Targeting HIV-1 RNase H: N’-(2-Hydroxy-benzylidene)-3,4,5-Trihydroxybenzoylhydrazone as Selective Inhibitor Active against NNRTIs-Resistant Variants

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and0 Methods
2.1. Biology
2.1.1. Cells Reagents and Viruses
2.1.2. Antiviral and Cytotoxicity Assay
2.1.3. Time of Addition Assay
2.2. Molecular Modeling
2.2.1. Ligand Preparation
2.2.2. Protein Preparation
2.2.3. Docking and Post-Docking
2.3. Pharmacophore Model Generation
2.4. Molecular Biology
2.4.1. Expression and Purification of Recombinant HIV-1 RTs Group M Subtype B wt and Mutants
2.4.2. Site-Directed Mutagenesis
2.4.3. HIV-1 DNA Polymerase-Independent RNase H Activity Determination
2.4.4. HIV-1 RNA-Dependent DNA Polymerase (RDDP) Activity Determination
2.5. Genetic Analysis
3. Results
3.1. Antiviral Activity of N-acylhydrazone Analogs against wt and NNRTI-Resistant HIV-1 Strains
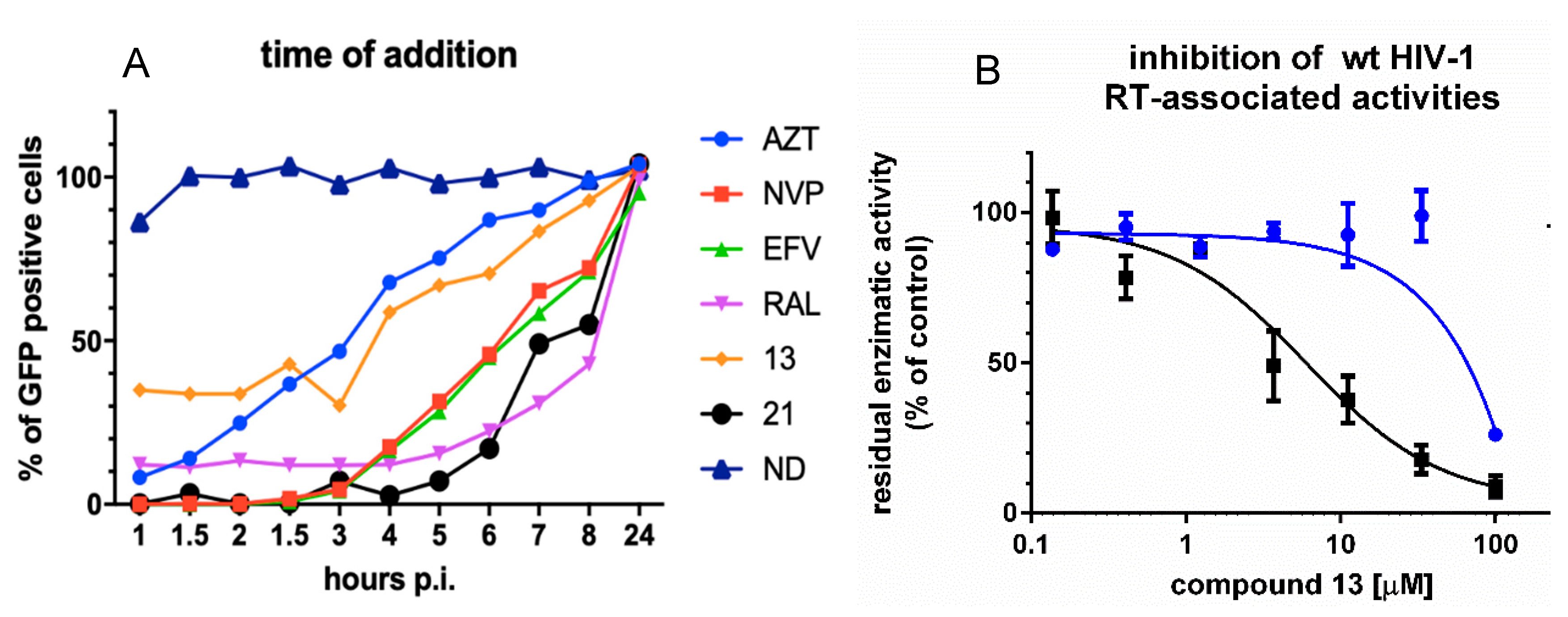
3.2. Analysis of Compound 13 Target of Inhibition by Time-of-Addition and Comparative Inhibition of HIV-1 RT Associated Activities
