Phage-Mediated Molecular Detection (PMMD): A Novel Rapid Method for Phage-Specific Bacterial Detection

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Growth and Phage Assay Conditions
2.2. Phage Lysates and CsCl Gradient Purification
2.3. RNA Preps, DNA Preps, Gel Electrophoresis and Sequencing
2.4. PCR and qPCR Reactions and Analysis
2.5. Oligonucleotides
2.6. Bacterial Strains and Phages
3. Results
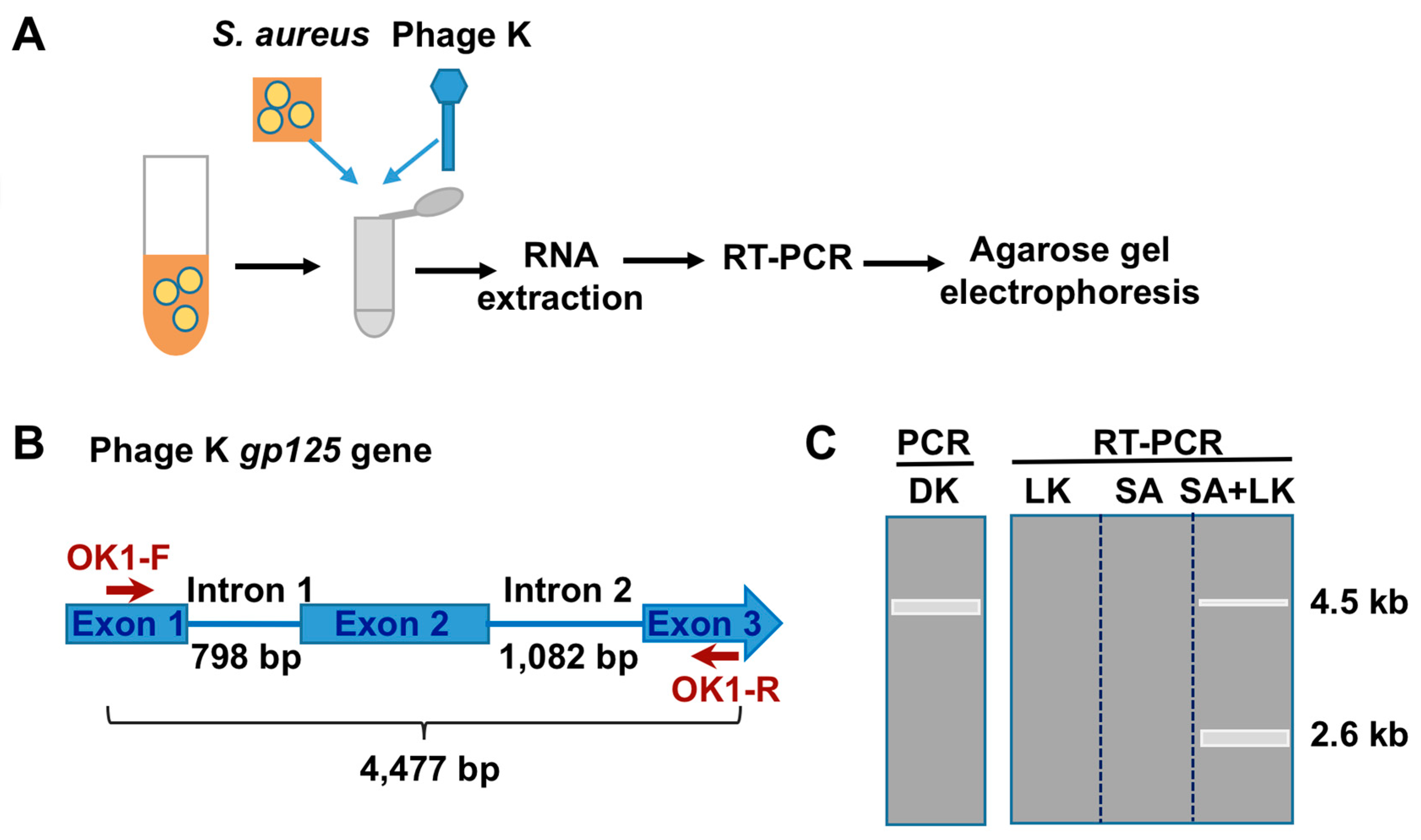
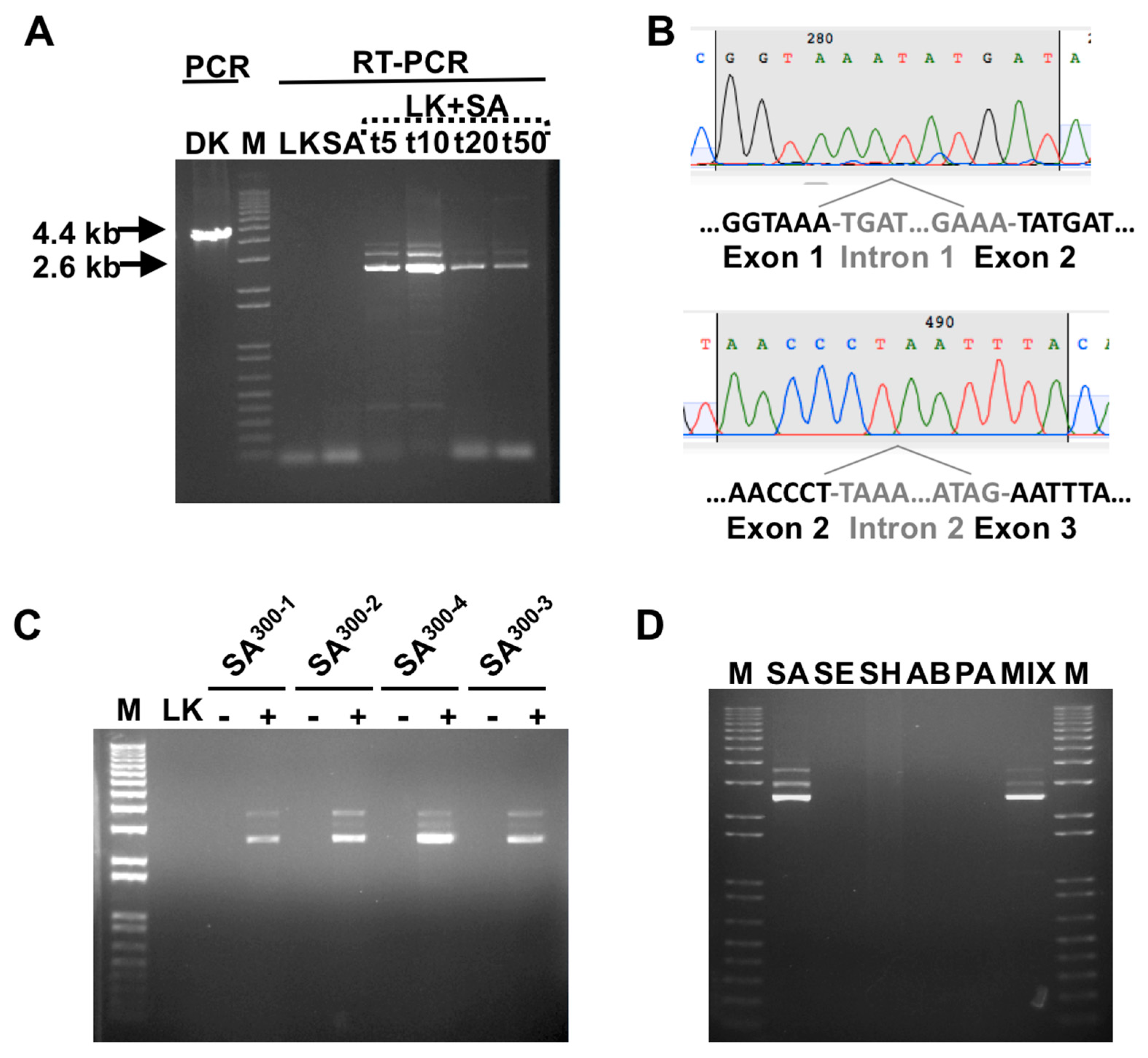
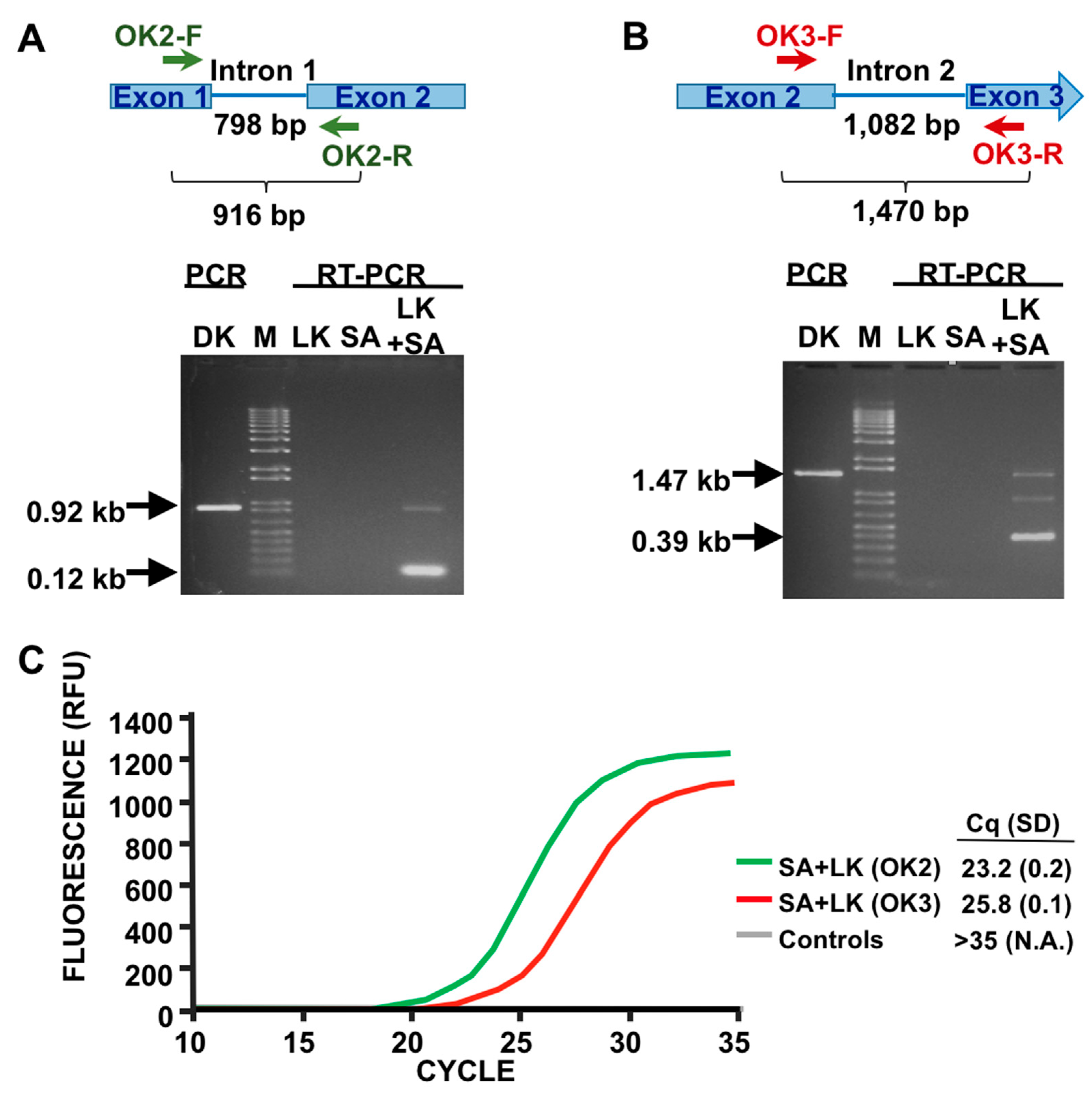
3.1. Detection of SA by Phage K RT-PCR Plus Gel Electrophoresis
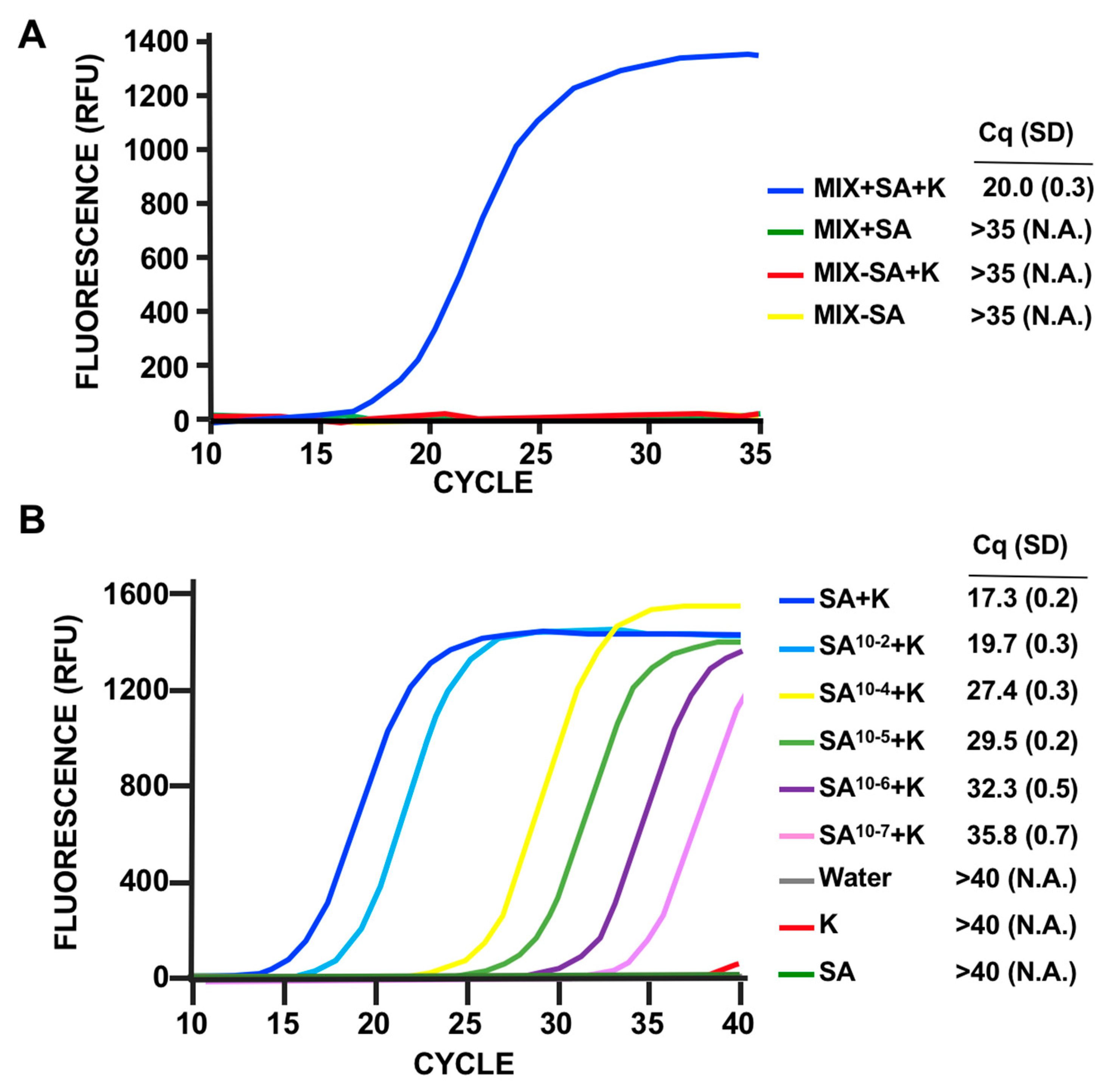
3.2. Phage Host-Range Specific Bacterial Detection
3.3. LOD Using Fluorescent Quantitative RT-PCR
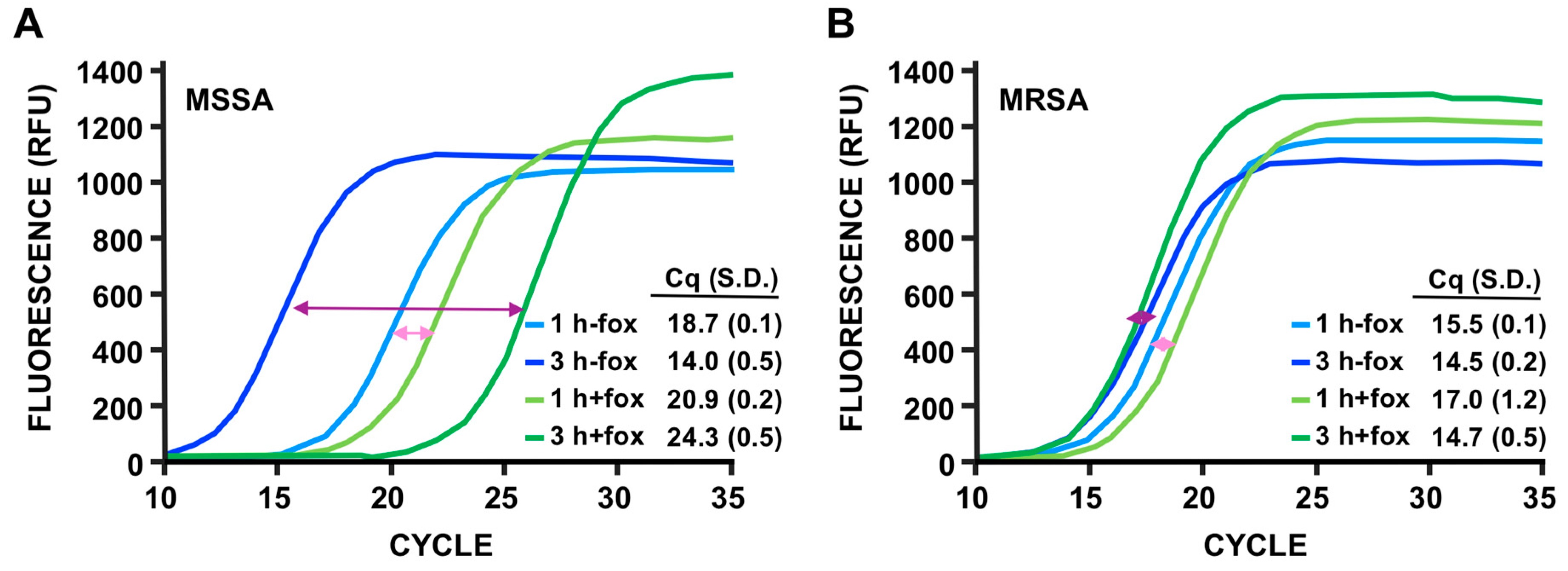
3.4. Discrimination of MSSA Versus MRSA
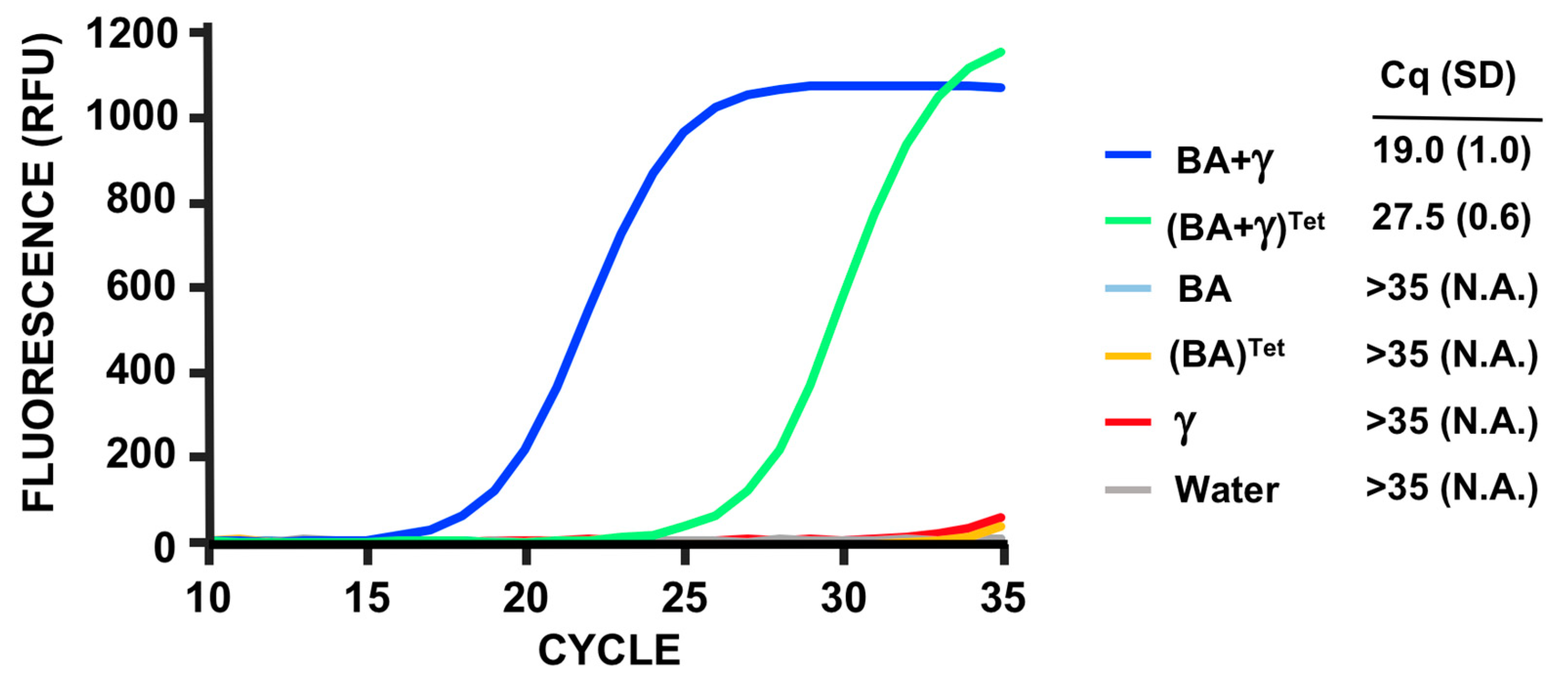
3.5. PMMD Applied to Bacillus Anthracis
4. Discussion
5. Patents
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lowy, F.D. Staphylococcus aureus infections. N. Engl. J. Med. 1988, 339, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.Y.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, V.S.; Dhaliwal, D.K.; Raju, L.; Kowalski, R.P. Antibiotic Resistance in the Treatment of Staphylococcus aureus Keratitis: A 20-Year Review. Cornea 2015, 34, 698–703. [Google Scholar] [CrossRef] [Green Version]
