A Targeted Metagenomics Approach to Study the Diversity of Norovirus GII in Shellfish Implicated in Outbreaks

Abstract
:1. Introduction
2. Material and Methods
2.1. Shellfish Samples
2.2. Nucleic Acids Extraction
2.3. Detection of Viral Genomes by rRT-PCR
2.4. Extraction Efficiency and Inhibition
2.5. Artificial Samples of Known Composition
2.6. Generation of RdRp and VP1 Amplicons
2.7. Classical Genotyping: Cloning and Sanger Sequencing
2.8. Targeted Metagenomics: Amplicon Tagging and Illumina Sequencing
2.9. Bioinformatics and Statistics
2.9.1. Quality Trimming
2.9.2. Clustering
2.9.3. Mapping
2.9.4. Analysis of Sequences and Phylogeny
2.10. Statistics
3. Results
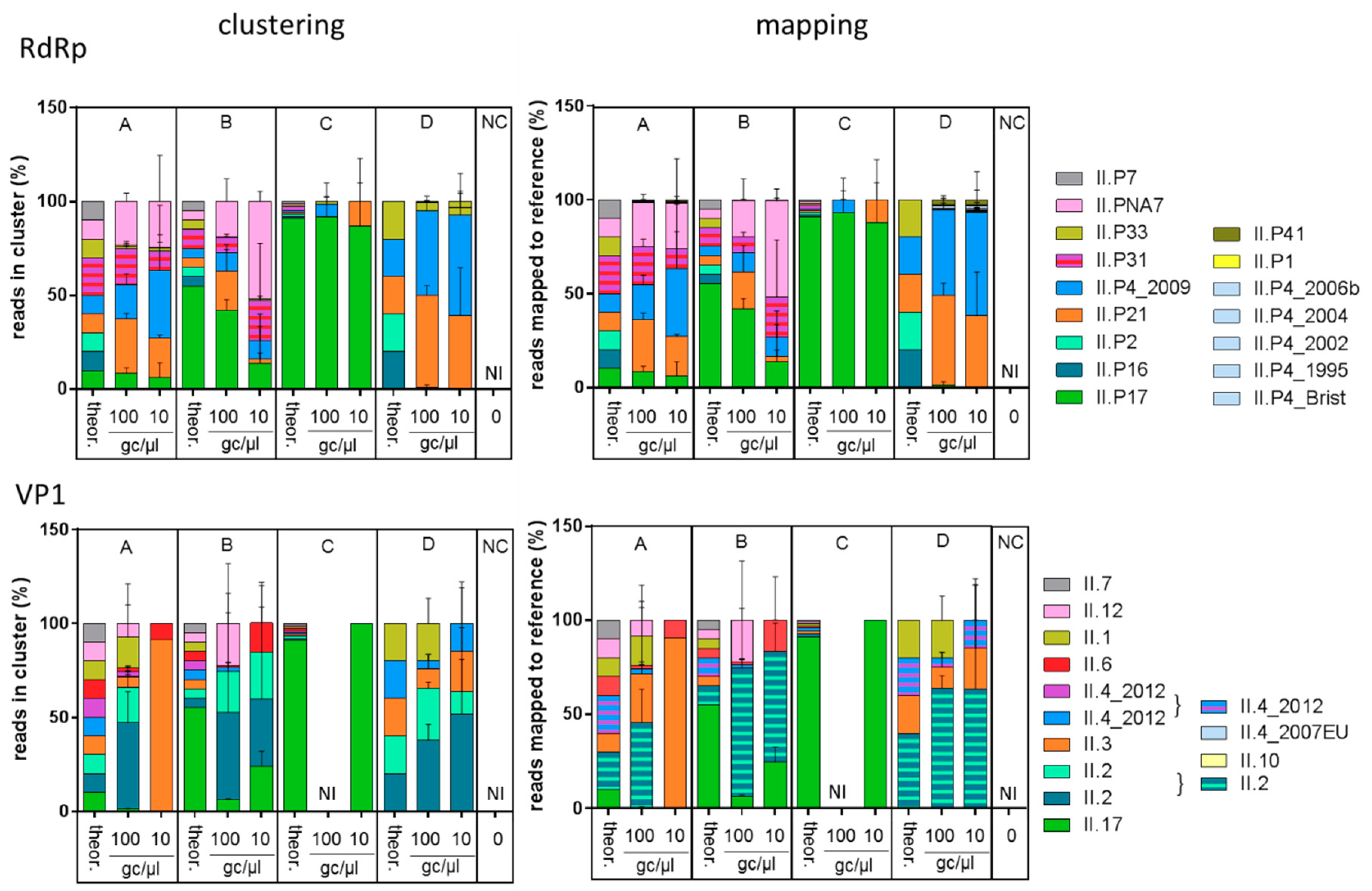
3.1. Assessing the Targeted Metagenomics Approach with Samples of Known Composition
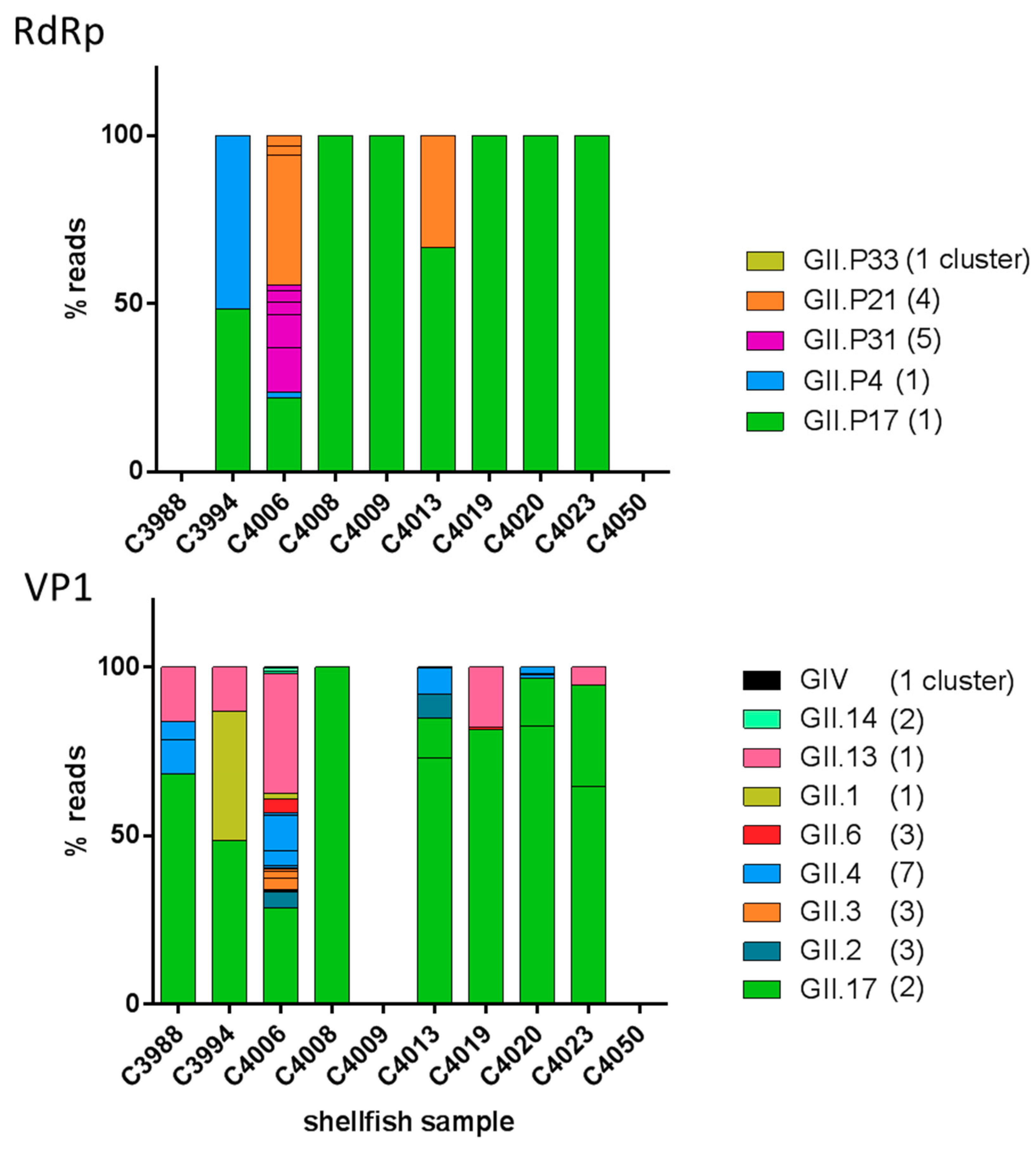
3.2. Diversity of NoV GII in Outbreak-Related Shellfish Samples
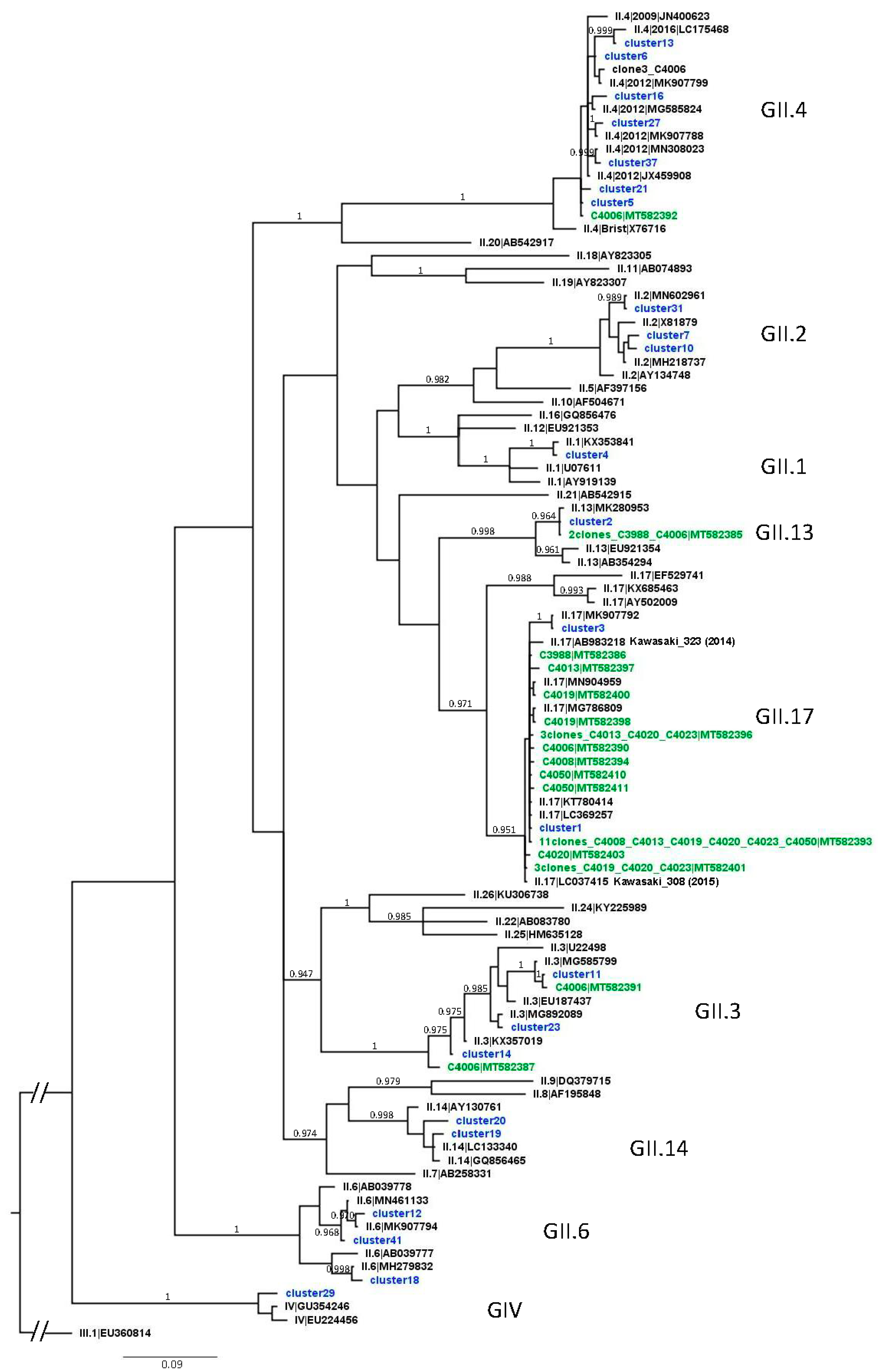
3.3. Analysis of NoV GII Sequences from Shellfish Samples
4. Discussion
4.1. Advantages and Limitations of the Deep-Sequencing Approach
Comparison of Methods for Bioinformatics Analysis
4.2. Impact of NoV Concentration
4.3. Selectivity
4.4. Comparison of Classical Genotyping and Targeted Metagenomics on Shellfish Samples
4.5. NoV Diversity in Shellfish Samples
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Graaf, M.; van Beek, J.; Koopmans, M.P.G. Human norovirus transmission and evolution in a changing world. Nat. Rev. Microbiol. 2016, 14, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Mathijs, E.; Stals, A.; Baert, L.; Botteldoorn, N.; Denayer, S.; Mauroy, A.; Scipioni, A.; Daube, G.; Dierick, K.; Herman, L.; et al. A Review of Known and Hypothetical Transmission Routes for Noroviruses. Food Environ. Virol. 2012, 4, 131–152. [Google Scholar] [CrossRef] [PubMed]
- Marsh, Z.; Shah, M.P.; Wikswo, M.E.; Barclay, L.; Kisselburgh, H.; Kambhampati, A.; Cannon, J.L.; Parashar, U.D.; Vinjé, J.; Hall, A.J. Epidemiology of Foodborne Norovirus Outbreaks—United States, 2009–2015. Food Saf. 2018, 6, 58–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardstaff, J.L.; Clough, H.E.; Lutje, V.; McIntyre, K.M.; Harris, J.P.; Garner, P.; O’Brien, S.J. Foodborne and Food-Handler Norovirus Outbreaks: A Systematic Review. Foodborne Pathog. Dis. 2018, 15, 589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teunis, P.F.M.; Sukhrie, F.H.A.; Vennema, H.; Bogerman, J.; Beersma, M.F.C.; Koopmans, M.P.G. Shedding of norovirus in symptomatic and asymptomatic infections. Epidemiol. Infect. 2015, 143, 1710–1717. [Google Scholar] [CrossRef] [Green Version]