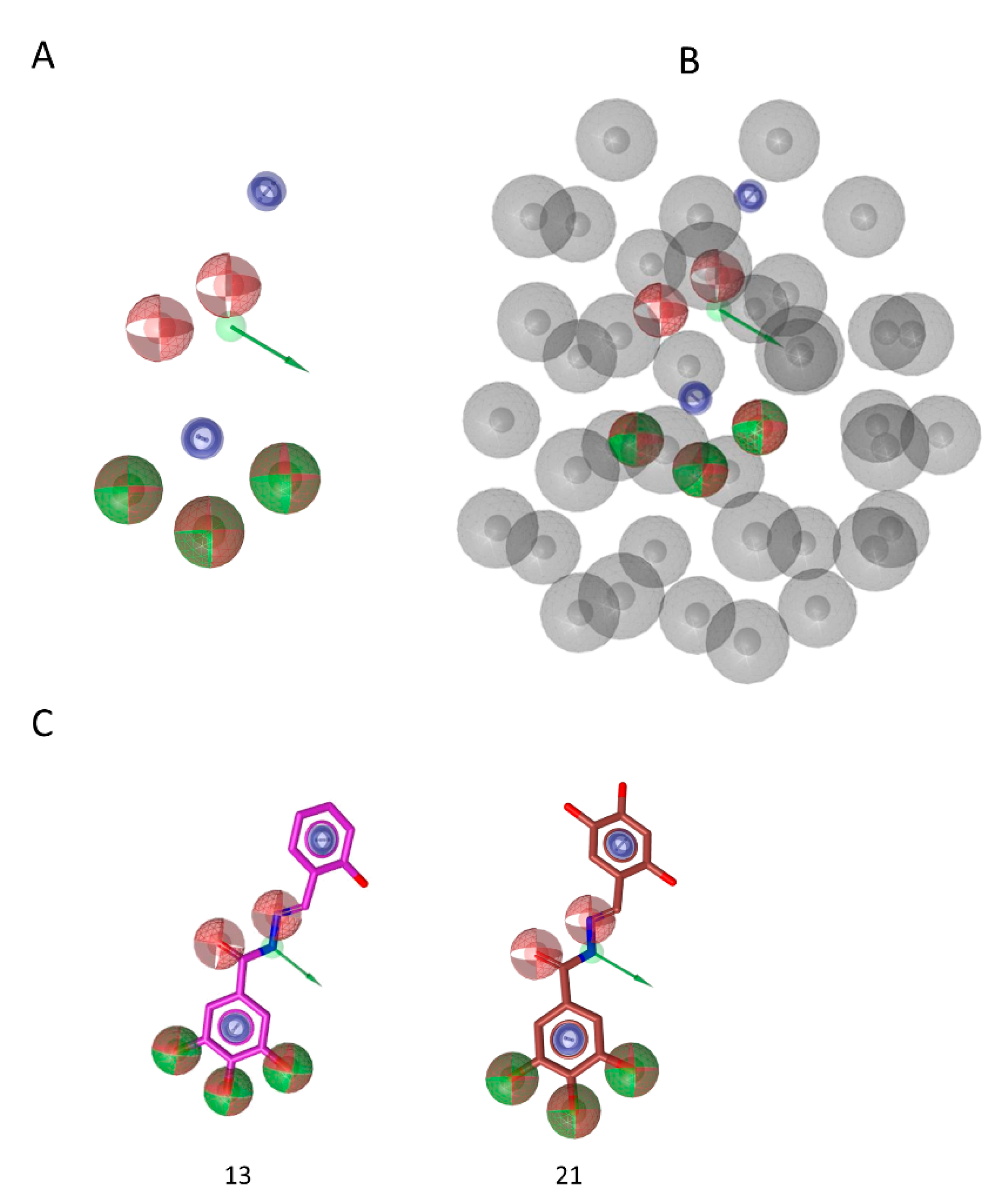
3.3. Analysis of the Binding of Compound 13 within the HIV-1 RT RNase H Domain
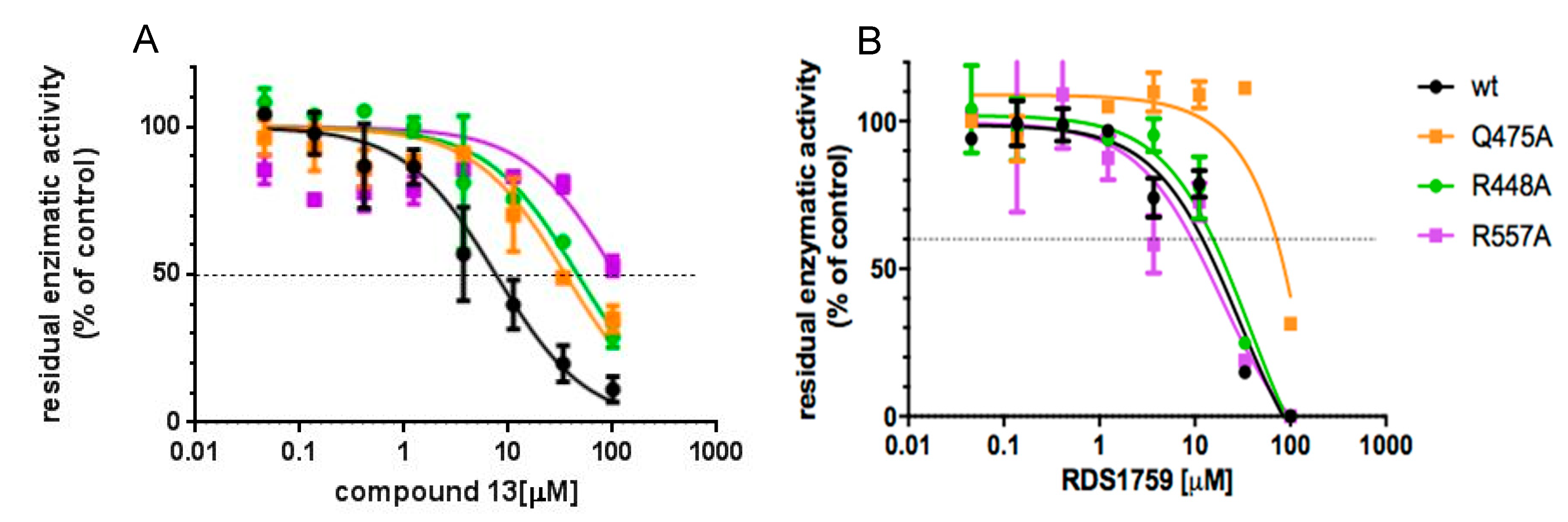
3.4. Site-Directed Mutagenesis Studies Detailing the Binding Mode of Compound 13
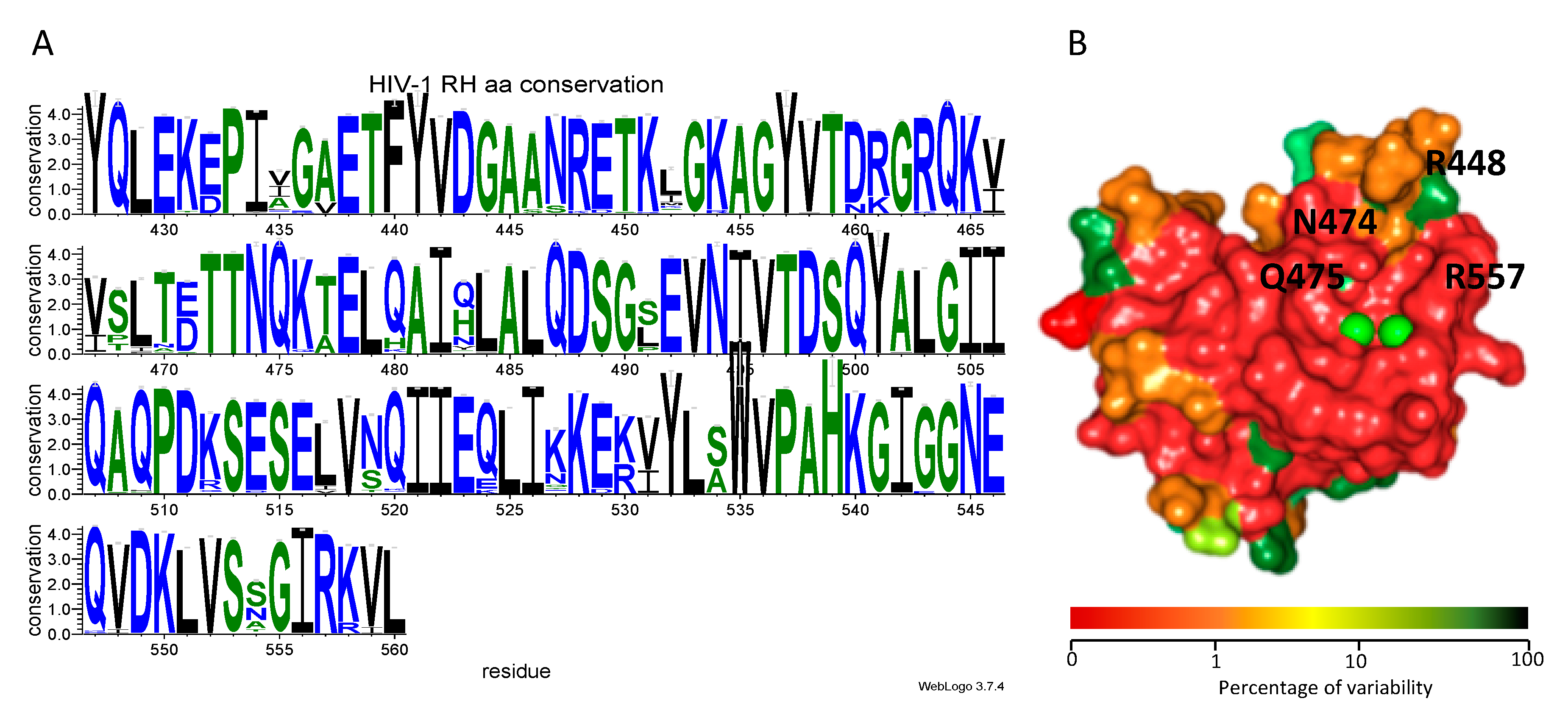
3.5. Genetic Analysis
3.6. Pharmacophore Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- UNAIDS 2018 Global HIV Statistics. Available online: https://www.unaids.org/sites/default/files/media_asset/UNAIDS_FactSheet_en.pdf (accessed on 17 February 2020).
