Divergent Influenza-Like Viruses of Amphibians and Fish Support an Ancient Evolutionary Association

 ,
,  and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Identification of Divergent Influenza-Like Viruses from Vertebrate De Novo Transcriptome Assemblies
2.2. Recovery of Additional Influenza-Like Virus Segments Through de Novo Assembly and Coverage Statistics
2.3. Influenza Virus Genome Annotation
2.4. Phylogenetic Analysis
3. Results
3.1. Discovery and Annotation of Novel Influenza-Like Viruses in Vertebrates
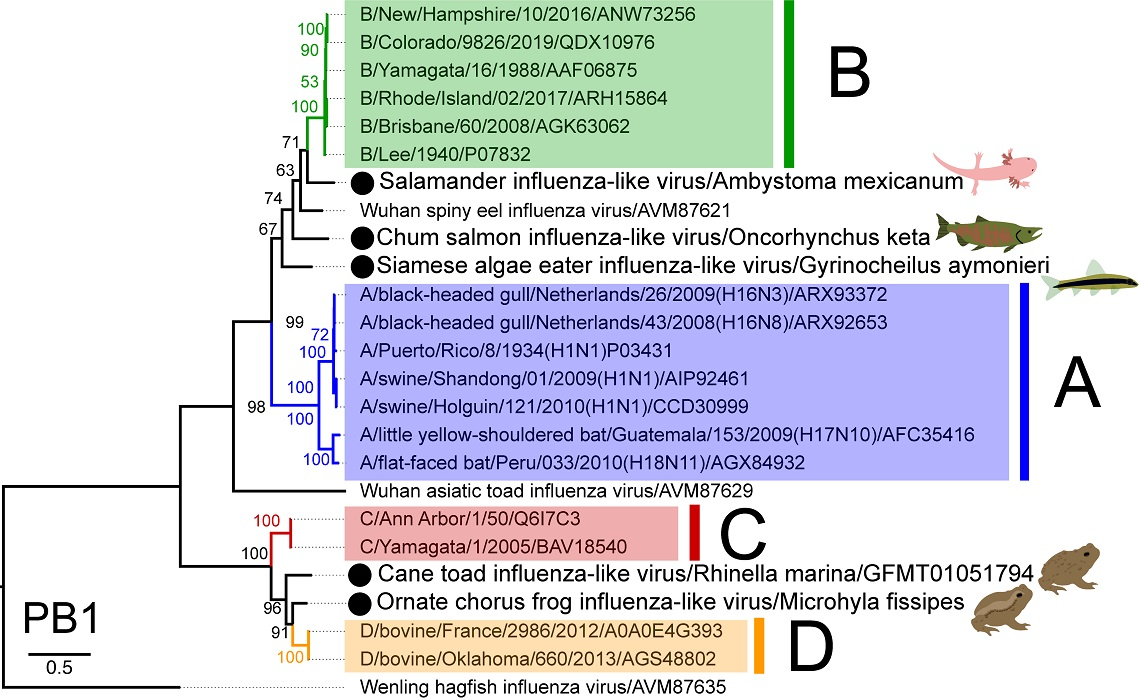
3.2. The Genome Organisation and Transcription of Novel Influenza-Like Virus Genes Are Highly Conserved
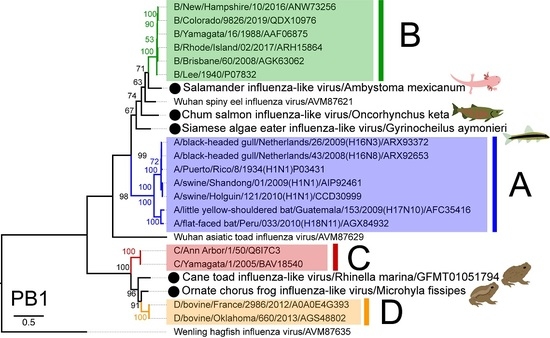
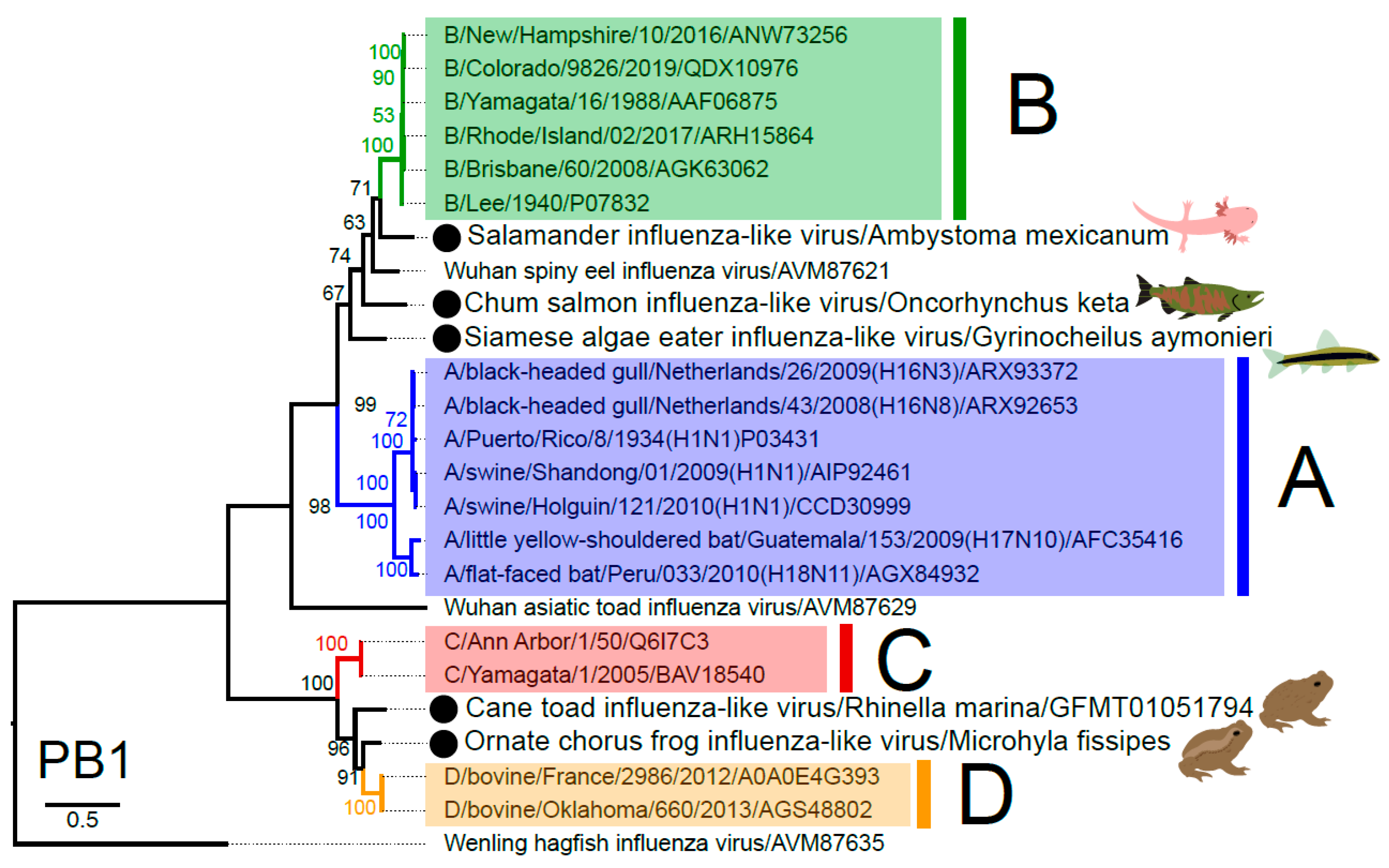
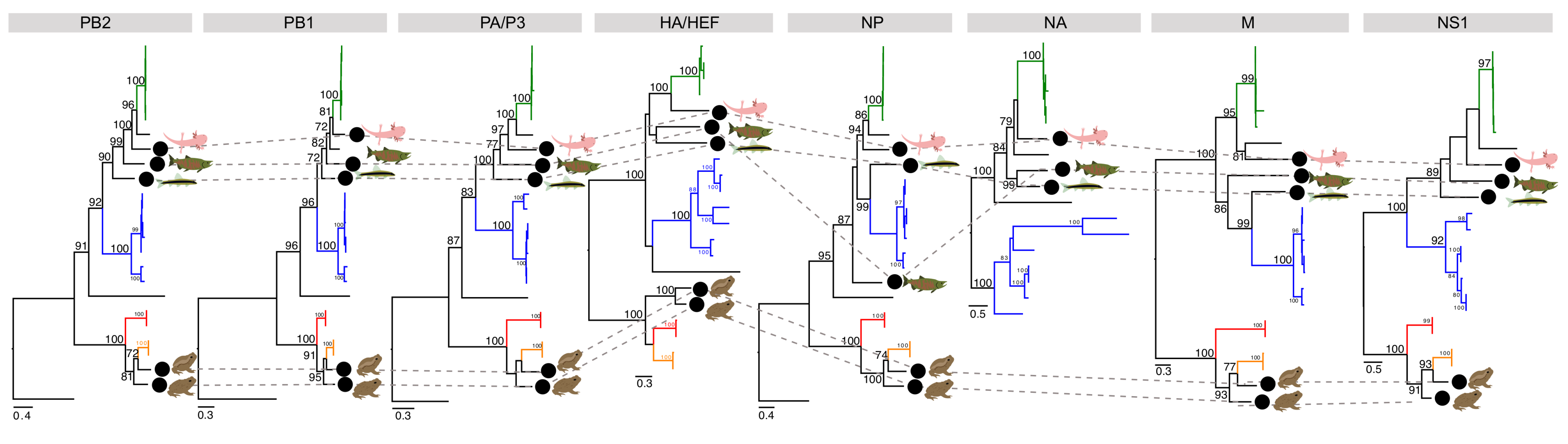
3.3. Phylogenetic Analysis of Novel Influenza-Like Viruses Suggests a History of Genomic Reassortment
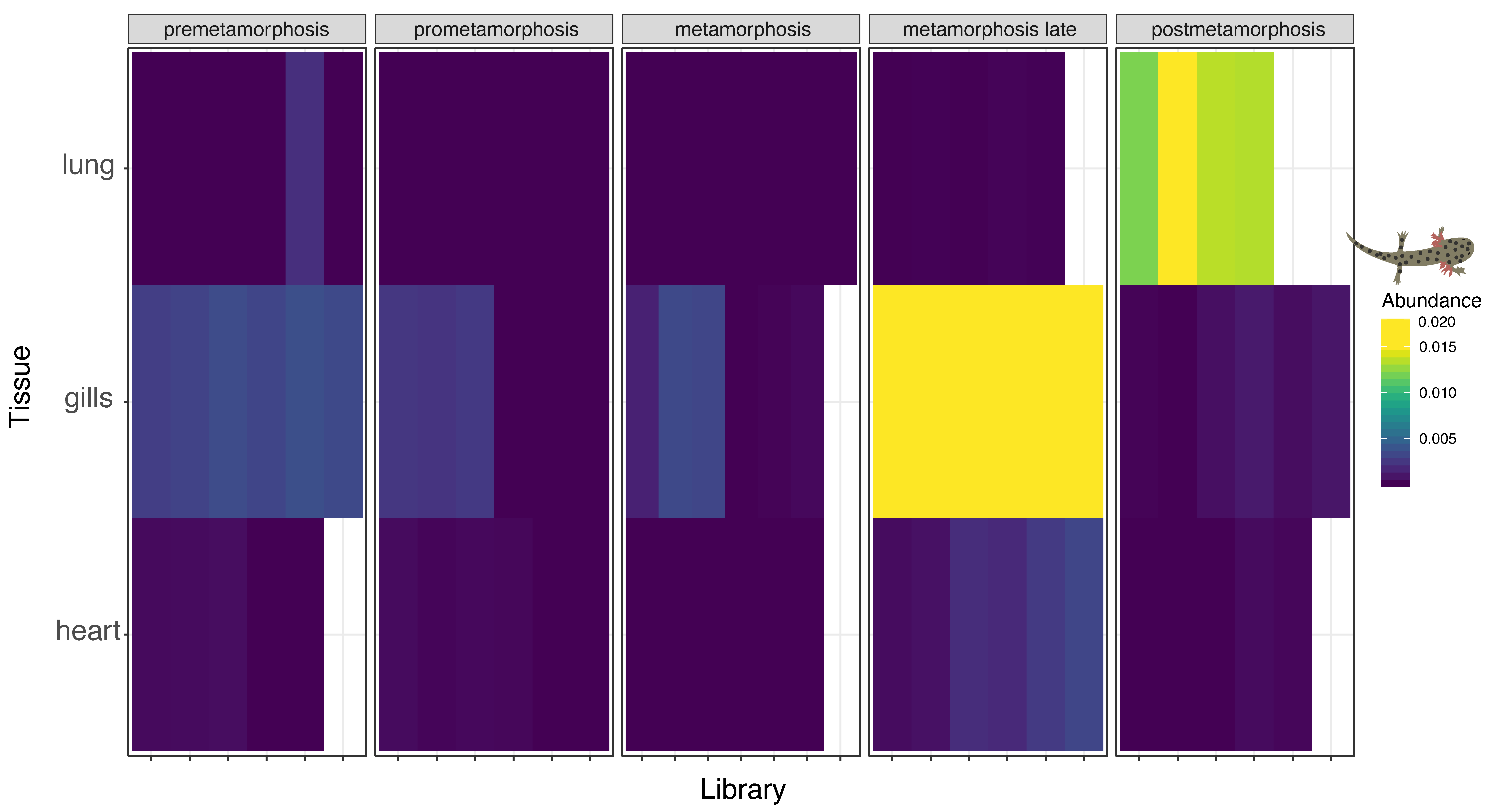
3.4. Multi-Tissue RNA-Sequencing of Developmental Plateau Tiger Salamander Libraries Suggests Conserved Influenza Virus Tropism in Vertebrates
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wille, M.; Holmes, E.C. The Ecology and Evolution of Influenza Viruses. Cold Spring Harb. Perspect. Med. 2019, 10, a038489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.D.; Chen, X.; Tian, J.H.; Chen, L.J.; Li, K.; Wang, W.; Eden, J.S.; Shen, J.J.; Liu, L.; et al. The evolutionary history of vertebrate RNA viruses. Nature 2018, 556, 197–202. [Google Scholar] [CrossRef] [PubMed]
- McCauley, J.W.; Hongo, S.; Kaverin, N.V.; Kochs, G.; Lamb, R.A.; Matrosovich, M.N.; Perez, D.R.; Palese, P.; Presti, R.M.; Rimstad, E.; et al. International Committee on the Taxonomy of Viruses: 2019; Negative Sense RNA Viruses: Orthomyxoviridae. Available online: https://talk.ictvonline.org/ictv-reports/ictv_9th_report/negative-sense-rna-viruses-2011/w/negrna_viruses/209/orthomyxoviridae. (accessed on 17 September 2020).