- Chambers, H.F.; DeLeo, F.R. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 2009, 7, 629–641. [Google Scholar] [CrossRef]
- Brakstad, O.G.; Aasbakk, K.; Maeland, J.A. Detection of Staphylococcus aureus by polymerase chain reaction amplification of the nuc gene. J. Clin. Microbiol. 1992, 30, 1654–1660. [Google Scholar] [CrossRef] [Green Version]
- Banada, P.P.; Chakravorty, S.; Shah, D.; Burday, M.; Mazzella, F.M.; Alland, D. Highly sensitive detection of Staphylococcus aureus directly from patient blood. PLoS ONE 2012, 7, e31126. [Google Scholar] [CrossRef] [Green Version]
- Gaibani, P.; Rossini, G.; Ambretti, S.; Gelsomino, F.; Pierro, A.M.; Varani, S.; Paolucci, M.; Landini, M.P.; Sambri, V. Blood culture systems: Rapid detection--how and why? Int. J. Antimicrob. Agents 2009, 34, S13–S15. [Google Scholar] [CrossRef]
- Keen, E.C. A century of phage research: Bacteriophages and the shaping of modern biology. Bioessays 2015, 37, 6–9. [Google Scholar] [CrossRef]
- Haq, I.U.; Chaudhry, W.N.; Akhtar, M.N.; Andleeb, S.; Qadri, I. Bacteriophages and their implications on future biotechnology: A review. Virol. J. 2012, 9, 9. [Google Scholar] [CrossRef] [Green Version]
- Mulvey, M.C.; Sacksteder, K.A.; Einck, L.; Nacy, C.A. Generation of a novel nucleic acid-based reporter system to detect phenotypic susceptibility to antibiotics in Mycobacterium tuberculosis. MBio 2012, 3, e00312-11. [Google Scholar] [CrossRef] [Green Version]