- Cahn, P.; Madero, J.S.; Arribas, J.R.; Antinori, A.; Ortiz, R.; Clarke, A.E.; Hung, C.C.; Rockstroh, J.K.; Girard, P.M.; Sievers, J.; et al. Durable Efficacy of Dolutegravir Plus Lamivudine in Antiretroviral Treatment-Naive Adults With HIV-1 Infection: 96-Week Results From the GEMINI-1 and GEMINI-2 Randomized Clinical Trials. J. Acquir. Immune Defic. Syndr. 2020, 83, 310–318. [Google Scholar] [CrossRef] [Green Version]
- Fauci, A.S.; Folkers, G.K. Toward an AIDS-free generation. JAMA J. Am. Med. Assoc. 2012, 308, 343–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeks, S.G.; Lewin, S.R.; Havlir, D.V. The end of AIDS: HIV infection as a chronic disease. Lancet 2013, 382, 1525–1533. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J. Combo of two HIV vaccines fails its big test. Science 2020, 367, 611–612. [Google Scholar] [CrossRef] [PubMed]
- Henderson, L.J.; Reoma, L.B.; Kovacs, J.A.; Nath, A. Advances toward Curing HIV-1 Infection in Tissue Reservoirs. J. Virol. 2019, 94, 1–21. [Google Scholar] [CrossRef]
- Stella-Ascariz, N.; Arribas, J.R.; Paredes, R.; Li, J.Z. The Role of HIV-1 Drug-Resistant Minority Variants in Treatment Failure. J. Infect. Dis. 2017, 216, S847–S850. [Google Scholar] [CrossRef]
- Gupta, R.K.; Gregson, J.; Parkin, N.; Haile-Selassie, H.; Tanuri, A.; Andrade Forero, L.; Kaleebu, P.; Watera, C.; Aghokeng, A.; Mutenda, N.; et al. HIV-1 drug resistance before initiation or re-initiation of first-line antiretroviral therapy in low-income and middle-income countries: A systematic review and meta-regression analysis. Lancet Infect. Dis. 2017, 18, 346–355. [Google Scholar] [CrossRef] [Green Version]
- Gregson, J.; Tang, M.; Ndembi, N.; Hamers, R.L.; Marconi, V.C.; Brooks, K.; Theys, K.; Arruda, M.; Garcia, F.; Monge, S.; et al. Global epidemiology of drug resistance after failure of WHO recommended first-line regimens for adult HIV-1 infection: A multicentre retrospective cohort study. Lancet Infect. Dis. 2016, 16, 565–575. [Google Scholar] [CrossRef] [Green Version]
- Schneider, A.; Corona, A.; Spöring, I.; Jordan, M.; Buchholz, B.; Maccioni, E.; Di Santo, R.; Bodem, J.; Tramontano, E.; Wöhrl, B.M. Biochemical characterization of a multi-drug resistant HIV-1 subtype AG reverse transcriptase: Antagonism of AZT discrimination and excision pathways and sensitivity to RNase H inhibitors. Nucleic Acids Res. 2016, 44, 2310–2322. [Google Scholar] [CrossRef] [Green Version]
- Schatz, O.; Cromme, F.V.; Naas, T.; Lindemann, D. Inactivation of the RNase H domain of HIV-1 reverse transcriptase blocks viral infectivity. In Gene Regulation and AIDS; Gulf Publishing Company: Houston, TX, USA, 1990; pp. 293–404. [Google Scholar]
- Davies, J.F.; Hostomska, Z.; Hostomsky, Z.; Jordan, S.R.; Matthews, D.A. Crystal structure of the ribonuclease H domain of HIV-1 reverse transcriptase. Science 1991, 252, 88–95. [Google Scholar] [CrossRef]
- Krupovic, M.; Blomberg, J.; Coffin, J.M.; Dasgupta, I.; Fan, H.; Geering, A.D.; Gifford, R.; Harrach, B.; Hull, R.; Johnson, W.; et al. Ortervirales: New Virus Order Unifying Five Families of Reverse-Transcribing Viruses. J. Virol. 2018, 92, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tadokoro, T.; Kanaya, S. Ribonuclease H: Molecular diversities, substrate binding domains, and catalytic mechanism of the prokaryotic enzymes. FEBS J. 2009, 276, 1482–1493. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.; Cherepanov, P. Retroviral integrase structure and dna recombination mechanism. Microbiol. Spectr. 2014, 2, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyjek, M.; Figiel, M.; Nowotny, M. RNases H: Structure and mechanism. DNA Repair. 2019, 84, 102672. [Google Scholar] [CrossRef] [PubMed]