- Sano, D.; Amarasiri, M.; Hata, A.; Watanabe, T.; Katayama, H. Risk management of viral infectious diseases in wastewater reclamation and reuse: Review. Environ. Int. 2016, 91, 220–229. [Google Scholar] [CrossRef] [Green Version]
- Kauppinen, A.; Miettinen, I. Persistence of Norovirus GII Genome in Drinking Water and Wastewater at Different Temperatures. Pathogens 2017, 6, 48. [Google Scholar] [CrossRef]
- Yu, Y.; Cai, H.; Hu, L.; Lei, R.; Pan, Y.; Yan, S.; Wang, Y. Molecular epidemiology of oyster-related human noroviruses and their global genetic diversity and temporal-geographical distribution from 1983 to 2014. Appl. Environ. Microbiol. 2015, 81, 7615–7624. [Google Scholar] [CrossRef] [Green Version]
- Le Guyader, F.S.; Atmar, R.L.; Le Pendu, J. Transmission of viruses through shellfish: When specific ligands come into play. Curr. Opin. Virol. 2012, 2, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Chhabra, P.; de Graaf, M.; Parra, G.I.; Chan, M.C.W.; Green, K.; Martella, V.; Wang, Q.; White, P.A.; Katayama, K.; Vennema, H.; et al. Updated classification of norovirus genogroups and genotypes. J. Gen. Virol. 2019, 100, 1393–1406. [Google Scholar] [CrossRef]
- Ludwig-Begall, L.F.; Mauroy, A.; Thiry, E. Norovirus recombinants: Recurrent in the field, recalcitrant in the lab—A scoping review of recombination and recombinant types of noroviruses. J. Gen. Virol. 2018, 99, 970–988. [Google Scholar] [CrossRef] [PubMed]
- Parra, G.I.; Squires, R.B.; Karangwa, C.K.; Johnson, J.A.; Lepore, C.; Sosnovtsev, S.V.; Green, K.Y. Static and Evolving Norovirus Genotypes: Implications for Epidemiology and Immunity. PLoS Pathog. 2017, 13, e1006136. [Google Scholar] [CrossRef]
- Van Beek, J.; de Graaf, M.; Al-Hello, H.; Allen, D.J.; Ambert-Balay, K.; Botteldoorn, N.; Brytting, M.; Buesa, J.; Cabrerizo, M.; Chan, M.; et al. Surveillance of norovirus, 2005–2016: An epidemiological analysis of data collected from the NoroNet network. Lancet Infect. Dis. 2018, 18, 545–553. [Google Scholar] [CrossRef]