- Aspehaug, V.; Mikalsen, A.B.; Snow, M.; Biering, E.; Villoing, S. Characterization of the infectious salmon anemia virus fusion protein. J. Virol. 2005, 79, 12544–12553. [Google Scholar] [CrossRef] [Green Version]
- Olsen, B.; Munster, V.J.; Wallensten, A.; Waldenstrom, J.; Osterhaus, A.D.; Fouchier, R.A. Global patterns of influenza a virus in wild birds. Science 2006, 312, 384–388. [Google Scholar] [CrossRef] [Green Version]
- Lewis, N.S.; Russell, C.A.; Langat, P.; Anderson, T.K.; Berger, K.; Bielejec, F.; Burke, D.F.; Dudas, G.; Fonville, J.M.; Fouchier, R.A.; et al. The global antigenic diversity of swine influenza A viruses. eLife 2016, 5, e12217. [Google Scholar] [CrossRef] [Green Version]
- Sack, A.; Cullinane, A.; Daramragchaa, U.; Chuluunbaatar, M.; Gonchigoo, B.; Gray, G.C. Equine influenza virus—A neglected, reemergent disease threat. Emerg. Infect. Dis 2019, 25, 1185–1191. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.J.; Bahl, J.; Vijaykrishna, D.; Zhang, J.; Poon, L.L.; Chen, H.; Webster, R.G.; Peiris, J.S.; Guan, Y. Dating the emergence of pandemic influenza viruses. Proc. Natl. Acad. Sci. USA 2009, 106, 11709–11712. [Google Scholar] [CrossRef] [Green Version]
- Vijaykrishna, D.; Holmes, E.C.; Joseph, U.; Fourment, M.; Su, Y.C.; Halpin, R.; Lee, R.T.; Deng, Y.M.; Gunalan, V.; Lin, X.; et al. The contrasting phylodynamics of human influenza B viruses. eLife 2015, 4, e05055. [Google Scholar] [CrossRef]
- Bodewes, R.; Morick, D.; de Mutsert, G.; Osinga, N.; Bestebroer, T.; van der Vliet, S.; Smits, S.L.; Kuiken, T.; Rimmelzwaan, G.F.; Fouchier, R.A.; et al. Recurring influenza B virus infections in seals. Emerg. Infect. Dis. 2013, 19, 511–512. [Google Scholar] [CrossRef]
- Ran, Z.; Shen, H.; Lang, Y.; Kolb, E.A.; Turan, N.; Zhu, L.; Ma, J.; Bawa, B.; Liu, Q.; Liu, H.; et al. Domestic pigs are susceptible to infection with influenza B viruses. J. Virol. 2015, 89, 4818–4826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzaki, Y.; Katsushima, N.; Nagai, Y.; Shoji, M.; Itagaki, T.; Sakamoto, M.; Kitaoka, S.; Mizuta, K.; Nishimura, H. Clinical features of influenza C virus infection in children. J. Infect. Dis. 2006, 193, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.; Gaunt, E.R.; Digard, P.; Templeton, K.; Simmonds, P. Detection of influenza C virus but not influenza D virus in Scottish respiratory samples. J. Clin. Virol. 2016, 74, 50–53. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.J.; Jin, F.G.; Wang, P.; Wang, M.; Zhu, J.M. Isolation of influenza C virus from pigs and experimental infection of pigs with influenza C virus. J. Gen. Virol 1983, 64 Pt 1, 177–182. [Google Scholar]
- Kimura, H.; Abiko, C.; Peng, G.; Muraki, Y.; Sugawara, K.; Hongo, S.; Kitame, F.; Mizuta, K.; Numazaki, Y.; Suzuki, H.; et al. Interspecies transmission of influenza C virus between humans and pigs. Virus Res. 1997, 48, 71–79. [Google Scholar] [CrossRef]
- Manuguerra, J.C.; Hannoun, C. Natural infection of dogs by influenza C virus. Res. Virol. 1992, 143, 199–204. [Google Scholar] [CrossRef]
- Manuguerra, J.C.; Hannoun, C.; Simon, F.; Villar, E.; Cabezas, J.A. Natural infection of dogs by influenza C virus: A serological survey in Spain. New Microbiol. 1993, 16, 367–371. [Google Scholar]
- Salem, E.; Cook, E.A.J.; Lbacha, H.A.; Oliva, J.; Awoume, F.; Aplogan, G.L.; Hymann, E.C.; Muloi, D.; Deem, S.L.; Alali, S.; et al. Serologic evidence for influenza C and D virus among ruminants and camelids, Africa, 1991–2015. Emerg. Infect. Dis. 2017, 23, 1556–1559. [Google Scholar] [CrossRef] [Green Version]
