
Genomic Characterization of a Novel Alphacoronavirus Isolated from Bats, Korea, 2020

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling, RNAExtraction and RT-PCR
2.2. Whole-Genome Sequencing, Genome Assembly, and Annotation
2.3. Sequence Alignment and Phylogenetic Construction
2.4. Potential Host Prediction
3. Results
3.1. Coronavirus Detection in Bat Samples
3.2. Whole-Genome Assembly and Annotation
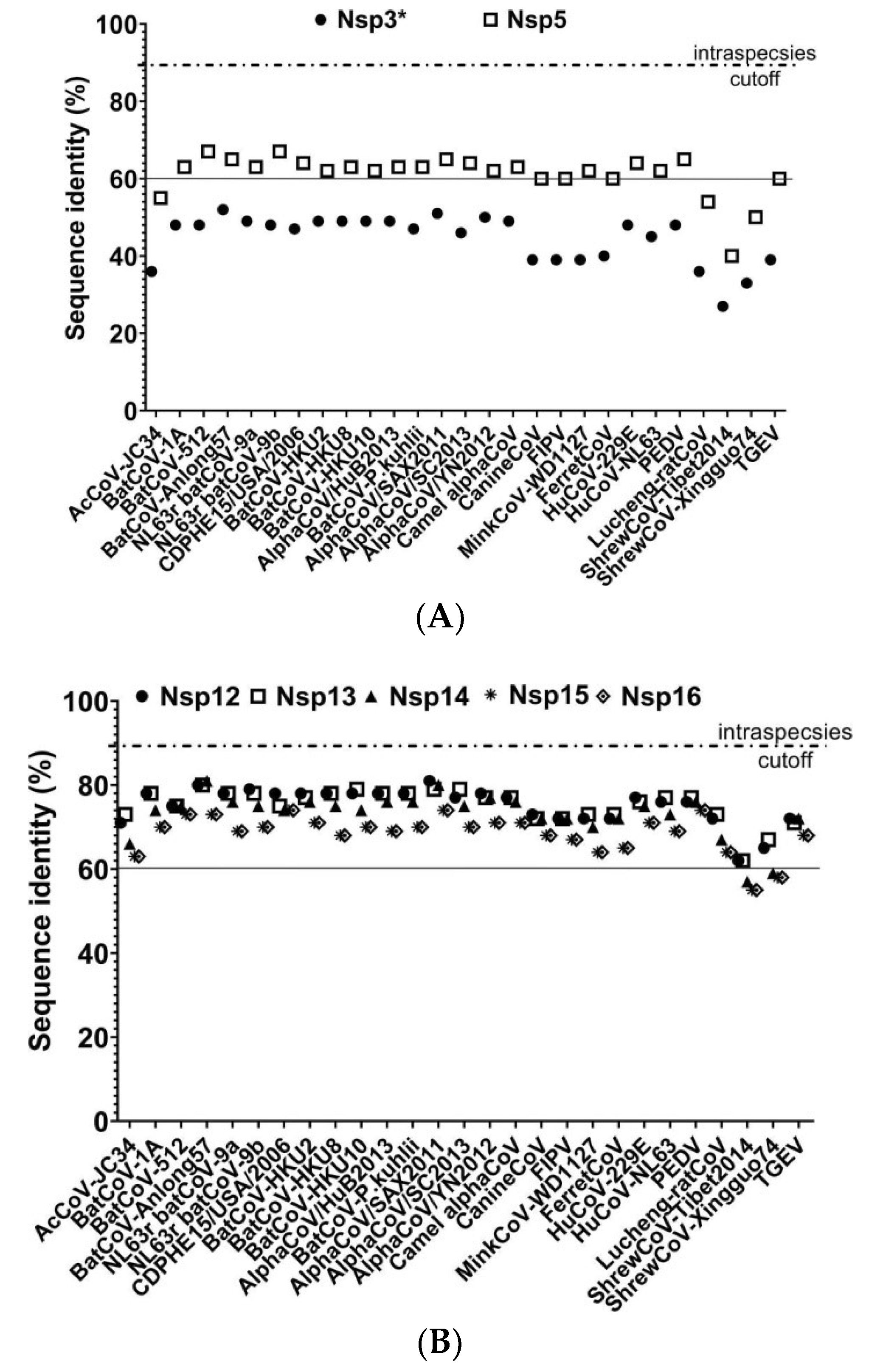
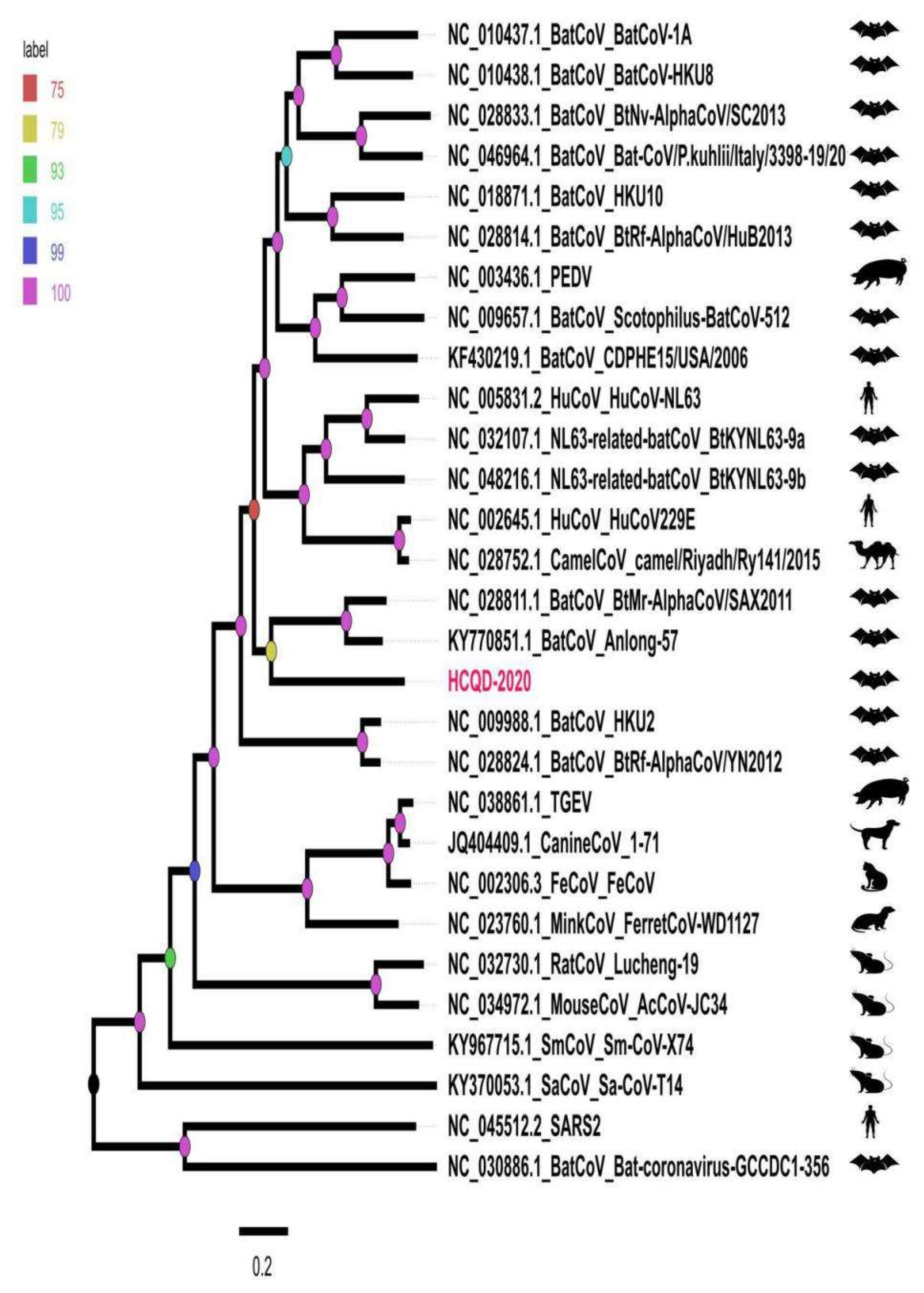
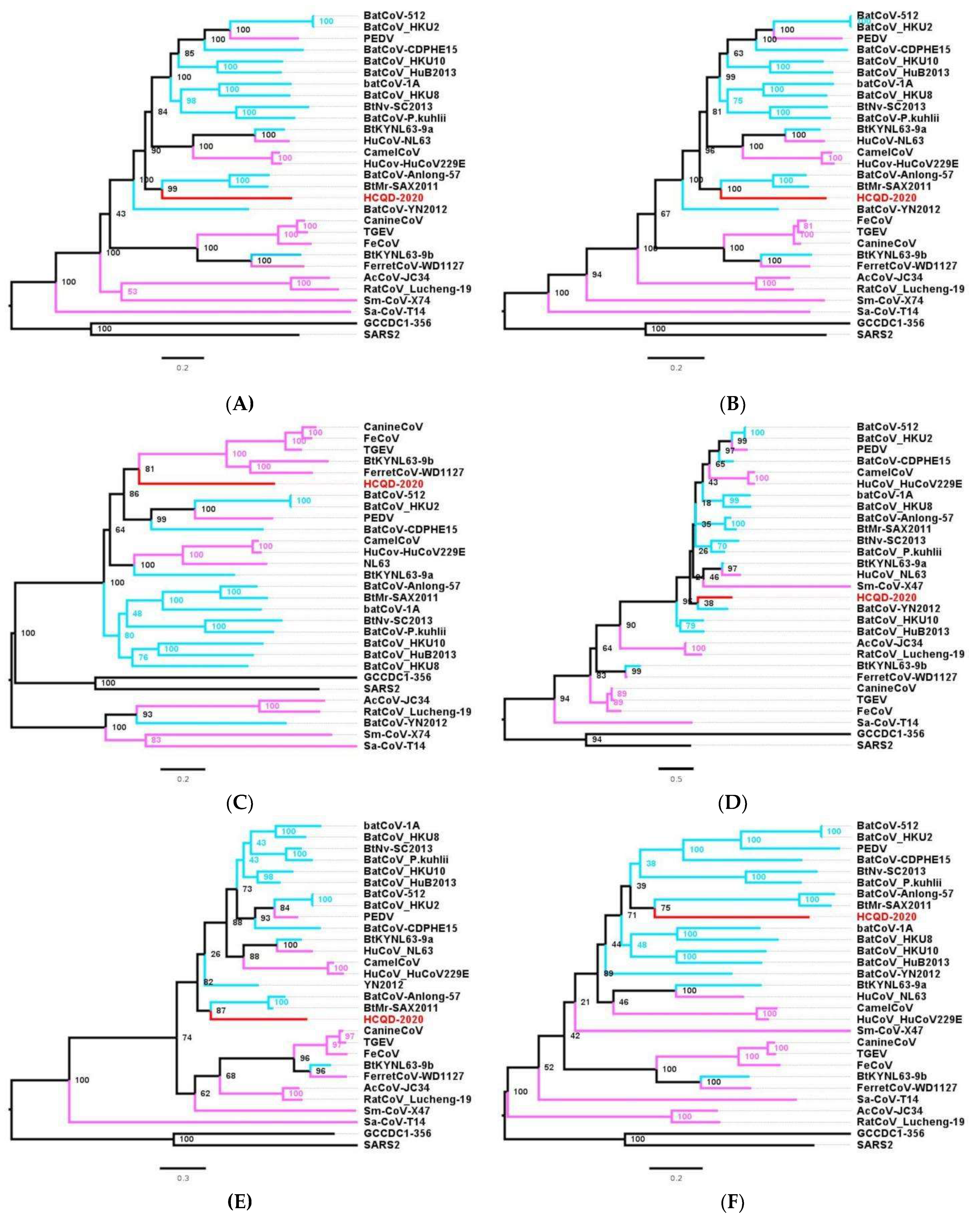
3.3. Phylogenetic Analysis Suggesting That HCQD-2020 Strain Might Be a Novel Species Belonging to the Alphacoronavirus Genus
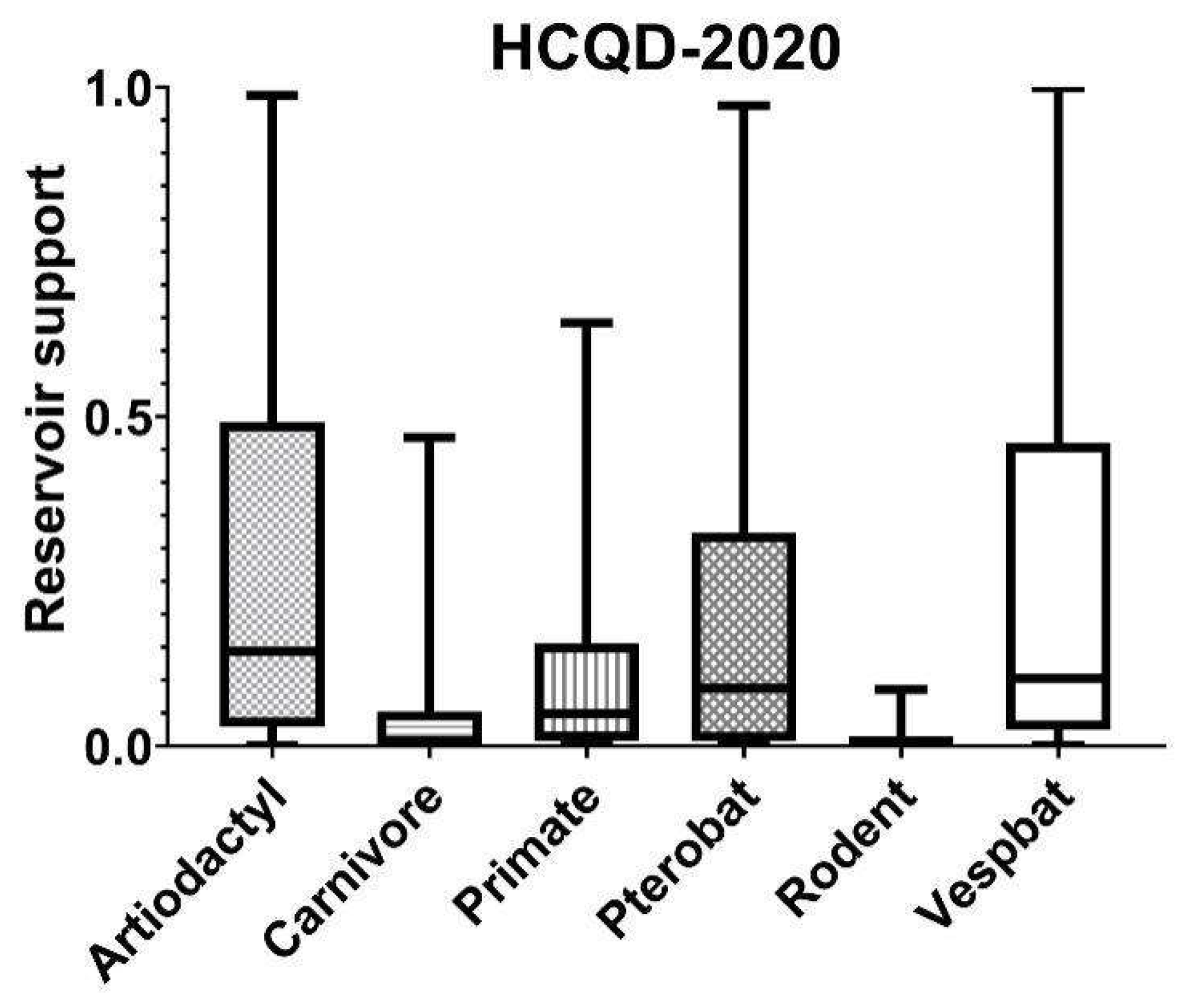
3.4. In Silico Cross-Species Infectious Ability Examination
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghai, R.R.; Carpenter, A.; Liew, A.Y.; Martin, K.B.; Herring, M.K.; Gerber, S.I.; Hall, A.J.; Sleeman, J.M.; VonDobschuetz, S.; Behravesh, C.B. Animal reservoirs and hosts for emerging alphacoronaviruses and betacoronaviruses. Emerg. Infect. Dis. 2021, 27, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.X.; Xie, Z.X.; Xie, L.J.; Deng, X.W.; Xie, Z.Q.; Luo, S.S.; Liu, J.B.; Pang, Y.S.; Khan, M.I. Simultaneous detection of eight swine reproductive and respiratory pathogens using a novel gexp analyser-based multiplex pcr assay. J. Virol. Methods 2015, 224, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Vlasova, A.N.; Wang, Q.; Jung, K.; Langel, S.N.; Malik, Y.S.; Saif, L.J. Porcine coronaviruses. Emerg. Transbound. Anim. Viruses 2020, 79–110. [Google Scholar] [CrossRef] [Green Version]
- Woo, P.C.; Lau, S.K.; Huang, Y.; Yuen, K.-Y. Coronavirus Diversity, Phylogeny and Interspecies Jumping. Exp. Biol. Med. 2009, 234, 1117–1127. [Google Scholar] [CrossRef] [Green Version]
- Anthony, S.J.; Johnson, C.K.; Greig, D.J.; Kramer, S.; Che, X.; Wells, H.; Hicks, A.L.; Joly, D.O.; Wolfe, N.D.; Daszak, P.; et al. Global patterns in coronavirus diversity. Virus Evol. 2017, 3, vex012. [Google Scholar] [CrossRef]