- Bidalot, M.; Théry, L.; Kaplon, J.; De Rougemont, A.; Ambert-Balay, K. Emergence of new recombinant noroviruses GII.p16-GII.4 and GII.p16-GII.2, France, winter 2016 to 2017. Eurosurveillance 2017, 22, 30508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Graaf, M.; van Beek, J.; Vennema, H.; Podkolzin, A.T.; Hewitt, J.; Bucardo, F.; Templeton, K.; Mans, J.; Nordgren, J.; Reuter, G.; et al. Emergence of a novel GII.17 norovirus—End of the GII.4 era? Eurosurveillance 2015, 20, 21178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhoef, L.; Hewitt, J.; Barclay, L.; Ahmed, S.; Lake, R.; Hall, A.J.; Lopman, B.; Kroneman, A.; Vennema, H. Norovirus Genotype Profiles Associated with Foodborne Transmission, 1999–2012. Emerg. Infect. Dis. 2015, 21, 592–599. [Google Scholar] [CrossRef]
- Okoh, A.I.; Sibanda, T.; Gusha, S.S. Inadequately Treated Wastewater as a Source of Human Enteric Viruses in the Environment. Int. J. Environ. Res. Public Health 2010, 7, 2620–2637. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Zhang, Z.; Li, S.; Wu, Q.; Tian, P.; Zhang, Z.; Wang, D. Fingerprinting of human noroviruses co-infections in a possible foodborne outbreak by metagenomics. Int. J. Food Microbiol. 2020, 333, 108787. [Google Scholar] [CrossRef]
- Rasmussen, L.D.; Schultz, A.C.; Uhrbrand, K.; Jensen, T.; Fischer, T.K. Molecular Evidence of Oysters as Vehicle of Norovirus GII.P17-GII.17. Emerg. Infect. Dis. 2016, 22, 2024–2025. [Google Scholar] [CrossRef]
- Le Guyader, F.S.; Bon, F.; DeMedici, D.; Parnaudeau, S.; Bertone, A.; Crudeli, S.; Doyle, A.; Zidane, M.; Suffredini, E.; Kohli, E.; et al. Detection of Multiple Noroviruses Associated with an International Gastroenteritis Outbreak Linked to Oyster Consumption Franc. J. Clin. Microbiol. 2006, 44, 3878–3882. [Google Scholar] [CrossRef] [Green Version]
- Desdouits, M.; De Graaf, M.; Strubbia, S.; Munnink, B.B.O.; Kroneman, A.; Le Guyader, F.S.; Koopmans, M.P. Novel opportunities for NGS-based one health surveillance of foodborne viruses. One Health Outlook 2020, 2, 14. [Google Scholar] [CrossRef]
- Suffredini, E.; Iaconelli, M.; Equestre, M.; Valdazo-González, B.; Ciccaglione, A.R.; Marcantonio, C.; Della Libera, S.; Bignami, F.; La Rosa, G. Genetic Diversity Among Genogroup II Noroviruses and Progressive Emergence of GII.17 in Wastewaters in Italy (2011–2016) Revealed by Next-Generation and Sanger Sequencing. Food Environ. Virol. 