YerA41, a Yersinia ruckeri Bacteriophage: Determination of a Non-Sequencable DNA Bacteriophage Genome via RNA-Sequencing

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Phage Propagation
2.2. Purification of Phage Particles
2.3. One Step Growth Curve
2.4. Total RNA Extraction
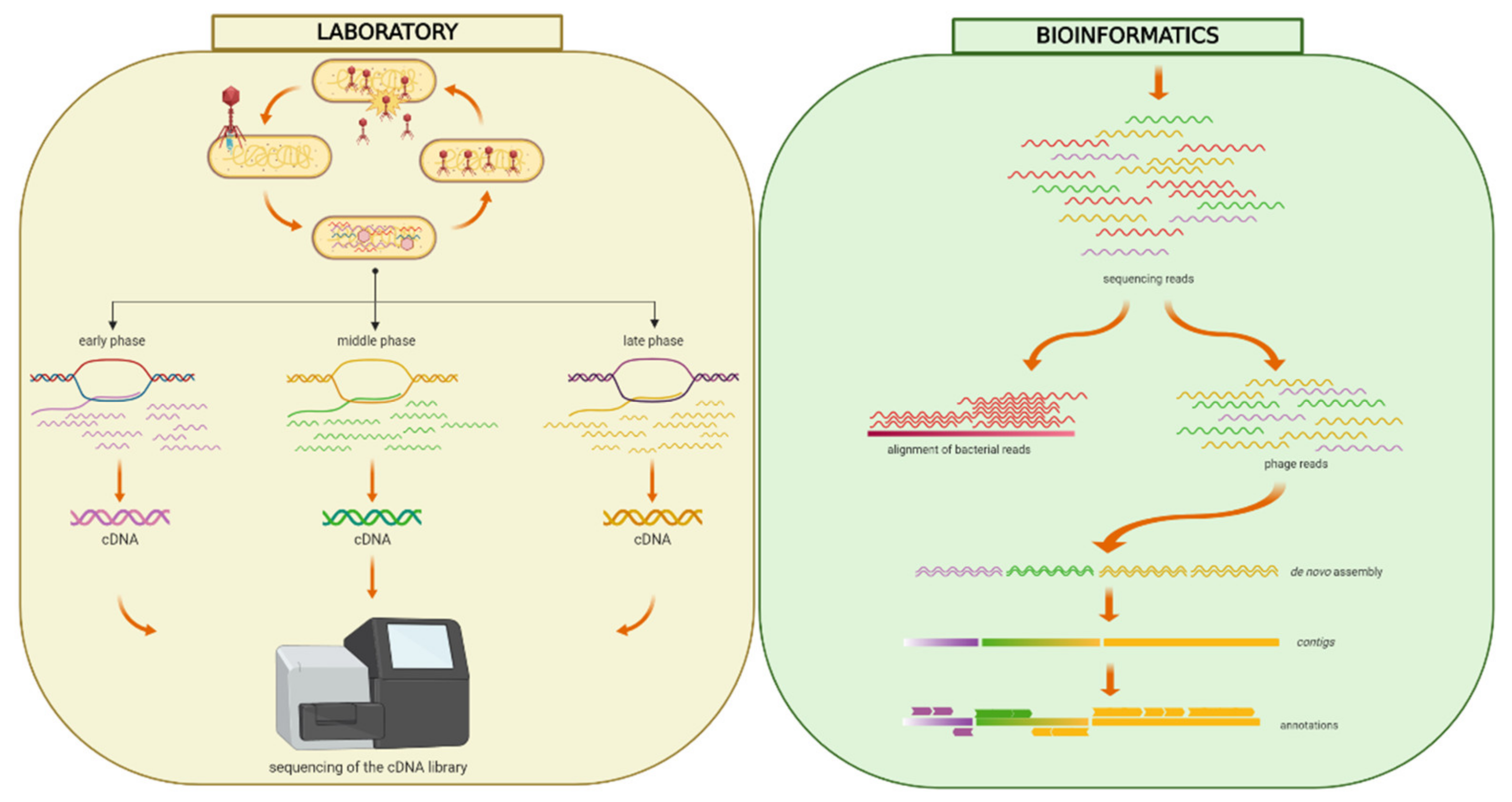
2.5. RNA Sequencing
2.6. Transcriptome Assembly
2.7. RNA-Sequencing Data Analysis
2.8. Proteome Analysis
2.9. DNA Isolation
2.10. Accession Numbers
3. Results
3.1. Characterization of YerA41
3.2. Genome Analysis
3.3. Proteomic Analysis of the Phage Structural Proteins
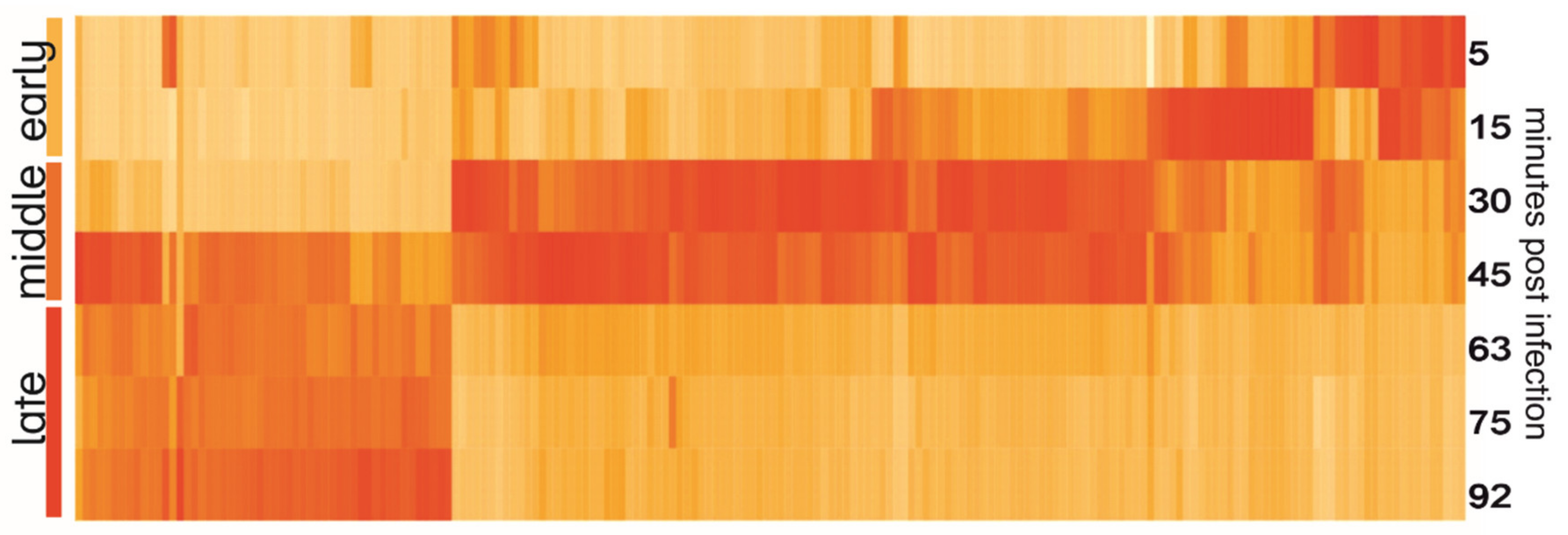
3.4. Temporal Expression of Phage Genes
3.5. Bacterial Response to Lytic Infection
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stevenson, R.M.W.; Airdrie, D.W. Isolation of Yersinia ruckeri bacteriophages. Appl. Environ. Microbiol. 1984, 47, 1201–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackermann, H.-W.; DuBow, M.S.; Gershman, M.; Karska-Wysocki, B.; Kasatiya, S.S.; Loessner, M.J.; Mamet-Bratley, M.D.; Regué, M. Taxonomic changes in tailed phages of enterobacteria. Arch. Virol. 1997, 142, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Gommers-Ampt, J.H.; Borst, P. Hypermodified bases in DNA. Faseb J. 1995, 9, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Weigele, P.; Raleigh, E.A. Biosynthesis and Function of Modified Bases in Bacteria and Their Viruses. Chem. Rev. 2016, 116, 12655–12687. [Google Scholar] [CrossRef] [PubMed]
- Warren, R.A. Modified bases in bacteriophage DNAs. Ann. Rev. Microbiol. 1980, 34, 137–158. [Google Scholar] [CrossRef]
- Song, H.K.; Sohn, S.H.; Suh, S.W. Crystal structure of deoxycytidylate hydroxymethylase from bacteriophage T4, a component of the deoxyribonucleoside triphosphate-synthesizing complex. Embo J. 1999, 18, 1104–1113. [Google Scholar] [CrossRef] [Green Version]
- Vrielink, A.; Ruger, W.; Driessen, H.P.; Freemont, P.S. Crystal structure of the DNA modifying enzyme beta-glucosyltransferase in the presence and absence of the substrate uridine diphosphoglucose. Embo J. 1994, 13, 3413–3422. [Google Scholar] [CrossRef]
- Kiljunen, S.; Hakala, K.; Pinta, E.; Huttunen, S.; Pluta, P.; Gador, A.; Lönnberg, H.; Skurnik, M. Yersiniophage fR1-37 is a tailed bacteriophage having a 270 kb DNA genome with thymidine replaced by deoxyuridine. Microbiology 2005, 151, 4093–4102. [Google Scholar] [CrossRef] [Green Version]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual, the Third Edition, 2nd ed.; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Ellis, E.L.; Delbruck, M. The Growth of Bacteriophage. J. Gen. Physiol. 1939, 22, 365–384. [Google Scholar] [CrossRef] [Green Version]