- Corona, A.; Schneider, A.; Schweimer, K.; Rösch, P.; Wöhrl, B.M.B.M.; Tramontano, E. Inhibition of foamy virus reverse transcriptase by human immunodeficiency virus type 1 ribonuclease H inhibitors. Antimicrob. Agents Chemother. 2014, 58, 4086–4093. [Google Scholar] [CrossRef] [Green Version]
- Corona, A.; Masaoka, T.; Tocco, G.; Tramontano, E.; Le Grice, S.F. Active site and allosteric inhibitors of the ribonuclease H activity of HIV reverse transcriptase. Futur. Med. Chem. 2013, 5, 2127–2139. [Google Scholar] [CrossRef]
- Wang, X.; Gao, P.; Menendez-Arias, L.; Liu, X.; Zhan, P.; Menéndez-Arias, L.; Liu, X.; Zhan, P. Update on Recent Developments in Small Molecular HIV-1 RNase H Inhibitors (2013–2016): Opportunities and Challenges. Curr. Med. Chem. 2018, 25, 1682–1702. [Google Scholar] [CrossRef]
- Tramontano, E.; Corona, A.; Menéndez-Arias, L. Ribonuclease H, an unexploited target for antiviral intervention against HIV and hepatitis B virus. Antivir. Res. 2019, 171, 104613. [Google Scholar] [CrossRef]
- Tramontano, E. HIV-1 RNase H: Recent progress in an exciting, yet little explored, drug target. Mini Rev. Med. Chem. 2006, 6, 727–737. [Google Scholar] [CrossRef]
- Wang, L.; Tang, J.; Huber, A.D.; Casey, M.C.; Kirby, K.A.; Wilson, D.J.; Kankanala, J.; Xie, J.; Parniak, M.A.; Sarafianos, S.G.; et al. 6-Arylthio-3-hydroxypyrimidine-2,4-diones potently inhibited HIV reverse transcriptase-associated RNase H with antiviral activity. Eur. J. Med. Chem. 2018, 156, 652–665. [Google Scholar] [CrossRef]
- Xi, Z.; Wang, Z.; Sarafianos, S.G.; Myshakina, N.S.; Ishima, R. Determinants of active-site inhibitor interaction with HIV-1 RNase H. ACS Infect. Dis. 2019, 5, 1963–1974. [Google Scholar] [CrossRef] [PubMed]
- Corona, A.; Esposito, F.; Tramontano, E. Can the ever-promising target HIV reverse transcriptase-associated RNase H become a success story for drug development? Future Virol. 2014, 9, 445–448. [Google Scholar] [CrossRef]
- Wang, L.; Sarafianos, S.G.; Wang, Z. Cutting into the Substrate Dominance: Pharmacophore and Structure-Based Approaches toward Inhibiting Human Immunodeficiency Virus Reverse Transcriptase-Associated Ribonuclease H. Acc. Chem. Res. 2020, 53, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Corona, A.; Di Leva, F.S.; Thierry, S.; Pescatori, L.; Cuzzucoli Crucitti, G.; Subra, F.; Delelis, O.; Esposito, F.; Rigogliuso, G.; Costi, R.; et al. Identification of Highly Conserved Residues Involved in Inhibition of HIV-1 RNase H Function by Diketo Acid Derivatives. Antimicrob. Agents Chemother. 2014, 58, 6101–6110. [Google Scholar] [CrossRef] [Green Version]
- Poongavanam, V.; Corona, A.; Steinmann, C.; Scipione, L.; Grandi, N.; Pandolfi, F.; Di Santo, R.; Costi, R.; Esposito, F.; Tramontano, E.; et al. Structure-guided approach identifies a novel class of HIV-1 ribonuclease H inhibitors: Binding mode insights through magnesium complexation and site-directed mutagenesis studies. Medchemcomm 2018, 9, 562–575. [Google Scholar] [CrossRef] [Green Version]
- Messore, A.; Corona, A.; Madia, V.N.; Saccoliti, F.; Tudino, V.; De Leo, A.; Scipione, L.; De Vita, D.; Amendola, G.; Di Maro, S.; et al. Pyrrolyl Pyrazoles as Non-Diketo Acid Inhibitors of the HIV-1 Ribonuclease H Function of Reverse Transcriptase. ACS Med. Chem. Lett. 2020, 11, 798–805. [Google Scholar] [CrossRef]