2018, 10, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Oshiki, M.; Miura, T.; Kazama, S.; Segawa, T.; Ishii, S.; Hatamoto, M.; Yamaguchi, T.; Kubota, K.; Iguchi, A.; Tagawa, T.; et al. Microfluidic PCR Amplification and MiSeq Amplicon Sequencing Techniques for High-Throughput Detection and Genotyping of Human Pathogenic RNA Viruses in Human Feces, Sewage, and Oysters. Front. Microbiol. 2018, 9, 830. [Google Scholar] [CrossRef] [PubMed]
- Imamura, S.; Kanezashi, H.; Goshima, T.; Haruna, M.; Okada, T.; Inagaki, N.; Uema, M.; Noda, M.; Akimoto, K. Next-Generation Sequencing Analysis of the Diversity of Human Noroviruses in Japanese Oysters. Foodborne Pathog. Dis. 2017, 14, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Le Guyader, F.S.; Le Saux, J.C.; Ambert-Balay, K.; Krol, J.; Serais, O.; Parnaudeau, S.; Giraudon, H.; Delmas, G.; Pommepuy, M.; Pothier, P.; et al. Aichi Virus, Norovirus, Astrovirus, Enterovirus, and Rotavirus Involved in Clinical Cases from a French Oyster-Related Gastroenteritis Outbreak. J. Clin. Microbiol. 2008, 46, 4011–4017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loisy, F.; Atmar, R.L.; Guillon, P.; Le Cann, P.; Pommepuy, M.; Le Guyader, F.S. Real-time RT-PCR for norovirus screening in shellfish. J. Virol. Methods 2005, 123, 1–7. [Google Scholar] [CrossRef]
- Kojima, S.; Kageyama, T.; Fukushi, S.; Hoshino, F.B.; Shinohara, M.; Uchida, K.; Natori, K.; Takeda, N.; Katayama, K. Genogroup-specific PCR primers for detection of Norwalk-like viruses. J. Virol. Methods 2002, 100, 107–114. [Google Scholar] [CrossRef]
- Polo, D.; Schaeffer, J.; Fournet, N.; Le Saux, J.C.; Parnaudeau, S.; McLeod, C.; Le Guyader, F.S. Digital PCR for Quantifying Norovirus in Oysters Implicated in Outbreaks, France. Emerg. Infect. Dis. 2016, 22, 2189–2191. [Google Scholar] [CrossRef]
- Le Guyader, F.; Estes, M.K.; Hardy, M.E.; Neill, F.H.; Green, J.; Brown, D.W.G.; Atmar, R.L. Evaluation of a degenerate primer for the PCR detection of human caliciviruses. Arch. Virol. 1996, 141, 2225–2235. [Google Scholar] [CrossRef]
