 21914 | 1405 | 5 |
| SARS-CoV-2_23_R | TGGATCACGGACAGCATCAG | 23299 | 23280 | ||
| SARS-CoV-2_24_F | TTGAACTTCTACATGCACCAGC | 23106 | 23127 | 1111 | 6 |
| SARS-CoV-2_24_R | CCAGAAGTGATTGTACCCGC | 24216 | 24197 | ||
| SARS-CoV-2_25_F | TTGCTGCTAGAGACCTCATTTG | 24093 | 24114 | 1190 | 1 |
| SARS-CoV-2_25_R | GCAACTGGTCATACAGCAAAG | 25282 | 25262 | ||
| SARS-CoV-2_26_F | GGTGACATCTCTGGCATTAATGC | 25061 | 25083 | 1163 | 2 |
| SARS-CoV-2_26_R | TGCTTACAAAGGCACGCTAG | 26223 | 26204 | ||
| SARS-CoV-2_27_F | ACCAGCTGTACTCAACTCAATTG | 26027 | 26049 | 1137 | 3 |
| SARS-CoV-2_27_R | CTGCTACTGGAATGGTCTGTG | 27163 | 27143 | ||
| SARS-CoV-2_28_F | TGACCAGACCGCTTCTAGAAAG | 26908 | 26929 | 1180 | 4 |
| SARS-CoV-2_28_R | GCCTCATCCACGCACAATTC | 28087 | 28068 | ||
| SARS-CoV-2_29_F | TGTCACGCCTAAACGAACATG | 27876 | 27896 | 1147 | 5 |
| SARS-CoV-2_29_R | GATTTCTTAGTGACAGTTTGGCC | 29022 | 29000 | ||
| SARS-CoV-2_30_F | CGAATTCGTGGTGGTGACG | 28550 | 28568 | 1173 | 6 |
| SARS-CoV-2_30_R | GGTGGCTCTTTCAAGTCCTC | 29722 | 29703 | ||
| Deletion Information | Affected Gene Information | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient Number | Start (bp) | End (bp) | Supporting Reads | Quality Score | SD Span | SD Pos | Gene Start | Gene End | Gene Name | Overlap (bp) | Gene2 Start | Gene2 End | Gene2 Name | Overlap2 (bp) |
| 22 | 19325 | 19380 | 9 | 10 | 4.42 | 58.42 | 266 | 21555 | Orf1ab | 55 | − | − | − | − |
| 22 | 20294 | 20429 | 8 | 10 | 0 | 0 | 266 | 21555 | Orf1ab | 135 | − | − | − | − |
| 22 | 25417 | 25796 | 10 | 12 | 0.79 | 0.86 | 25393 | 26220 | Orf3a | 379 | − | − | − | − |
| 22 | 27578 | 27624 | 16 | 19 | 2.49 | 2.45 | 27394 | 27759 | Orf7a | 46 | − | − | − | − |
| 22 | 28756 | 28884 | 7 | 8 | 2.27 | 3.4 | 28274 | 29533 | N | 128 | − | − | − | − |
| 23 | 2143 | 2198 | 5 | 5 | 4.36 | 73.69 | 266 | 21555 | Orf1ab | 55 | − | − | − | − |
| 23 | 2375 | 2421 | 8 | 8 | 5.54 | 82.85 | 266 | 21555 | Orf1ab | 46 | − | − | − | − |
| 23 | 2589 | 2642 | 6 | 6 | 3.2 | 41.78 | 266 | 21555 | Orf1ab | 53 | − | − | − | − |
| 23 | 2654 | 2741 | 5 | 5 | 8.73 | 51.88 | 266 | 21555 | Orf1ab | 87 | − | − | − | − |
| 23 | 2859 | 2904 | 9 | 10 | 2.42 | 88.48 | 266 | 21555 | Orf1ab | 45 | − | − | − | − |
| 25 | 20274 | 20383 | 7 | 8 | 6.2 | 7.5 | 266 | 21555 | Orf1ab | 109 | − | − | − | − |
| 25 | 20279 | 20340 | 10 | 12 | 3.83 | 2.57 | 266 | 21555 | Orf1ab | 61 | − | − | − | − |
| 25 | 27594 | 27640 | 10 | 11 | 2.37 | 31.77 | 27394 | 27759 | Orf7a | 46 | − | − | − | − |
| 26 | 27386 | 27699 | 91 | 99 | 0.86 | 1.66 | 27394 | 27759 | Orf7a | 305 | 27202 | 27387 | ORF6 | 1 |
| 26 | 20274 | 20338 | 9 | 11 | 1.5 | 1.02 | 266 | 21555 | Orf1ab | 64 | − | − | − | − |
| 27 | 27020 | 27073 | 11 | 13 | 0.6 | 0.3 | 26523 | 27191 | M | 53 | − | − | − | − |
| 27 | 27522 | 27761 | 24 | 29 | 1.09 | 2.04 | 27394 | 27759 | Orf7a | 237 | − | − | − | − |
| 27 | 27689 | 27763 | 23 | 27 | 3.58 | 29.9 | 27394 | 27759 | Orf7a | 70 | ||||