- Hause, B.M.; Ducatez, M.; Collin, E.A.; Ran, Z.G.; Liu, R.X.; Sheng, Z.Z.; Armien, A.; Kaplan, B.; Chakravarty, S.; Hoppe, A.D.; et al. Isolation of a novel swine influenza virus from Oklahoma in 2011 which is distantly related to human influenza C viruses. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef] [Green Version]
- Flynn, O.; Gallagher, C.; Mooney, J.; Irvine, C.; Ducatez, M.; Hause, B.; McGrath, G.; Ryan, E. Influenza D virus in cattle, Ireland. Emerg. Infect. Dis. 2018, 24, 389–391. [Google Scholar] [CrossRef] [Green Version]
- Collin, E.A.; Sheng, Z.; Lang, Y.; Ma, W.; Hause, B.M.; Li, F. Cocirculation of two distinct genetic and antigenic lineages of proposed influenza D virus in cattle. J. Virol. 2015, 89, 1036–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asha, K.; Kumar, B. Emerging influenza D virus threat: What we know so far! J. Clin. Med. 2019, 8, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, S.K.; Ma, W.J.; McDaniel, C.J.; Gray, G.C.; Lednicky, J.A. Serologic evidence of exposure to influenza D virus among persons with occupational contact with cattle. J. Clin. Virol. 2016, 81, 31–33. [Google Scholar] [CrossRef] [PubMed]
- Longdon, B.; Murray, G.G.; Palmer, W.J.; Day, J.P.; Parker, D.J.; Welch, J.J.; Obbard, D.J.; Jiggins, F.M. The evolution, diversity, and host associations of rhabdoviruses. Virus Evol. 2015, 1, vev014. [Google Scholar] [CrossRef] [Green Version]
- Parry, R.; Asgari, S. Discovery of novel crustacean and cephalopod flaviviruses: Insights into the evolution and circulation of flaviviruses between marine invertebrate and vertebrate hosts. J. Virol. 2019, 93, e00432-19. [Google Scholar] [CrossRef] [Green Version]
- Francois, S.; Filloux, D.; Roumagnac, P.; Bigot, D.; Gayral, P.; Martin, D.P.; Froissart, R.; Ogliastro, M. Discovery of parvovirus-related sequences in an unexpected broad range of animals. Sci. Rep. 2016, 6, 30880. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Huang, Y.; Li, X.; Baldwin, C.C.; Zhou, Z.; Yan, Z.; Crandall, K.A.; Zhang, Y.; Zhao, X.; Wang, M.; et al. Fish-T1K (Transcriptomes of 1,000 Fishes) Project: Large-scale transcriptome data for fish evolution studies. Gigascience 2016, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Marin, A. Characterization and prediction of alternative splice sites. Gene 2006, 366, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Dubois, J.; Terrier, O.; Rosa-Calatrava, M. Influenza viruses and mRNA splicing: Doing more with less. mBio 2014, 5, e00070-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Le, S.Q.; Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef] [Green Version]
- Whelan, S.; Goldman, N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 2001, 18, 691–699. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.C.; Le, Q.S.; Gascuel, O.; Le, V.S. FLU, an amino acid substitution model for influenza proteins. BMC Evol. Biol. 2010, 10, 99. [Google Scholar] [CrossRef] [Green Version]
- Soubrier, J.; Steel, M.; Lee, M.S.; Der Sarkissian, C.; Guindon, S.; Ho, S.Y.; Cooper, A. The influence of rate heterogeneity among sites on the time dependence of molecular rates. Mol. Biol. Evol. 2012, 29, 3345–3358. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Nguyen, M.A.T.; von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Dwaraka, V.B.; Smith, J.J.; Woodcock, M.R.; Voss, S.R. Comparative transcriptomics of limb regeneration: Identification of conserved expression changes among three species of Ambystoma. Genomics 2019, 111, 1216–1225. [Google Scholar] [CrossRef]