- Pajunen, M.; Kiljunen, S.; Skurnik, M. Bacteriophage fYeO3-12, specific for Yersinia enterocolitica serotype O:3, is related to coliphages T3 and T7. J. Bacteriol. 2000, 182, 5114–5120. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Rutherford, K.; Parkhill, J.; Crook, J.; Horsnell, T.; Rice, P.; Rajandream, M.A.; Barrell, B. Artemis: Sequence visualization and annotation. Bioinformatics 2000, 16, 944–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Varjosalo, M.; Keskitalo, S.; Van Drogen, A.; Nurkkala, H.; Vichalkovski, A.; Aebersold, R.; Gstaiger, M. The protein interaction landscape of the human CMGC kinase group. Cell Rep. 2013, 3, 1306–1320. [Google Scholar] [CrossRef] [Green Version]
- Skurnik, M.; Bengoechea, J.A. The biosynthesis and biological role of lipopolysaccharide O-antigens of pathogenic Yersiniae. Carbohydr. Res. 2003, 338, 2521–2529. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Blasdel, B.G.; Chevallereau, A.; Monot, M.; Lavigne, R.; Debarbieux, L. Comparative transcriptomics analyses reveal the conservation of an ancestral infectious strategy in two bacteriophage genera. ISME J. 2017, 11, 1988–1996. [Google Scholar] [CrossRef] [Green Version]
- Sacher, J.C.; Flint, A.; Butcher, J.; Blasdel, B.; Reynolds, H.M.; Lavigne, R.; Stintzi, A.; Szymanski, C.M. Transcriptomic Analysis of the Campylobacter jejuni Response to T4-Like Phage NCTC 12673 Infection. Viruses 2018, 10, 332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, D.C.; Schmelcher, M.; Rodriguez-Rubio, L.; Klumpp, J.; Pritchard, D.G.; Dong, S.L.; Donovan, D.M. Endolysins as Antimicrobials. Adv. Virus Res. Vol 83 Bacteriophages Pt B 2012, 83, 299–365. [Google Scholar] [CrossRef] [Green Version]
- Schmelcher, M.; Donovan, D.M.; Loessner, M.J. Bacteriophage endolysins as novel antimicrobials. Future Microbiol. 2012, 7, 1147–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Bacterial Species | YerA41 Sensitive Serotypes | YerA41 Sensitive Serotypes at 37 °C | YerA41 Resistant Serotypes or Strains |
|---|---|---|---|