- Yosef, I.; Manor, M.; Kiro, R.; Qimron, U. Temperate and lytic bacteriophages programmed to sensitize and kill antibiotic-resistant bacteria. Proc. Natl. Acad. Sci. USA 2015, 112, 7267–7272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smartt, A.E.; Ripp, S. Bacteriophage reporter technology for sensing and detecting microbial targets. Anal. Bioanal. Chem. 2011, 400, 991–1007. [Google Scholar] [CrossRef]
- Bhowmick, T.; Mirrett, S.; Reller, L.B.; Price, C.; Qi, C.; Weinstein, M.P.; Kirn, T.J. Controlled multicenter evaluation of a bacteriophage-based method for rapid detection of Staphylococcus aureus in positive blood cultures. J. Clin. Microbiol. 2013, 51, 1226–1230. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, K.V.; Turner, N.N.; Roundtree, S.S.; McGowan, K.L. Rapid detection of methicillin-resistant Staphylococcus aureus (MRSA) and methicillin-susceptible Staphylococcus aureus (MSSA) using the KeyPath MRSA/MSSA blood culture test and the BacT/ALERT system in a pediatric population. Arch. Pathol. Lab. Med. 2013, 137, 1103–1105. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Liu, S.; Ren, H.; Yang, M.; Ke, Y.; Huang, L.; Liu, C.; Liu, B.; Chen, Z. Ultrasensitive detection of bacteria by targeting abundant transcripts. Sci. Rep. 2016, 6, 20393. [Google Scholar] [CrossRef] [Green Version]