| 28 | 2025 | 2088 | 5 | 5 | 7.16 | 80.94 | 266 | 21555 | Orf1ab | 63 | ||||
| 28 | 25508 | 25568 | 6 | 6 | 8.31 | 61.45 | 25393 | 26220 | Orf3a | 60 | ||||
| 28 | 27884 | 27934 | 6 | 6 | 7.19 | 27.43 | 27894 | 28259 | Orf8 | 40 | ||||
| 29 | 27546 | 27650 | 7 | 8 | 14.61 | 64.8 | 27394 | 27759 | Orf7a | 104 | ||||
| 29 | 28454 | 28731 | 6 | 7 | 26.89 | 58.79 | 28274 | 29533 | N | 277 | ||||
| 30 | 25398 | 25776 | 12 | 14 | 1.56 | 69.87 | 25393 | 26220 | Orf3a | 378 | ||||
| 30 | 27555 | 27625 | 31 | 38 | 1.94 | 5.71 | 27394 | 27759 | Orf7a | 70 | ||||
| 30 | 8676 | 8723 | 11 | 12 | 1.79 | 38.61 | 266 | 21555 | Orf1ab | 47 | ||||
| 31 | 28444 | 28775 | 62 | 77 | 1.29 | 1.31 | 28274 | 29533 | N | 331 | ||||
| 32 | 25480 | 25551 | 97 | 99 | 1.62 | 2.04 | 25393 | 26220 | Orf3a | 71 | ||||
| 36 | 20274 | 20339 | 11 | 13 | 1.43 | 0.87 | 266 | 21555 | Orf1ab | 65 | ||||
| 37 | 25429 | 25641 | 11 | 13 | 17 | 7.89 | 25393 | 26220 | Orf3a | 212 | ||||
| 37 | 25432 | 25808 | 10 | 12 | 5.24 | 44.62 | 25393 | 26220 | Orf3a | 376 | ||||
| 37 | 28444 | 28776 | 8 | 9 | 0.71 | 0.65 | 28274 | 29533 | N | 332 | ||||
| 43 | 27426 | 27559 | 5 | 5 | 19.77 | 50.25 | 27394 | 27759 | Orf7a | 133 | ||||
| 43 | 27690 | 27732 | 6 | 6 | 2.25 | 49.85 | 27394 | 27759 | Orf7a | 42 | ||||
| 43 | 28011 | 28062 | 13 | 14 | 5.36 | 77.82 | 27894 | 28259 | Orf8 | 51 | ||||
| 43 | 28196 | 28238 | 5 | 5 | 2 | 96.73 | 27894 | 28259 | Orf8 | 42 | ||||
| 43 | 28481 | 28536 | 5 | 5 | 3 | 64.73 | 28274 | 29533 | N | 55 | ||||
| 28601 | 28718 | 5 | 5 | 4.82 | 43.76 | 28274 | 29533 | N | 117 | |||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moore, S.C.; Penrice-Randal, R.; Alruwaili, M.; Randle, N.; Armstrong, S.; Hartley, C.; Haldenby, S.; Dong, X.; Alrezaihi, A.; Almsaud, M.; et al. Amplicon-Based Detection and Sequencing of SARS-CoV-2 in Nasopharyngeal Swabs from Patients With COVID-19 and Identification of Deletions in the Viral Genome That Encode Proteins Involved in Interferon Antagonism. Viruses 2020, 12, 1164. https://0-doi-org.brum.beds.ac.uk/10.3390/v12101164
Moore SC, Penrice-Randal R, Alruwaili M, Randle N, Armstrong S, Hartley C, Haldenby S, Dong X, Alrezaihi A, Almsaud M, et al. Amplicon-Based Detection and Sequencing of SARS-CoV-2 in Nasopharyngeal Swabs from Patients With COVID-19 and Identification of Deletions in the Viral Genome That Encode Proteins Involved in Interferon Antagonism. Viruses. 2020; 12(10):1164. https://0-doi-org.brum.beds.ac.uk/10.3390/v12101164
Chicago/Turabian StyleMoore, Shona C., Rebekah Penrice-Randal, Muhannad Alruwaili, Nadine Randle, Stuart Armstrong, Catherine Hartley, Sam Haldenby, Xiaofeng Dong, Abdulrahman Alrezaihi, Mai Almsaud, and et al. 2020. "Amplicon-Based Detection and Sequencing of SARS-CoV-2 in Nasopharyngeal Swabs from Patients With COVID-19 and Identification of Deletions in the Viral Genome That Encode Proteins Involved in Interferon Antagonism" Viruses 12, no. 10: 1164. https://0-doi-org.brum.beds.ac.uk/10.3390/v12101164