- Marshall, J.A.; Yuen, L.K.W.; Catton, M.G.; Gunesekere, I.C.; Wright, P.J.; Bettelheim, K.A.; Griffith, J.M.; Lightfoot, D.; Hogg, G.G.; Gregory, J.; et al. Multiple outbreaks of Norwalk-like virus gastro-enteritis associated with a Mediterranean-style restaurant. J. Med. Microbiol. 2001, 50, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Kroneman, A.; Vennema, H.; Deforche, K.V.D.; Avoort, H.V.D.; Peñaranda, S.; Oberste, M.S.; Vinjé, J.; Koopmans, M. An automated genotyping tool for enteroviruses and noroviruses. J. Clin. Virol. 2011, 51, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Afgan, E.; Baker, D.; Batut, B.; Van Den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escudié, F.; Auer, L.; Bernard, M.; Mariadassou, M.; Cauquil, L.; Vidal, K.; Maman, S.; Hernandez-Raquet, G.; Combes, S.; Pascal, G. FROGS: Find, Rapidly, OTUs with Galaxy Solution. Berger, B., éditeur. Bioinformatics 2018, 34, 1287–1294. [Google Scholar]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef] [PubMed]
- Gregori, J.; Perales, C.; Rodriguez-Frias, F.; Esteban, J.I.; Quer, J.; Domingo, E. Viral quasispecies complexity measures. Virology 2016, 493, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Caro-Pérez, N.; Martínez-Rebollar, M.; Gregori, J.; Quer, J.; González, P.; Gambato, M.; García-González, N.; González-Candelas, F.; Costa, J.; Esteban, J.I.; et al. Phylogenetic analysis of an epidemic outbreak of acute hepatitis C in HIV-infected patients by ultra-deep pyrosequencing. J. Clin. Virol. 2017, 92, 42–47. [Google Scholar] [CrossRef] [Green Version]
- Uyaguari-Diaz, M.I.; Chan, M.; Chaban, B.L.; Croxen, M.A.; Finke, J.F.; Hill, J.E.; Peabody, M.A.; Van Rossum, T.; Suttle, C.A.; Brinkman, F.S.; et al. A comprehensive method for amplicon-based and metagenomic characterization of viruses, bacteria, and eukaryotes in freshwater samples. Microbiome 2016, 4, 20. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.; Sykes, S.; Young, S.; Kota, K.; Sanka, R.; Sheth, N.; Orvis, J.; Sodergren, E.; Wang, Z.; Weinstock, G.M.; et al. Optimizing Read Mapping to Reference Genomes to Determine Composition and Species Prevalence in Microbial Communities. PLoS ONE 2012, 7, e36427. [Google Scholar] [CrossRef]
- EFSA. Analysis of the European baseline survey of norovirus in oysters. EFSA J. 2019, 17, 1–99. [Google Scholar]
- Pinto, A.J.; Raskin, L. PCR Biases Distort Bacterial and Archaeal Community Structure in Pyrosequencing Datasets. Bertilsson, S., éditeur. PLoS ONE 2012, 7, e43093. [Google Scholar] [CrossRef] [Green Version]
- Imamura, S.; Haruna, M.; Goshima, T. Application of next-generation sequencing to investigation of norovirus diversity in shellfish collected from two coastal sites in Japan from 2013 to 2014. J. Vet. Res. 2016, 64, 113–122. [Google Scholar]