- Monchatre-Leroy, E.; Boué, F.; Boucher, J.M.; Renault, C.; Moutou, F.; Ar Gouilh, M.; Umhang, G. Identification of alpha and beta coronavirus in wildlife species in france: Bats, rodents, rabbits, and hedgehogs. Viruses 2017, 9, 364. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Lin, X.D.; Zhang, H.L.; Wang, M.R.; Guan, X.Q.; Holmes, E.C.; Zhang, Y.Z. Extensive genetic diversity and host range of rodent-borne coronaviruses. Virus Evol. 2020, 6, veaa078. [Google Scholar] [CrossRef]
- Smith, C.S.; de Jong, C.E.; Meers, J.; Henning, J.; Wang, L.F.; Field, H.E. Coronavirus infection and diversity in bats in the australasian region. EcoHealth 2016, 13, 72–82. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Lin, X.-D.; Guo, W.-P.; Zhou, R.-H.; Wang, M.-R.; Wang, C.-Q.; Ge, S.; Mei, S.-H.; Li, M.-H.; Shi, M.; et al. Discovery, diversity and evolution of novel coronaviruses sampled from rodents in china. Virology 2015, 474, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.D.; Wang, W.; Hao, Z.Y.; Wang, Z.X.; Guo, W.P.; Guan, X.Q.; Wang, M.R.; Wang, H.W.; Zhou, R.H.; Li, M.H.; et al. Extensive diversity of coronaviruses in bats from china. Virology 2017, 507, 1–10. [Google Scholar] [CrossRef]
- Wang, W.; Lin, X.D.; Liao, Y.; Guan, X.Q.; Guo, W.P.; Xing, J.G.; Holmes, E.C.; Zhang, Y.Z. Discovery of a highly divergent coronavirus in the asian house shrew from china illuminates the origin of the alphacoronaviruses. J. Virol. 2017, 91, e00764-17. [Google Scholar] [CrossRef] [Green Version]
- Woo, P.C.; Lau, S.K.; Li, K.S.; Poon, R.W.; Wong, B.H.; Tsoi, H.W.; Yip, B.C.; Huang, Y.; Chan, K.H.; Yuen, K.Y. Molecular diversity of coronaviruses in bats. Virology 2006, 351, 180–187. [Google Scholar] [CrossRef] [Green Version]
- So, R.T.Y.; Chu, D.K.W.; Miguel, E.; Perera, R.; Oladipo, J.O.; Fassi-Fihri, O.; Aylet, G.; Ko, R.L.W.; Zhou, Z.; Cheng, M.S.; et al. Diversity of Dromedary Camel Coronavirus HKU23 in African Camels Revealed Multiple Recombination Events among Closely Related Betacoronaviruses of the Subgenus Embecovirus. J. Virol. 2019, 93, e01236-19. [Google Scholar] [CrossRef] [Green Version]