- Mulvey, M.C.; Lemmon, M.; Rotter, S.; Lees, J.; Einck, L.; Nacy, C.A. Optimization of a nucleic acid-based reporter system to detect Mycobacterium tuberculosis antibiotic sensitivity. Antimicrob. Agents Chemother. 2015, 59, 407–413. [Google Scholar] [CrossRef] [Green Version]
- Krupovic, M.; Prangishvili, D.; Hendrix, R.W.; Bamford, D.H. Genomics of bacterial and archaeal viruses: Dynamics within the prokaryotic virosphere. Microbiol. Mol. Biol. Rev. 2011, 75, 610–635. [Google Scholar] [CrossRef] [Green Version]
- Xia, G.; Wolz, C. Phages of Staphylococcus aureus and their impact on host evolution. Infect. Genet. Evol. 2014, 21, 593–601. [Google Scholar] [CrossRef]
- Estrella, L.A.; Quinones, J.; Henry, M.; Hannah, R.M.; Pope, R.K.; Hamilton, T.; Teneza-Mora, N.; Hall, E.; Biswajit, B. Characterization of novel Staphylococcus aureus lytic phage and defining their combinatorial virulence using the OmniLog system. Bacteriophage 2016, 6, e1219440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köhrer, K.; Domdey, H. Preparation of high molecular weight RNA. Methods Enzymol. 1991, 194, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.W. Ape, a Plasmid Editor. 2017. Available online: http://biologylabs.utah.edu/jorgensen/wayned/ape (accessed on 19 May 2017).