- Corona, A.; Di Leva, F.S.; Rigogliuso, G.; Pescatori, L.; Noemi, V.; Subra, F.; Delelis, O.; Esposito, F.; Cadeddu, M.; Costi, R.; et al. New insights into the interaction between pyrrolyl diketoacids and HIV-1 integrase active site and comparison with RNase H. Antivir. Res. 2016, 134, 236–243. [Google Scholar] [CrossRef]
- Carcelli, M.; Rogolino, D.; Gatti, A.; Pala, N.; Corona, A.; Caredda, A.; Tramontano, E.; Pannecouque, C.; Naesens, L.; Esposito, F. Chelation motifs affecting metal-dependent viral enzymes: N-acylhydrazone ligands as dual target inhibitors of HIV-1 integrase and reverse transcriptase ribonuclease Hdomain. Front. Microbiol. 2017, 8, 440. [Google Scholar] [CrossRef]
- Rausch, J.W.; Lener, D.; Miller, J.T.; Julias, J.G.; Hughes, S.H.; Le Grice, S.F.J. Altering the RNase H primer grip of human immunodeficiency virus reverse transcriptase modifies cleavage specificity. Biochemistry 2002, 41, 4856–4865. [Google Scholar] [CrossRef]
- Clouser, C.L.; Patterson, S.E.; Mansky, L.M. Exploiting Drug Repositioning for Discovery of a Novel HIV Combination Therapy. J. Virol. 2010, 84, 9301–9309. [Google Scholar] [CrossRef] [Green Version]
- Pannecouque, C.; Daelemans, D.; De Clercq, E. Tetrazolium-based colorimetric assay for the detection of HIV replication inhibitors: Revisited 20 years later. Nat. Protoc. 2008, 3, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Badia, R.; Grau, J.; Riveira-Muñoz, E.; Ballana, E.; Giannini, G.; Esté, J.A. The thioacetate-ω(γ-lactam carboxamide) HDAC inhibitor ST7612AA1 as HIV-1 latency reactivation agent. Antivir. Res. 2015, 123, 62–69. [Google Scholar] [CrossRef]
- Reed, L.J.; Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Daelemans, D.; Pauwels, R.; De Clercq, E.; Pannecouque, C. A time-of-drug addition approach to target identification of antiviral compounds. Nat. Protoc. 2011, 6, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Mohamadi, F.; Richards, N.G.J.; Guida, W.C.; Liskamp, R.; Lipton, M.; Caufield, C.; Chang, G.; Hendrickson, T.; Still, W.C.; Giuda, W.C.; et al. Macromodel—An integrated software system for modeling organic and bioorganic molecules using molecular mechanics. J. Comput. Chem. 1990, 11, 440–467. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. II. MMFF94 van der Waals and electrostatic parameters for intermolecular interactions. J. Comput. Chem. 1996, 17, 520–552. [Google Scholar] [CrossRef]
- Still, W.C.C.; Tempczyk, A.; Hawley, R.C.C.; Hendrickson, T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 1990, 112, 6127–6129. [Google Scholar] [CrossRef]
- Su, H.-P.; Yan, Y.; Prasad, G.S.; Smith, R.F.; Daniels, C.L.; Abeywickrema, P.D.; Reid, J.C.; Loughran, H.M.; Kornienko, M.; Sharma, S.; et al. Structural basis for the inhibition of RNase H activity of HIV-1 reverse transcriptase by RNase H active site-directed inhibitors. J. Virol. 2010, 84, 7625–7633. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger LLC. Maestro GUI; Schrödinger LLC: New York, NY, USA, 2013. [Google Scholar]
- Jorgensen, W.L.; Jorgensen, W.L.; Maxwell, D.S.; Maxwell, D.S.; Tirado-Rives, J.; Tirado-Rives, J. Development and Testing of the OLPS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Schrödinger LLC. QMPolarized Protocol; Schrödinger LLC: New York, NY, USA.