- Borucki, M.K.; Allen, J.E.; Chen-Harris, H.; Zemla, A.; Vanier, G.; Mabery, S.; Torres, C.; Hullinger, P.; Slezak, T. The Role of Viral Population Diversity in Adaptation of Bovine Coronavirus to New Host Environments. PLoS ONE 2013, 8, e52752. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, L.; Ren, X.; He, G.; Zhang, J.; Yang, J.; Qian, Z.; Dong, J.; Sun, L.; Zhu, Y.; et al. Deciphering the bat virome catalog to better understand the ecological diversity of bat viruses and the bat origin of emerging infectious diseases. ISME J. 2016, 10, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Boni, M.F.; Lemey, P.; Jiang, X.; Lam, T.T.; Perry, B.W.; Castoe, T.A.; Rambaut, A.; Robertson, D.L. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat. Microbiol. 2020, 5, 1408–1417. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Lau, S.K.P.; Lam, C.S.F.; Lau, C.C.Y.; Tsang, A.K.L.; Lau, J.H.N.; Bai, R.; Teng, J.L.L.; Tsang, C.C.C.; Wang, M.; et al. Discovery of Seven Novel Mammalian and Avian Coronaviruses in the Genus Deltacoronavirus Supports Bat Coronaviruses as the Gene Source of Alphacoronavirus and Betacoronavirus and Avian Coronaviruses as the Gene Source of Gammacoronavirus and Deltacoronavirus. J. Virol. 2012, 86, 3995–4008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letko, M.; Seifert, S.N.; Olival, K.J.; Plowright, R.K.; Munster, V.J. Bat-borne virus diversity, spillover and emergence. Nat. Rev. Microbiol. 2020, 18, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Lo, V.T.; Yoon, S.W.; Noh, J.Y.; Kim, Y.; Choi, Y.G.; Jeong, D.G.; Kim, H.K. Long-term surveillance of bat coronaviruses in korea: Diversity and distribution pattern. Transbound Emerg. Dis. 2020, 67, 2839–2848. [Google Scholar] [CrossRef] [PubMed]
- Maganga, G.D.; Pinto, A.; Mombo, I.M.; Madjitobaye, M.; Mbeang Beyeme, A.M.; Boundenga, L.; Ar Gouilh, M.; N’Dilimabaka, N.; Drexler, J.F.; Drosten, C.; et al. Genetic diversity and ecology of coronaviruses hosted by cave-dwelling bats in gabon. Sci. Rep. 2020, 10, 7314. [Google Scholar] [CrossRef]