- O’Flaherty, S.; Coffey, A.; Edwards, R.; Meaney, W.; Fitzgerald, G.F.; Ross, R.P. Genome of staphylococcal phage K: A new lineage of Myoviridae infecting gram-positive bacteria with a low G+C content. J. Bacteriol. 2004, 186, 2862–2871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, J.J. Revised genome sequence of Staphylococcus aureus bacteriophage K. Genome Announc. 2014, 2, e01173-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuch, R.; Fischetti, V.A. Detailed genomic analysis of the Wbeta and gamma phages infecting Bacillus anthracis: Implications for evolution of environmental fitness and antibiotic resistance. J. Bacteriol. 2006, 188, 3037–3051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, A.P.; Northrop, J.H. The kinetics of the bacterium-bacteriophage reaction. J. Gen. Physiol. 1930, 14, 223–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raiston, D.J.; Krueger, A.P. The isolation of a staphylococcal phage variant susceptible to an unusual host control. J. Gen. Physiol. 1954, 37, 685–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hugh, R.; Ellis, M.A. The neotype strain for Staphylococcus epidermidis (Winslow and Winslow 1908) Evans 1916. Int. J. Syst. Evol. Microbiol. 1968, 18, 231–239. [Google Scholar] [CrossRef] [Green Version]
- Holloway, B.W. Genetic recombination in Pseudomonas aeruginosa. J. Gen. Microbiol. 1955, 13, 572–581. [Google Scholar] [CrossRef] [Green Version]