| Yersinia enterocolitica | O:8 (5) | O:8 (5) | O:1(2),O:1,2,3(1), O:2(2), O:3(2), O:4(1), O:4,32(2), O:5(3), O:5,27(2), O:6(2), O:6,30(2), O:6,31(2), O:7,8(8), O:9(2), O:13,17(1), O:13,7(2), O:13a,13b(2), O:14(1), O:15(2) O:20(2), O:21(2), O:25(1), O:25,26(1), O:26,44(1), O:28,50(1), O:34 (1), O:35,36 (1), O:35,52(1), O:41(27)43(2), O:41(27)42 K1(1), O:50(2), O:41(27)K1(1),O:41,43(1), K1 non-typable (2), non-typable(4) |
| Yersinia pseudo-tuberculosis | - | - | O:1(1), O:1a(1), O:1b(1), O:2 (2), O:2a(1), O:2b(1), O:2c(1), O:3 (2), O:4a (1), O:4b (1), O:5a (1), O:5b(1), O:6 (1), O:7 (1), O:8 (1), O:9(1), |
| Yersinia frederikseni | - | O:16(1),O:35(1) | O:48 (1), non-typable (4) |
| Yersinia intermedia | O:16,21(1), O:52,54 (1) | O:16,21(1) | - |
| Yersinia kristenseni | O:12,25(1), O:16(2), non-typable (1) | O:12.25(1), O:16(2), non-typable (1) | non-typable(1) |
| Yersinia mollareti | - | - | O:59(20,36,7) (1) |
| Yersinia pestis | - | - | (2) |
| Yersinia bercoveri | - | - | O:58,16(1), non-typable(1) |
| Yersinia ruckeri | (2) | (2) | - |
| Providencia rettgeri | - | - | (1) |
| Salmonella typhimurium | - | - | (1) |
| Shigella flexneri | (1) | (1) | - |
| ID | Size [bp] | GC% |
|---|---|---|
| scaffold_1 | 42 987 | 34.5 |
| scaffold_2 | 27 377 | 31.6 |
| scaffold_3 | 26 591 | 31.7 |
| scaffold_4 | 15 134 | 32.0 |
| scaffold_5 | 11 502 | 32.8 |
| scaffold_6 | 8 240 | 29.6 |
| scaffold_7 | 5 287 | 28.9 |
| scaffold_8 | 3 672 | 29.6 |
| scaffold_9 | 2 506 | 29.6 |
| TOTAL/Average: | 143 296 | 32.3 |
| Temporal Expression | Gene ID | Scaffold | Putative Functions of Gene Products Based on Database Similarity |
| Early | g054 | 2 | DNA directed RNA polymerase, subunit* |
| g055 | 2 | DNA-directed RNA polymerase, subunit* | |
| g056 | 2 | DNA-directed RNA polymerase* | |
| g078 | 2 | Lytic transglycosylase | |
| g097 | 3 | DNA polymerase III, subunit | |
| g116 | 3 | RNA 2’-phosphotransferase | |
| g126 | 3 | Endonuclease-like protein | |
| g135 | 4 | DNA topoisomerase* | |
| g161 | 5 | Tail fiber protein | |
| g162 | 5 | DNA directed RNA polymerase, subunit / Putative DNA helicase | |
| g189 | 7 | Endonuclease-like protein | |
| g190 | 7 | DNA topoisomerase* | |
| g193 | 7 | Helicase* | |
| g195 | 8 | DNA polymerase | |
| g199 | 9 | DNA ligase* | |
| Middle | g060 | 2 | 5’-deoxynucleotidase |
| g061 | 2 | DNA polymerase* | |
| g064 | 2 | UDP-GlcNAc 2-epimerase | |
| g065 | 2 | Oxidoreductase | |
| g066 | 2 | SDR family oxidoreductase | |
| g067 | 2 | Polysaccharide deacetylase | |