- Góes, L.G.B.; Campos, A.C.A.; Carvalho, C.; Ambar, G.; Queiroz, L.H.; Cruz-Neto, A.P.; Munir, M.; Durigon, E.L. Genetic diversity of bats coronaviruses in the atlantic forest hotspot biome, brazil. Infect. Genet. Evol. 2016, 44, 510–513. [Google Scholar] [CrossRef]
- Tang, X.C.; Zhang, J.X.; Zhang, S.Y.; Wang, P.; Fan, X.H.; Li, L.F.; Li, G.; Dong, B.Q.; Liu, W.; Cheung, C.L.; et al. Prevalence and Genetic Diversity of Coronaviruses in Bats from China. J. Virol. 2006, 80, 7481–7490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, P.C.; Lau, S.K.; Yuen, K.Y. Infectious diseases emerging from chinese wet-markets: Zoonotic origins of severe respiratory viral infections. Curr. Opin. Virol. 2006, 19, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Gloza-Rausch, F.; Ipsen, A.; Seebens, A.; Gottsche, M.; Panning, M.; Drexler, J.F.; Petersen, N.; Annan, A.; Grywna, K.; Muller, M.; et al. Detection and prevalence patterns of group I coronaviruses in bats, northern Germany. Emerg. Infect. Dis. 2008, 14, 626–631. [Google Scholar] [CrossRef]
- Lau, S.K.; Li, K.S.; Tsang, A.K.; Shek, C.T.; Wang, M.; Choi, G.K.; Guo, R.; Wong, B.H.; Poon, R.W.; Lam, C.S.; et al. Recent transmission of a novel alphacoronavirus, bat coronavirus hku10, from leschenault’s rousettes to pomona leaf-nosed bats: First evidence of interspecies transmission of coronavirus between bats of different suborders. J. Virol. 2012, 86, 11906–11918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Ji, J.; Chen, X.; Bi, Y.; Li, J.; Wang, Q.; Hu, T.; Song, H.; Zhao, R.; Chen, Y.; et al. Identification of novel bat coronaviruses sheds light on the evolutionary origins of sars-cov-2 and related viruses. Cell 2021, 184, 4380–4391.e4314. [Google Scholar] [CrossRef]
- Lee, S.; Jo, S.D.; Son, K.; An, I.; Jeong, J.; Wang, S.J.; Kim, Y.; Jheong, W.; Oem, J.K. Genetic characteristics of coronaviruses from korean bats in 2016. Microb. Ecol. 2018, 75, 174–182. [Google Scholar] [CrossRef]