- La Bella, G.; Martella, V.; Basanisi, M.G.; Nobili, G.; Terio, V.; La Salandra, G. Food-Borne Viruses in Shellfish: Investigation on Norovirus and HAV Presence in Apulia (SE Italy). Food Environ. Virol. 2017, 9, 179–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, T.; Parnaudeau, S.; Grodzki, M.; Okabe, S.; Atmar, R.L.; Le Guyader, F.S. Environmental Detection of Genogroup I, II, and IV Noroviruses by Using a Generic Real-Time Reverse Transcription-PCR Assay. Appl. Environ. Microbiol. 2013, 79, 6585–6592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pu, J.; Kazama, S.; Miura, T.; Azraini, N.D.; Konta, Y.; Ito, H.; Ueki, Y.; Cahyaningrum, E.E.; Omura, T.; Watanabe, T. Pyrosequencing Analysis of Norovirus Genogroup II Distribution in Sewage and Oysters: First Detection of GII.17 Kawasaki 2014 in Oysters. Food Environ. Virol. 2016, 8, 310–312. [Google Scholar] [CrossRef]
- Rosa, G.L.; Libera, S.D.; Proroga MIYTR. Detection of Norovirus GII. 17 Kawasaki 2014 in Shellfish, Marine Water and Underwater Sewage Discharges in Italy. Food Environ. Virol. 2017, 9, 326–333. [Google Scholar] [CrossRef]
- Koo, E.S.; Kim, M.S.; Choi, Y.S.; Park, K.-S.; Jeong, Y.S. Occurrence of novel GII.17 and GII.21 norovirus variants in the coastal environment of South Korea in 2015. Sekaran SD, éditeur. PLoS ONE 2017, 12, e0172237. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Sample | Shellfish Species | Sampling Date | Country of Origin/Coastal Area | Cases/Exposed | NoV in Patients Stool | Mean NoV GII Ct Value |
|---|---|---|---|---|---|---|
| C3988 | Mussel | 20/01/2016 | Spain | 2/2 | GI.1, GII.17 | 32 |
| C3994 | Mussel | 28/01/2016 | Spain | 2/10 | GII.17 | 35 |
| C4006 | Clam | 24/02/2016 | Spain | 2/3 | NA | 35 |
| C4008 | Oyster | 01/03/2016 | France/Channel | 4/13 | NA | 36 |
| C4009 | Oyster | 35 | ||||
| C4013 | Oyster | 02/03/2016 | France/Atlantic | 5/8 | NA | 36 |
| C4019 | Oyster | 26/02/2016 | France/Atlantic | 4/4 | NA | 36 |
| C4020 | Oyster | 36 | ||||
| C4023 | Oyster | 10/03/2016 | France/Atlantic | 4/5 | NA | 36 |
| C4050 | Oyster | 04/04/2016 | France/Atlantic | 3/4 | NA | 34 |
| Strain | Year | GII. Genotype | Proportion of NoV Strain in Artificial Samples | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| VP1 | RdRp | A100 | A10 | B100 | B10 | C100 | C10 | D100 | D10 | ||