| g068 | 2 | 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase (EC 2.7.7.60) | |
| g070 | 2 | Glycerophosphodiester phosphodiesterase | |
| g109 | 3 | Ribonucleotide reductase | |
| g110 | 3 | Ribonucleoside-diphosphate reductase subunit alpha* | |
| g112 | 3 | Ribosomal protein modification protein* | |
| g114 | 3 | dCTP deaminase | |
| g127 | 3 | Thymidylate synthetase | |
| g133 | 4 | Transglycosylase | |
| g136 | 4 | DNA topoisomerase* | |
| g137 | 4 | DNA polymerase III, subunit* | |
| g140 | 4 | Structural protein | |
| g141 | 4 | dUTP diphosphatase* | |
| g143 | 4 | ATP-dependent DNA helicase* | |
| g145 | 4 | Replicative helicase | |
| g149 | 4 | Exodeoxyribonuclease* | |
| g167 | 5 | Endonuclease* | |
| g174 | 5 | Phage baseplate assembly protein | |
| g179 | 6 | Exonuclease | |
| g180 | 6 | Exonuclease* | |
| Late | g007 | 1 | DNA packaging terminase* |
| g012 | 1 | Prohead core protein protease* | |
| g014 | 1 | Capsid protein* | |
| g016 | 1 | Sugar binding protein* | |
| g019 | 1 | RNA polymerase sigma factor* | |
| g024 | 1 | Phage tail sheath protein* | |
| g025 | 1 | Tail protein | |
| g028 | 1 | Tail family protein* | |
| g031 | 1 | Tail protein | |
| g032 | 1 | Tail protein | |
| g034 | 1 | Tail-associated lysozyme | |
| g035 | 1 | Tail-associated lysozyme | |
| g037 | 1 | Baseplate wedge protein* | |
| g040 | 1 | Capsid protein | |
| g041 | 1 | Virion structural protein | |
| g042 | 1 | Baseplate wedge protein* | |
| g043 | 1 | Tail fiber protein* | |
| g044 | 1 | Phage tail fiber assembly protein* | |
| g045 | 1 | Tail fiber protein* | |
| g049 | 1 | Endolysin* |
| Gene ID | Function | 15 min | 30 min | ||
| logFC | FDR | logFC | FDR | ||
| CSF007_17485 | Dihydrolipoamide dehydrogenase | 7.07 | 1.97 × 10−146 | 1.28 | 2.16 × 10−07 |
| CSF007_17480 | Peroxiredoxin family protein/glutaredoxin | 6.95 | 4.94 × 10−128 | 1.12 | 7.36 × 10−05 |
| CSF007_6300 | Non-specific DNA-binding protein Dps / Iron-binding ferritin-like antioxidant protein / Ferroxidase | 4.39 | 4.13 × 10−66 | 1.90 | 5.48 × 10−11 |
| CSF007_12285 | Catalase | 4.10 | 3.26 × 10−59 | 1.54 | 9.40 × 10−09 |
| CSF007_9590 | Pyruvate kinase | 1.95 | 7.27 × 10−15 | 2.11 | 9.00 × 10−13 |
| CSF007_11760 | Putrescine importer | 1.43 | 1.44 × 10−07 | 1.71 | 4.96 × 10−05 |
| CSF007_5840 | Cytochrome d ubiquinol oxidase subunit I | 1.21 | 1.89 × 10−06 | 1.49 | 0.00011 |