- Poon, L.L.; Chu, D.K.; Chan, K.H.; Wong, O.K.; Ellis, T.M.; Leung, Y.H.; Lau, S.K.; Woo, P.C.; Suen, K.Y.; Yuen, K.Y.; et al. Identification of a novel coronavirus in bats. J. Virol. 2005, 79, 2001–2009. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. Spades: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Scotto–Lavino, E.; Du, G.; Frohman, M.A. 5′ end cdna amplification using classic race. Nat. Protoc. 2006, 1, 2555. [Google Scholar] [CrossRef]
- Chen, L.-L.; Ou, H.-Y.; Zhang, R.; Zhang, C.-T. Zcurve_cov: A new system to recognize protein coding genes in coronavirus genomes, and its applications in analyzing sars-cov genomes. Biochem. Biophys. Res. Commun. 2003, 307, 382–388. [Google Scholar] [CrossRef]
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridge, A.; Brown, S.D.; Chang, H.Y.; El-Gebali, S.; Fraser, M.I.; et al. Interpro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2019, 47, D351–D360. [Google Scholar] [CrossRef] [Green Version]
- Kalvari, I.; Nawrocki, E.P.; Ontiveros-Palacios, N.; Argasinska, J.; Lamkiewicz, K.; Marz, M.; Griffiths-Jones, S.; Toffano-Nioche, C.; Gautheret, D.; Weinberg, Z.; et al. Rfam 14: Expanded coverage of metagenomic, viral and microrna families. Nucleic Acids Res. 2021, 49, D192–D200. [Google Scholar] [CrossRef]
- Graham, R.L.; Baric, R.S. Recombination, Reservoirs, and the Modular Spike: Mechanisms of Coronavirus Cross-Species Transmission. J. Virol. 2010, 84, 3134–3146. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Babayan, S.A.; Orton, R.J.; Streicker, D.G. Predicting reservoir hosts and arthropod vectors from evolutionary signatures in rna virus genomes. Science 2018, 362, 577–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Family—Coronaviridae. In Virus Taxonomy; King, A.M.Q.; Adams, M.J.; Carstens, E.B.; Lefkowitz, E.J. (Eds.) Elsevier: San Diego, CA, USA, 2012; pp. 806–828. [Google Scholar]
- Tan, C.W.; Yang, X.; Anderson, D.E.; Wang, L.F. Bat virome research: The past, the present and the future. Curr. Opin. Virol. 2021, 49, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Halpin, K.; Young, P.L.; Field, H.E.; Mackenzie, J.S. Isolation of hendra virus from pteropid bats: A natural reservoir of hendra virus. J. Gen. Virol. 2000, 81, 1927–1932. [Google Scholar] [CrossRef] [PubMed]