| S582 | 2016 | 17 | [P17] | 10% | 55% | 91% | - | ||||
| S587 | 2009 | 3 | [P21] | 10% | 5% | 1% | 20% | ||||
| S596 | 2017 | 1 | [P41/P33] * | 10% | 5% | 1% | 20% | ||||
| S597 | 2017 | 4_2012 a | [P4_2009] | 10% | 5% | 1% | 20% | ||||
| S598 | 2017 | 2 b | [P16] | 10% | 5% | 1% | 20% | ||||
| S570 | 2015 | 4_2012 a | [P31] c | 10% | 5% | 1% | - | ||||
| S555 | 2014 | 6 | [P31] c | 10% | 5% | 1% | - | ||||
| S514 | 2013 | 2 b | [P2] | 10% | 5% | 1% | 20% | ||||
| S510 | 2012 | 7 | [P7] | 10% | 5% | 1% | - | ||||
| S23 | 2007 | 12 | [PNA7] | 10% | 5% | 1% | - | ||||
| Total NoV GII concentration, gc/µl | Target | 100 | 10 | 100 | 10 | 100 | 10 | 100 | 10 | ||
| Measured | 149 | 9.08 | 122 | 16.3 | 66.2 | 18.5 | 90.6 | 41.8 | |||
| Strain | S570 | S597 | S598 | S514 | S510 | S287 | S555 | S582 | S596 | S23b | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| GII P-Type | P31 | P4 | P16 | P2 | P7 | P21 | P31 | P17 | P33/41 | PNA7 | |
| GII Genotype | 4_2012 | 4_2012 | 2 | 2 | 7 | 3 | 6 | 17 | 1 | 12 | |
| RdRp | Cluster1 | 72.80 | 76.92 | 73.84 | 79.81 | NA | 75.50 | 75.47 | 100.00 | 75.82 | 76.24 |
| Cluster2 | 83.20 | 83.76 | 79.14 | 83.65 | NA | 100.00 | 83.02 | 75.50 | 79.92 | 87.13 | |
| Cluster3 | 85.60 | 100.00 | 79.14 | 77.89 | NA | 82.78 | 87.74 | 75.50 | 78.28 | 78.22 | |
| Cluster4 | 79.20 | 80.34 | 77.15 | 79.81 | NA | 83.44 | 79.25 | 76.16 | 78.28 | 100.00 | |
| Cluster5 | 100.00 | 86.33 | 78.48 | 77.89 | NA | 82.78 | 100.00 | 72.85 | 78.28 | 85.15 | |
| Cluster6 | 79.20 | 81.20 | 77.82 | 75.00 | NA | 80.80 | 82.08 | 76.82 | 97.13 | 79.21 | |
| Cluster9 | 80.00 | 82.05 | 77.15 | 77.89 | NA | 79.47 | 83.02 | 77.48 | 91.39 | 81.19 | |
| Cluster10 | 82.40 | 82.91 | 78.48 | 78.85 | NA | 80.13 | 83.96 | 74.17 | 92.21 | 79.21 | |
| VP1 | Cluster1 | 74.17 | 76.38 | 100.00 | 98.09 | 77.76 | 79.86 | 77.18 | 78.09 | 79.88 | 79.58 |
| Cluster2 | 74.17 | 74.17 | 76.37 | 74.16 | 76.94 | 80.92 | 77.59 | 100.00 | 84.02 | 82.08 | |
| Cluster3 | 72.52 | 74.72 | 97.89 | 100.00 | 76.94 | 79.86 | 76.35 | 76.33 | 79.29 | 80.83 | |
| Cluster4 | 77.48 | 76.93 | 78.06 | 77.03 | 82.65 | 99.65 | 78.01 | 81.27 | 81.66 | 76.25 | |
| Cluster5 | 71.85 | 73.62 | 79.75 | 79.90 | 75.71 | 78.09 | 79.67 | 83.04 | 91.72 | 100.00 | |
| Cluster6 | 96.36 | 99.59 | 71.31 | 71.77 | 77.14 | 75.97 | 75.93 | 75.27 | 78.11 | 69.58 | |