| CSF007_5845 | Cytochrome d ubiquinol oxidase subunit II | 1.14 | 1.98 × 10−06 | 1.48 | 2.78 × 10−05 |
| CSF007_13405 | Inosine-5-monophosphate dehydrogenase | 0.91 | 0.00034 | 1.19 | 0.00017 |
| CSF007_12920 | hypothetical protein | 0.88 | 0.00014 | 1.11 | 0.00015 |
| CSF007_5505 | hypothetical protein | −0.78 | 1.77 × 10−05 | −1.18 | 1.44 × 10−05 |
| CSF007_14715 | Glycine cleavage system H protein | −0.81 | 0.00044 | −1.88 | 3.38 × 10−07 |
| CSF007_9025 | Alkyl sulfatase | −0.93 | 2.94 × 10−06 | −1.32 | 1.03 × 10−05 |
| CSF007_13885 | D-ribulokinase | −0.94 | 6.94 × 10−07 | −2.04 | 9.33 × 10−14 |
| CSF007_13880 | Phosphosugar isomerase/binding protein | −1.01 | 2.19 × 10−06 | −1.98 | 6.52 × 10−09 |
| CSF007_1760 | Aspartate ammonia-lyase | −1.02 | 6.88 × 10−05 | −1.98 | 3.86 × 10−12 |
| CSF007_0675 | Oligopeptidase A | −1.03 | 0.00043 | −1.29 | 0.00043 |
| CSF007_9680 | Hemin transport protein HmuS | −1.06 | 1.77 × 10−05 | −1.38 | 0.00097 |
| CSF007_17975 | Glutamine synthetase type I | −1.09 | 0.00014 | 1.92 | 4.07 × 10−05 |
| CSF007_14720 | Aminomethyltransferase (glycine cleavage system T protein) | −1.11 | 3.59 × 10−09 | −1.69 | 7.18 × 10−10 |
| CSF007_11035 | Transcriptional repressor of PutA and PutP / Proline dehydrogenase (Proline oxidase) / Delta-1-pyrroline-5-carboxylate dehydrogenase | −1.12 | 9.24 × 10−06 | −2.14 | 5.30 × 10−14 |
| CSF007_13080 | NADP-dependent malic enzyme | −1.12 | 9.98 × 10−08 | −1.68 | 2.82 × 10−09 |
| CSF007_6400 | Galactose/methyl galactoside ABC transport system ATP-binding protein MglA | −1.13 | 1.92 × 10−07 | −1.72 | 6.99 × 10−09 |
| CSF007_0690 | Universal stress protein A | −1.20 | 1.55 × 10−07 | −1.56 | 5.98 × 10−06 |
| CSF007_0605 | Aerobic C4-dicarboxylate transporter for fumarate/L-malate/D-malate/succunate | −1.23 | 1.07 × 10−09 | −1.09 | 0.00046 |
| CSF007_1210 | Cyclic AMP receptor protein | −1.32 | 5.98 × 10−08 | −1.42 | 1.30 × 10−05 |
| CSF007_0245 | 16 kDa heat shock protein A | −1.37 | 0.00033 | −1.80 | 6.55 × 10−06 |
| CSF007_5820 | Dihydrolipoamide succinyltransferase component (E2) of 2-oxoglutarate dehydrogenase complex | −1.41 | 7.97 × 10−08 | −2.87 | 1.10 × 10−12 |
| CSF007_18075 | Ribose ABC transport system periplasmic ribose-binding protein RbsB | −1.41 | 1.96 × 10−11 | −1.57 | 8.42 × 10−07 |
| CSF007_16000 | hypothetical protein | −1.42 | 2.65 × 10−06 | −1.99 | 6.12 × 10−06 |
| CSF007_11865 | Mannonate dehydratase | −1.46 | 8.78 × 10−10 | −2.13 | 6.04 × 10−12 |