- Yob, J.M.; Field, H.; Rashdi, A.M.; Morrissy, C.; van der Heide, B.; Rota, P.; bin Adzhar, A.; White, J.; Daniels, P.; Jamaluddin, A.; et al. Nipah virus infection in bats (order Chiroptera) in peninsular Malaysia. Emerg. Infect. Dis. 2001, 7, 439–441. [Google Scholar] [CrossRef]
- Li, H.; Mendelsohn, E.; Zong, C.; Zhang, W.; Hagan, E.; Wang, N.; Li, S.; Yan, H.; Huang, H.; Zhu, G.; et al. Human-animal interactions and bat coronavirus spillover potential among rural residents in Southern China. Biosaf Health 2019, 1, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Li, S.Y.; Yang, X.L.; Huang, H.M.; Zhang, Y.J.; Guo, H.; Luo, C.M.; Miller, M.; Zhu, G.; Chmura, A.A.; et al. Serological Evidence of Bat SARS-Related Coronavirus Infection in Humans, China. Virol. Sin. 2018, 33, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Bergner, L.M.; Orton, R.J.; Streicker, D.G. Complete Genome Sequence of an Alphacoronavirus from Common Vampire Bats in Peru. Microbiol. Resour. Announc. 2020, 9, e00742-20. [Google Scholar] [CrossRef] [PubMed]
- Lazov, C.M.; Belsham, G.J.; Botner, A.; Rasmussen, T.B. Full-Genome Sequences of Alphacoronaviruses and Astroviruses from Myotis and Pipistrelle Bats in Denmark. Viruses 2021, 13, 1073. [Google Scholar] [CrossRef]
- Prada, D.; Boyd, V.; Baker, M.L.; O’Dea, M.; Jackson, B. Viral Diversity of Microbats within the South West Botanical Province of Western Australia. Viruses Basel 2019, 11, 1157. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Fang, B.; Liu, Y.; Cai, M.; Jun, J.; Ma, J.; Bu, D.; Wang, L.; Zhou, P.; Wang, H.; et al. Newly emerged porcine enteric alphacoronavirus in southern China: Identification, origin and evolutionary history analysis. Infect. Genet. Evol. 2018, 62, 179–187. [Google Scholar] [CrossRef]