- Janet, P.-M.; Juan, C.-P.; Annie, E.-C.; Gilberto, M.-C.; Hilda, L.; Enrique, S.-V.; Denhi, S.; Jesus, C.-M.; Alfredo, C.-R. Multi-organ transcriptomic landscape of Ambystoma velasci metamorphosis. bioRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Sabin, K.Z.; Jiang, P.; Gearhart, M.D.; Stewart, R.; Echeverri, K. AP-1(cFos/JunB)/miR-200a regulate the pro-regenerative glial cell response during axolotl spinal cord regeneration. Commun. Biol. 2019, 2, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, S.L.; Baselga-Garriga, C.; Melton, D.A. Midkine is a dual regulator of wound epidermis development and inflammation during the initiation of limb regeneration. eLife 2020, 9, e50765. [Google Scholar] [CrossRef] [PubMed]
- Huggins, P.; Johnson, C.K.; Schoergendorfer, A.; Putta, S.; Bathke, A.C.; Stromberg, A.J.; Voss, S.R. Identification of differentially expressed thyroid hormone responsive genes from the brain of the Mexican Axolotl (Ambystoma mexicanum). Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2012, 155, 128–135. [Google Scholar] [CrossRef] [Green Version]
- King, B.L.; Yin, V.P. A Conserved MicroRNA Regulatory circuit is differentially controlled during limb/appendage regeneration. PLoS ONE 2016, 11, e0157106. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Lee, H.J.; Kim, Y.K. Comparative transcriptome profiling of selected osmotic regulatory proteins in the gill during seawater acclimation of chum salmon (Oncorhynchus keta) fry. Sci. Rep. 2020, 10, 1987. [Google Scholar] [CrossRef]
- Edwards, R.J.; Tuipulotu, D.E.; Amos, T.G.; O’Meally, D.; Richardson, M.F.; Russell, T.L.; Vallinoto, M.; Carneiro, M.; Ferrand, N.; Wilkins, M.R.; et al. Draft genome assembly of the invasive cane toad, Rhinella marina. Gigascience 2018, 7, giy095. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, L.; Wang, S.; Wang, H.; Jiang, J. Transcriptome profiles of metamorphosis in the ornamented pygmy frog Microhyla fissipes clarify the functions of thyroid hormone receptors in metamorphosis. Sci. Rep. 2016, 6, 27310. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Wang, S.; Zhao, L.; Jiang, J. De novo transcriptome assembly for the lung of the ornamented pygmy frog (Microhyla fissipes). Genom. Data 2017, 13, 44–45. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.G.; Eden, J.S.; Enosi Tuipulotu, D.; Shi, M.; Selechnik, D.; Shine, R.; Rollins, L.A.; Holmes, E.C.; White, P.A. Viral discovery in the invasive Australian Cane Toad (Rhinella marina) using metatranscriptomic and genomic approaches. J. Virol. 2018, 92, e00768-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betakova, T.; Nermut, M.V.; Hay, A.J. The NB protein is an integral component of the membrane of influenza B virus. J. Gen. Virol. 1996, 77 Pt 11, 2689–2694. [Google Scholar] [CrossRef]
- Demircan, T.; Ilhan, A.E.; Ayturk, N.; Yildirim, B.; Ozturk, G.; Keskin, I. A histological atlas of the tissues and organs of neotenic and metamorphosed axolotl. Acta Histochem. 2016, 118, 746–759. [Google Scholar] [CrossRef] [PubMed]
- Ibricevic, A.; Pekosz, A.; Walter, M.J.; Newby, C.; Battaile, J.T.; Brown, E.G.; Holtzman, M.J.; Brody, S.L. Influenza virus receptor specificity and cell tropism in mouse and human airway epithelial cells. J. Virol. 2006, 80, 7469–7480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wille, M.; van Run, P.; Waldenstrom, J.; Kuiken, T. Infected or not: Are PCR-positive oropharyngeal swabs indicative of low pathogenic influenza A virus infection in the respiratory tract of Mallard Anas platyrhynchos? Vet. Res. 2014, 45, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciminski, K.; Schwemmle, M. Bat-borne influenza A viruses: An awakening. Cold Spring Harb. Perspect. Med. 2019. [Google Scholar] [CrossRef] [Green Version]