- Abshire, T.G.; Brown, J.E.; Ezzell, W. Production and validation of the use of gamma phage for identification of Bacillus anthracis. J. Clin. Microbiol. 2005, 43, 4780–4788. [Google Scholar] [CrossRef] [Green Version]
- Brown, E.R.; Cherry, W.B. Specific identification of Bacillus anthracis by means of a variant bacteriophage. J. Infect. Dis. 1955, 96, 34–39. [Google Scholar] [CrossRef]
- Kreiswirth, B.N.; Löfdahl, S.; Betley, M.J.; O’reilly, M.; Schlievert, P.M.; Bergdoll, M.S.; Novick, R.P. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 1983, 305, 709–712. [Google Scholar] [CrossRef]
- Zurawski, D.V.; Thompson, M.G.; McQueary, C.N.; Matalka, M.N.; Sahl, J.W.; Craft, D.W.; Rasko, D.A. Genome sequences of four divergent multidrug-resistant Acinetobacter baumannii strains isolated from patients with sepsis or osteomyelitis. J. Bacteriol. 2012, 194, 1619–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterne, M. Avirulent anthrax vaccine. Onderstepoort J. Vet. Sci. Anim. Ind. 1946, 21, 41–43. [Google Scholar] [PubMed]
- Carrel, M.; Perencevich, E.N.; David, M.Z. USA300 methicillin-resistant Staphylococcus aureus, United States, 2000–2013. Emerg. Infect. Dis. 2015, 21, 1973–1980. [Google Scholar] [CrossRef]