- Huang, Y.W.; Dickerman, A.W.; Pineyro, P.; Li, L.; Fang, L.; Kiehne, R.; Opriessnig, T.; Meng, X.J. Origin, evolution, and genotyping of emergent porcine epidemic diarrhea virus strains in the united states. mBio 2013, 4, e00737-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, L.; Li, J.; Zhou, Q.; Xu, Z.; Chen, L.; Zhang, Y.; Xue, C.; Wen, Z.; Cao, Y. A new bat-HKU2–like coronavirus in swine, China, 2017. Emerg. Infect. Dis. 2017, 23, 1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alagaili, A.N.; Briese, T.; Mishra, N.; Kapoor, V.; Sameroff, S.C.; Burbelo, P.D.; de Wit, E.; Munster, V.J.; Hensley, L.E.; Zalmout, I.S.; et al. Middle East respiratory syndrome coronavirus infection in dromedary camels in Saudi Arabia. mBio 2014, 5, e00884-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alshukairi, A.N.; Zheng, J.; Zhao, J.; Nehdi, A.; Baharoon, S.A.; Layqah, L.; Bokhari, A.; Al Johani, S.M.; Samman, N.; Boudjelal, M.; et al. High Prevalence of MERS-CoV Infection in Camel Workers in Saudi Arabia. mBio 2018, 9, e01985-18. [Google Scholar] [CrossRef] [Green Version]
- Sabir, J.S.M.; Lam, T.T.-Y.; Ahmed, M.M.M.; Li, L.; Shen, Y.; Abo-Aba, S.E.M.; Qureshi, M.I.; Abu-Zeid, M.; Zhang, Y.; Khiyami, M.A.; et al. Co-circulation of three camel coronavirus species and recombination of mers-covs in saudi arabia. Science 2016, 351, 81–84. [Google Scholar] [CrossRef] [Green Version]
- Vlasova, A.N.; Diaz, A.; Damtie, D.; Xiu, L.; Toh, T.H.; Lee, J.S.; Saif, L.J.; Gray, G.C. Novel Canine Coronavirus Isolated from a Hospitalized Pneumonia Patient, East Malaysia. Clin. Infect. Dis. 2021. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ORFs | Length (nt/aa) | TRS Location | TRS Sequence(s) (Distance to ATG) 1 |
|---|---|---|---|
| ORF1ab | 20,477/6824 | 36 | CCCCTCAACTAAACGAA(215)ATG |
| S | 4035/1344 | 20,731 | GTTTCAACCAAATGAAAAA |
| ORF3 | 675/224 | 24,723 | AGTCGAACTrAAACTCA(34)ATG |
| E | 246/81 | 25,342 | TATTGAACTAAGTGAC(61)ATG |
| M | 678/225 | 25,658 | TGTCTAACTAAATCAA(1)ATG |
| N | 1155/384 | 26,530 | TAATCAATTAAACAAA(4)ATG |
| ORF7 | 837/278 | 27,514 | ACTCAACTAAACATG |
| RNA Structural Elements | Position | Rfam | Note |
|---|---|---|---|
| 5′ UTR | 1–289 | RF03116 | |
| -1 frameshift element | 12,762–12,768 | Conservative heptamer TTTAAAC | |