- Percino-Daniel, R. New record of Ambystoma velasci (Dugès, 1888) from Western Mexico. Herpetol. Notes 2019, 12, 351–352. [Google Scholar]
- Contreras, V.; Martinez-Meyer, E.; Valiente, E.; Zambrano, L. Recent decline and potential distribution in the last remnant area of the microendemic Mexican axolotl (Ambystoma mexicanum). Biol. Conserv. 2009, 142, 2881–2885. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus Name | Host | Tissue Sampled | Reference | BLASTp Output of Predicted Amino Acid Identity of Polymerase Genes | |||
|---|---|---|---|---|---|---|---|
| Segment | GenBank Accession | Coverage (%); Identity (%) | E-value | ||||
| Salamander influenza-like virus | Mexican walking fish (Ambystoma mexicanum) | Various | [43,45,46,47,48] | PB1 Influenza B virus (B/California/24/2016) PB2 Influenza B virus (B/Sydney/19/2011) PA Influenza B virus (B/Indiana/07/2016) | QHI05420 AZY32600 ANW74127 | 100%; 75.70% 99%; 61.83% 97%; 59.64% | 0.0 0.0 0.0 |
| Plateau tiger salamander (Ambystoma velasci) | Various | [44] | |||||
| Siamese algae-eater influenza-like virus | Siamese algae-eater (Gyrinocheilus aymonieri) | Gills | [28] | PB1 Influenza B virus (B/California/24/2016) PB2 Influenza B virus (B/New York/1121/2007) PA Wuhan spiny eel influenza virus | ANW79211 AHL92298 AVM87622 | 99%; 69.46% 99%; 48.58% 96%; 53.89% | 0.0 0.0 0.0 |
| Chum salmon influenza-like virus | Chum salmon (Oncorhynchus keta) | Gills | [49] | PB1 Influenza B virus (B/Iowa/14/2017) PB2 Influenza B virus (B/Memphis/5/93) PA Influenza B virus (B/Taiwan/45/2007) | QHI05420 AAU94860 ACO06009 | 100%; 69.50% 99%; 57.05% 98%; 51.88% | 0.0 0.0 0.0 |
| Cane toad influenza-like virus | Cane Toad (Rhinella marina) | Tissue unknown, Larval | [50] | PB1 Influenza D virus (bovine/Mexico/S7/2015) PB2 Influenza D virus (bovine/Kansas/14-22/2012) P3 Influenza D virus (bovine/Yamagata/10710/2016) | AMN87903 AIO11621 BBC14929 | 99%; 77.03% 99%; 62.87% 100%; 62.66% | 0.0 0.0 0.0 |
| Ornate chorus frog influenza-like virus | Ornate chorus frog (Microhyla fissipes) | Larval, Lung | [51,52] | PB1 Influenza D virus (bovine/Mississippi/C00046N/2014) PB2 Influenza D virus (swine/Italy/173287-4/2016) P3 Influenza D virus (D/bovine/Shandong/Y217/2014) | ALE66333 AON76692 AIE52099 | 98%; 81.91% 99%; 68.22% 99%; 63.78% | 0.0 0.0 0.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parry, R.; Wille, M.; Turnbull, O.M.H.; Geoghegan, J.L.; Holmes, E.C. Divergent Influenza-Like Viruses of Amphibians and Fish Support an Ancient Evolutionary Association. Viruses 2020, 12, 1042. https://0-doi-org.brum.beds.ac.uk/10.3390/v12091042
Parry R, Wille M, Turnbull OMH, Geoghegan JL, Holmes EC. Divergent Influenza-Like Viruses of Amphibians and Fish Support an Ancient Evolutionary Association. Viruses. 2020; 12(9):1042. https://0-doi-org.brum.beds.ac.uk/10.3390/v12091042
Chicago/Turabian StyleParry, Rhys, Michelle Wille, Olivia M. H. Turnbull, Jemma L. Geoghegan, and Edward C. Holmes. 2020. "Divergent Influenza-Like Viruses of Amphibians and Fish Support an Ancient Evolutionary Association" Viruses 12, no. 9: 1042. https://0-doi-org.brum.beds.ac.uk/10.3390/v12091042