- Schrödinger LLC. The PyMOL Molecular Graphics System, Version 1.7.2.1 2014; Schrödinger LLC: New York, NY, USA.
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Wolber, G.; Dornhofer, A.A.; Langer, T. Efficient Overlay of Small Organic Molecules Using 3D Pharmacophores. J. Comput. Aided. Mol. Des. 2006, 20, 773–788. [Google Scholar] [CrossRef]
- Corona, A.; Meleddu, R.; Esposito, F.; Distinto, S.; Bianco, G.; Masaoka, T.; Maccioni, E.; Menéndez-Arias, L.; Alcaro, S.; Le Grice, S.F.J.; et al. Ribonuclease H/DNA Polymerase HIV-1 Reverse Transcriptase Dual Inhibitor: Mechanistic Studies on the Allosteric Mode of Action of Isatin-Based Compound RMNC6. PLoS ONE 2016, 11, e0147225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pala, N.; Esposito, F.; Rogolino, D.; Carcelli, M.; Sanna, V.; Palomba, M.; Naesens, L.; Corona, A.; Grandi, N.; Tramontano, E.; et al. Inhibitory effect of 2,3,5,6-tetrafluoro-4-[4-(Aryl)-1H-1,2,3-triazol-1-yl]benzenesulfonamide derivatives on HIV reverse transcriptase associated RNase H activities. Int. J. Mol. Sci. 2016, 17, 1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, G.; Rocca, R.; Corona, A.; Grandi, N.; Moraca, F.; Romeo, I.; Talarico, C.; Giovanna, M.; Alessandra, F.; Ortuso, F.; et al. Novel natural non-nucleoside inhibitors of HIV-1 reverse transcriptase identified by shape- and structure-based virtual screening techniques. Eur. J. Med. Chem. 2019, 161, 1–10. [Google Scholar] [CrossRef]
- Rhee, S.-Y.; Kantor, R.; Katzenstein, D.A.; Camacho, R.; Morris, L.; Sirivichayakul, S.; Jorgensen, L.; Brigido, L.F.; Schapiro, J.M.; Shafer, R.W.; et al. HIV-1 pol mutation frequency by subtype and treatment experience: Extension of the HIVseq program to seven non-B subtypes. AIDS 2006, 20, 643–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, S.-Y.; Gonzales, M.J.; Kantor, R.; Betts, B.J.; Ravela, J.; Shafer, R.W. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 2003, 31, 298–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Distinto, S.; Maccioni, E.; Meleddu, R.; Corona, A.; Alcaro, S.; Tramontano, E. Molecular Aspects of the RT/drug Interactions. Perspective of Dual Inhibitors. Curr. Pharm. Des. 2013, 19, 1850–1859. [Google Scholar] [CrossRef]
- Himmel, D.M.; Maegley, K.A.; Pauly, T.A.; Bauman, J.D.; Das, K.; Dharia, C.; Clark, A.D.; Ryan, K.; Hickey, M.J.; Love, R.A.; et al. Structure of HIV-1 reverse transcriptase with the inhibitor beta-Thujaplicinol bound at the RNase H active site. Structure 2009, 17, 1625–1635. [Google Scholar] [CrossRef] [Green Version]
- Kirby, K.A.; Myshakina, N.A.; Christen, M.T.; Chen, Y.L.; Schmidt, H.A.; Huber, A.D.; Xi, Z.; Kim, S.; Rao, R.K.; Kramer, S.T.; et al. A 2-hydroxyisoquinoline-1,3-dione active-site RNase H inhibitor binds in multiple modes to HIV-1 reverse transcriptase. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tramontano, E.; Esposito, F.; Badas, R.; Di Santo, R.; Costi, R.; La Colla, P. 6-[1-(4-Fluorophenyl)methyl-1H-pyrrol-2-yl)]-2,4-dioxo-5-hexenoic acid ethyl ester a novel diketo acid derivative which selectively inhibits the HIV-1 viral replication in cell culture and the ribonuclease H activity in vitro. Antivir. Res. 2005, 65, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Boyer, P.L.; Smith, S.J.; Zhao, X.Z.; Das, K.; Gruber, K.; Arnold, E.; Burke, T.R.; Hughes, S.H. Developing and Evaluating Inhibitors against the RNase H Active Site of HIV-1 Reverse Transcriptase. J. Virol. 2018, 92, 1–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arion, D.; Sluis-Cremer, N.; Min, K.-L.L.; Abram, M.E.; Fletcher, R.S.; Parniak, M.A. Mutational analysis of Tyr-501 of HIV-1 reverse transcriptase. Effects on ribonuclease H activity and inhibition of this activity by N-acylhydrazones. J. Biol. Chem. 2002, 277, 1370–1374. [Google Scholar] [CrossRef] [Green Version]