| CSF007_13895 | Ribose ABC transport system permease protein RbsC | −1.48 | 1.29 × 10−12 | −2.07 | 7.16 × 10−12 |
| CSF007_0935 | Transcriptional activator of maltose regulon MalT | −1.49 | 7.07 × 10−14 | −1.48 | 8.42 × 10−07 |
| CSF007_16315 | Maltose operon periplasmic protein MalM | −1.51 | 6.85 × 10-06 | −2.03 | 0.00043 |
| CSF007_18085 | Ribose ABC transport system ATP-binding protein RbsA | −1.51 | 7.72 × 10−10 | −1.85 | 2.56 × 10−06 |
| CSF007_9675 | TonB-dependent hemin ferrichrome receptor | −1.55 | 9.46 × 10−16 | −1.13 | 0.00018 |
| CSF007_5825 | Succinyl-CoA ligase [ADP-forming] beta chain | −1.60 | 7.19 × 10−08 | −2.86 | 5.80 × 10−10 |
| CSF007_5830 | Succinyl-CoA ligase [ADP-forming] alpha chain | −1.63 | 1.56 × 10−09 | −3.01 | 4.78 × 10−14 |
| CSF007_18090 | Ribose ABC transport system high affinity permease RbsD | −1.66 | 1.10 × 10−11 | −2.16 | 4.82 × 10−08 |
| CSF007_16340 | Maltose/maltodextrin ABC transporter substrate binding periplasmic protein MalE | −1.68 | 5.91 × 10−08 | −2.03 | 1.45 × 10−06 |
| CSF007_3355 | Aconitate hydratase 2 | −1.69 | 3.40 × 10−12 | −1.66 | 1.56 × 10−06 |
| CSF007_5815 | 2-oxoglutarate dehydrogenase E1 component | −1.80 | 2.19 × 10−13 | −2.96 | 3.74 × 10−18 |
| CSF007_9550 | Putative transport protein | −1.80 | 4.34 × 10−12 | −1.51 | 2.19 × 10−06 |
| CSF007_16325 | Maltose/maltodextrin transport ATP-binding protein MalK | −1.81 | 6.80 × 10−06 | −2.93 | 3.65 × 10−08 |
| CSF007_9650 | Phosphoenolpyruvate synthase | −1.82 | 6.56 × 10−15 | −2.54 | 1.16 × 10−09 |
| CSF007_11875 | D-mannonate oxidoreductase | −1.87 | 3.62 × 10−13 | −2.57 | 4.08 × 10−14 |
| CSF007_12965 | Sialic acid transporter (permease) NanT | −1.92 | 2.90 × 10−12 | −2.72 | 4.08 × 10−14 |
| CSF007_13900 | Ribose/xylose/arabinose/galactoside ABC-type transport system ATP-binding protein | −2.08 | 2.79 × 10−28 | −2.06 | 7.97 × 10−05 |
| CSF007_6395 | Galactose/methyl galactoside ABC transport system galactose-binding periplasmic protein MglB | −2.08 | 2.72 × 10−14 | −2.79 | 1.40 × 10−17 |
| CSF007_11455 | hypothetical protein | −2.12 | 8.23 × 10−28 | −1.57 | 4.48 × 10−05 |
| CSF007_0865 | Gluconokinase | −2.12 | 9.37 × 10−17 | −1.80 | 7.28 × 10−06 |
| CSF007_12460 | membrane protein | −2.21 | 3.34 × 10−18 | −2.35 | 7.75 × 10−09 |
| CSF007_15720 | Hexuronate transporter | −2.38 | 7.15 × 10−28 | −2.40 | 1.25 × 10−10 |
| CSF007_17650 | Glycerol uptake facilitator protein | −2.39 | 1.55 × 10−28 | −2.08 | 5.48 × 10−11 |