- Palavecino, E.L. Clinical, epidemiologic, and laboratory aspects of methicillin-resistant Staphylococcus aureus infections. Methods Mol. Biol. 2020, 269, 1–28. [Google Scholar] [CrossRef]
- Swenson, J.M.; Tenover, F.C.; Cefoxitin Disk Study Group. Results of disk diffusion testing with cefoxitin correlate with presence of mecA in Staphylococcus spp. J. Clin. Microbiol. 2005, 43, 3818–3823. [Google Scholar] [CrossRef] [Green Version]
- Sergueev, K.V.; He, Y.; Borschel, R.H.; Nikolich, M.P.; Filippov, A.A. Rapid and sensitive detection of Yersinia pestis using amplification of plague diagnostic bacteriophages monitored by real-time PCR. PLoS ONE 2010, 5, e11337. [Google Scholar] [CrossRef] [Green Version]
- Sergueev, K.V.; Filippov, A.A.; Nikolich, M.P. Highly Sensitive Bacteriophage-Based Detection of Brucella abortus in Mixed Culture and Spiked Blood. Viruses 2017, 9, E144. [Google Scholar] [CrossRef]
- Stanley, E.C.; Mole, R.J.; Smith, R.J.; Glenn, S.M.; Barer, M.R.; McGowan, M.; Rees, C.E. Development of a new, combined rapid method using phage and PCR for detection and identification of viable Mycobacterium paratuberculosis bacteria within 48 hours. Appl. Environ. Microbiol. 2007, 73, 1851–1857. [Google Scholar] [CrossRef] [Green Version]