- Sluis-Cremer, N. Destabilization of the HIV-1 Reverse Transcriptase Dimer upon Interaction with N-Acyl Hydrazone Inhibitors. Mol. Pharmacol. 2002, 62, 398–405. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Wang, X.; Sun, L.; Cheng, X.; Poongavanam, V.; Kongsted, J.; Álvarez, M.; Luczkowiak, J.; Pannecouque, C.; De Clercq, E.; et al. Design, synthesis, and biologic evaluation of novel galloyl derivatives as HIV-1 RNase H inhibitors. Chem. Biol. Drug Des. 2019, 93, 582–589. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Tang, J.; Huber, A.D.; Casey, M.C.; Kirby, K.A.; Wilson, D.J.; Kankanala, J.; Parniak, M.A.; Sarafianos, S.G.; Wang, Z. 6-Biphenylmethyl-3-hydroxypyrimidine-2,4-diones potently and selectively inhibited HIV reverse transcriptase-associated RNase H. Eur. J. Med. Chem. 2018, 156, 680–691. [Google Scholar] [CrossRef]
- Corona, A.; Desantis, J.; Massari, S.; Distinto, S.; Masaoka, T.; Sabatini, S.; Esposito, F.; Manfroni, G.; Maccioni, E.; Cecchetti, V.; et al. Studies on Cycloheptathiophene-3-carboxamide Derivatives as Allosteric HIV-1 Ribonuclease H Inhibitors. ChemMedChem 2016, 11, 1709–1720. [Google Scholar] [CrossRef]
- Klumpp, K. Two-metal ion mechanism of RNA cleavage by HIV RNase H and mechanism-based design of selective HIV RNase H inhibitors. Nucleic Acids Res. 2003, 31, 6852–6859. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HIV-1 wt MT EC50 1 (µM) | HIV-1 K103N-Y181C MT4 EC50 1 (µM) | MT4 CC50 2 (µM) | SI 3 | |
|---|---|---|---|---|
| 13 | 10.1 ± 4.7 | 12.4 ± 0.8 | 61.5 ± 2.2 | 6.1 |
| 21 | 5.0 ± 1.4 | 8.7 ± 0.1 | 31.8 ± 0.4 | 6.4 |
| RDS1759 | 8.2 ± 0.9 | 4.6 ± 1.1 | 39 ± 4.4 | 4.7 |
| AZT | 0.007 ± 0.005 | 0.0031 ± 0.0007 | >3.7 | >500 |
| EFV | 0.050 ± 0.012 | >1.58 | >0.31 | >6.25 |
| NVP | 0.67 ± 0.22 | >3.7 | >3.7 | >5.5 |
| RAL | 0.0022 ± 0.0001 | 0.004 ± 0.001 | >2.25 | >1987 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corona, A.; Ballana, E.; Distinto, S.; Rogolino, D.; Del Vecchio, C.; Carcelli, M.; Badia, R.; Riveira-Muñoz, E.; Esposito, F.; Parolin, C.; et al. Targeting HIV-1 RNase H: N’-(2-Hydroxy-benzylidene)-3,4,5-Trihydroxybenzoylhydrazone as Selective Inhibitor Active against NNRTIs-Resistant Variants. Viruses 2020, 12, 729. https://0-doi-org.brum.beds.ac.uk/10.3390/v12070729
Corona A, Ballana E, Distinto S, Rogolino D, Del Vecchio C, Carcelli M, Badia R, Riveira-Muñoz E, Esposito F, Parolin C, et al. Targeting HIV-1 RNase H: N’-(2-Hydroxy-benzylidene)-3,4,5-Trihydroxybenzoylhydrazone as Selective Inhibitor Active against NNRTIs-Resistant Variants. Viruses. 2020; 12(7):729. https://0-doi-org.brum.beds.ac.uk/10.3390/v12070729
Chicago/Turabian StyleCorona, Angela, Ester Ballana, Simona Distinto, Dominga Rogolino, Claudia Del Vecchio, Mauro Carcelli, Roger Badia, Eva Riveira-Muñoz, Francesca Esposito, Cristina Parolin, and et al. 2020. "Targeting HIV-1 RNase H: N’-(2-Hydroxy-benzylidene)-3,4,5-Trihydroxybenzoylhydrazone as Selective Inhibitor Active against NNRTIs-Resistant Variants" Viruses 12, no. 7: 729. https://0-doi-org.brum.beds.ac.uk/10.3390/v12070729