| CSF007_16005 | Trehalose-6-phosphate hydrolase | −2.46 | 3.37 × 10−14 | −3.86 | 3.31 × 10−24 |
| CSF007_5810 | Succinate dehydrogenase iron-sulfur protein | −2.47 | 1.33 × 10−16 | −3.06 | 6.67 × 10−15 |
| CSF007_13910 | Ribose/xylose/arabinose/galactoside ABC-type transport system periplasmic sugar binding protein | −2.48 | 1.26 × 10−19 | −3.70 | 1.55 × 10−33 |
| CSF007_5800 | Succinate dehydrogenase hydrophobic membrane anchor protein | −2.49 | 4.39 × 10−19 | −2.64 | 1.15 × 10−10 |
| CSF007_17655 | Glycerol kinase | −2.56 | 1.20 × 10−19 | −3.31 | 2.81 × 10−15 |
| CSF007_5795 | Succinate dehydrogenase cytochrome b-556 subunit | −2.64 | 4.76 × 10−26 | −2.04 | 9.68 × 10−13 |
| CSF007_15715 | hypothetical protein | −2.64 | 2.61 × 10−10 | −3.74 | 1.21 × 10−17 |
| CSF007_5805 | Succinate dehydrogenase flavoprotein subunit | −2.65 | 2.01 × 10−22 | −3.18 | 3.94 × 10−18 |
| CSF007_12450 | Ascorbate-specific PTS system, EIIA component | −3.07 | 6.74 × 10−23 | −3.17 | 6.76 × 10−10 |
| CSF007_12455 | Putative sugar phosphotransferase component II B | −3.21 | 1.62 × 10−22 | −3.52 | 1.76 × 10−09 |
| CSF007_5790 | Citrate synthase | −3.23 | 8.51 × 10−22 | −3.12 | 8.36 × 10−10 |
| CSF007_13905 | hypothetical protein | −3.28 | 2.14 × 10−13 | −5.24 | 6.87 × 10−10 |
| CSF007_16010 | PTS system, trehalose-specific IIB component-PTS system | −3.39 | 3.53 × 10−41 | −3.85 | 1.29 × 10−27 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leskinen, K.; Pajunen, M.I.; Vilanova, M.V.G.-R.; Kiljunen, S.; Nelson, A.; Smith, D.; Skurnik, M. YerA41, a Yersinia ruckeri Bacteriophage: Determination of a Non-Sequencable DNA Bacteriophage Genome via RNA-Sequencing. Viruses 2020, 12, 620. https://0-doi-org.brum.beds.ac.uk/10.3390/v12060620
Leskinen K, Pajunen MI, Vilanova MVG-R, Kiljunen S, Nelson A, Smith D, Skurnik M. YerA41, a Yersinia ruckeri Bacteriophage: Determination of a Non-Sequencable DNA Bacteriophage Genome via RNA-Sequencing. Viruses. 2020; 12(6):620. https://0-doi-org.brum.beds.ac.uk/10.3390/v12060620
Chicago/Turabian StyleLeskinen, Katarzyna, Maria I. Pajunen, Miguel Vincente Gomez-Raya Vilanova, Saija Kiljunen, Andrew Nelson, Darren Smith, and Mikael Skurnik. 2020. "YerA41, a Yersinia ruckeri Bacteriophage: Determination of a Non-Sequencable DNA Bacteriophage Genome via RNA-Sequencing" Viruses 12, no. 6: 620. https://0-doi-org.brum.beds.ac.uk/10.3390/v12060620