- Pournaras, S.; Sabat, A.J.; Grundmann, H.; Hendrix, R.; Tsakris, A.; Friedrich, A.W. Driving forces of mechanisms regulating oxacillin-resistance phenotypes of MRSA: Truly oxacillin-susceptible mecA-positive Staphylococcus aureus clinical isolates also exist. Curr. Pharm. Des. 2015, 21, 2048–2053. [Google Scholar] [CrossRef]
- El Haddad, L.; Abdallah, N.B.; Plante, P.L.; Dumaresq, J.; Katsarava, R.; Labrie, S.; Corbeil, J.; St-Gelais, D.; Moineau, S. Improving the safety of Staphylococcus aureus polyvalent phages by their production on a Staphylococcus xylosus strain. PLoS ONE 2014, 9, e102600. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malagon, F.; Estrella, L.A.; Stockelman, M.G.; Hamilton, T.; Teneza-Mora, N.; Biswas, B. Phage-Mediated Molecular Detection (PMMD): A Novel Rapid Method for Phage-Specific Bacterial Detection. Viruses 2020, 12, 435. https://0-doi-org.brum.beds.ac.uk/10.3390/v12040435
Malagon F, Estrella LA, Stockelman MG, Hamilton T, Teneza-Mora N, Biswas B. Phage-Mediated Molecular Detection (PMMD): A Novel Rapid Method for Phage-Specific Bacterial Detection. Viruses. 2020; 12(4):435. https://0-doi-org.brum.beds.ac.uk/10.3390/v12040435
Chicago/Turabian StyleMalagon, Francisco, Luis A. Estrella, Michael G. Stockelman, Theron Hamilton, Nimfa Teneza-Mora, and Biswajit Biswas. 2020. "Phage-Mediated Molecular Detection (PMMD): A Novel Rapid Method for Phage-Specific Bacterial Detection" Viruses 12, no. 4: 435. https://0-doi-org.brum.beds.ac.uk/10.3390/v12040435