| Cluster7 | 73.18 | 80.80 | 76.37 | 74.64 | 80.82 | 77.39 | 100.00 | 76.68 | 81.07 | 79.17 | |
| Cluster8 | 73.51 | 76.38 | 78.48 | 78.47 | 76.94 | 81.27 | 78.84 | 80.92 | 100.00 | 86.25 | |
| Cluster9 | 100.00 | 98.48 | 72.15 | 71.29 | 77.14 | 78.09 | 75.93 | 75.27 | 76.92 | 69.58 | |
| Gene | RdRp | VP1 | ||||
|---|---|---|---|---|---|---|
| Sample | Amplicon | P-types of Clones (n. of Clones) | P-types Identified by Deep Sequencing | Amplicon | Genotypes of Clones (n. of Clones) | Genogroups/Genotypes Identified by Deep Sequencing |
| C3988 | No | NA | NA | Yes | GII.17 (1), GII.13 (1) | GII.17, GII.4, GII.13 |
| C3994 | Yes | GII.P17 (6), GII.P21 (1) | GII.P17, GII.P4 | Yes | NA | GII.17, GII.1, GII.13 |
| C4006 | Yes | GII.P31 (4), GII.P21 (2), GII.P17 (1) | GII.P17, GII.P4, GII.P21, GII.P31, GII.P33 | Yes | GII.3 (2), GII.4 (2), GII.13 (1), GII.17 (1) | GII.17, GII.2, GII.3, GII.4, GII.6, GII.1, GII.13, GII.14, GIV |
| C4008 | Yes | GII.P17 (7) | GII.P17 | Yes | GII.17 (2) | GII.17 |
| C4009 | Yes | GII.P17 (5) | GII.P17 | No | NA | NA |
| C4013 | Yes | GII.P17 (7) | GII.P17, GII.P21 | Yes | GII.17 (3) | GII.17, GII.2, GII.4, GII.6 |
| C4019 | Yes | GII.P17 (6) | GII.P17 | Yes | GII.17 (4) | GII.17, GII.13, GII.6 |
| C4020 | Yes | GII.P17 (6) | GII.P17 | Yes | GII.17 (4) | GII.17, GII.4 |
| C4023 | Yes | GII.P17 (8) | GII.P17 | Yes | GII.17 (6) | GII.17, GII.13 |
| C4050 | Yes | GII.P17 (7) | - | Yes | GII.17 (5) | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desdouits, M.; Wacrenier, C.; Ollivier, J.; Schaeffer, J.; Le Guyader, F.S. A Targeted Metagenomics Approach to Study the Diversity of Norovirus GII in Shellfish Implicated in Outbreaks. Viruses 2020, 12, 978. https://0-doi-org.brum.beds.ac.uk/10.3390/v12090978
Desdouits M, Wacrenier C, Ollivier J, Schaeffer J, Le Guyader FS. A Targeted Metagenomics Approach to Study the Diversity of Norovirus GII in Shellfish Implicated in Outbreaks. Viruses. 2020; 12(9):978. https://0-doi-org.brum.beds.ac.uk/10.3390/v12090978
Chicago/Turabian StyleDesdouits, Marion, Candice Wacrenier, Joanna Ollivier, Julien Schaeffer, and Françoise S. Le Guyader. 2020. "A Targeted Metagenomics Approach to Study the Diversity of Norovirus GII in Shellfish Implicated in Outbreaks" Viruses 12, no. 9: 978. https://0-doi-org.brum.beds.ac.uk/10.3390/v12090978