| Frameshifting stimulation | 12,770–12,848 | RF00507 | |
| 3′ UTR | 28,408–28,753 | RF03121 |
| Nsp | First–Last Amino Acid Residues 1 | Protein Size | Cleavage Sequence | Putative Functional Domains |
|---|---|---|---|---|
| Nsp1 | M1-G282 | 282 | GNVEAG|DVVFTS | Unknown function, PFAM: PF19211 |
| Nsp2 | D283-G890 | 608 | FKRGGG|VTFGGD | Unknown function, PFAM: PF19212 |
| Nsp3 | V891–G2580 | 1690 | IVQKSG|SGPQFP | Papain-like protease, PFAM: PF08715 |
| Nsp4 | S2581–Q3058 | 478 | SSLQ|AGLR | Membrane spanning domain, PFAM: PF16348 |
| Nsp5 | A3059-Q3360 | 302 | VTLQ|GGRK | 3C-like protease, PFAM: PF05409 |
| Nsp6 | G3361–Q36439 | 279 | SSVQ|SKLT | Membrane spanning protein, PFAM: PF19213 |
| Nsp7 | S3640–Q3722 | 83 | AMLQ|SIAS | RNA replicate protein complex, PFAM: PF08716 |
| Nsp8 | S3723–Q3917 | 195 | VKLQ|NNEV | Transferase activity, PFAM: PF08717 |
| Nsp9 | N3918–Q4026 | 109 | IRLQ|AGKQ | Single strain RNA binding protein, PFAM: PF08710 |
| Nsp10 | A4027–Q4161 | 135 | ANVQ|SFDQ | Nucleic-binding protein, PFAM: PF09401 |
| Nsp11 | A4162-D4178 | 17 | - | Short peptide in C-terminate of ORF1a |
| Nsp12 | A4162-Q5062 | 901 | TVLQ|ASGM | RNA-depend RNA polymerase, PFAM: PF06478 |
| Nsp13 | A5063–Q5659 | 597 | TDLQ|ATEG | Helicase, Interpro: IRP027351 |
| Nsp14 | A5660–Q6178 | 519 | TKIQ|GLEN | Exoribonuclease and Guanine-N7 methyltransferase, Interpro: IPR009466 |
| Nsp15 | G6179-Q6526 | 347 | PQLQ|SAEW | EndoU-like endoribonuclease, PFAM: PF19215 |
| Nsp16 | S6526–K6825 | 300 | - | O-methyltransferase, PFAM: PF06460 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Do, H.-Q.; Nguyen, V.-G.; Chung, C.-U.; Jeon, Y.-S.; Shin, S.; Jang, K.-C.; Pham, L.B.H.; Kong, A.; Kim, C.-U.; Park, Y.-H.; et al. Genomic Characterization of a Novel Alphacoronavirus Isolated from Bats, Korea, 2020. Viruses 2021, 13, 2041. https://0-doi-org.brum.beds.ac.uk/10.3390/v13102041
Do H-Q, Nguyen V-G, Chung C-U, Jeon Y-S, Shin S, Jang K-C, Pham LBH, Kong A, Kim C-U, Park Y-H, et al. Genomic Characterization of a Novel Alphacoronavirus Isolated from Bats, Korea, 2020. Viruses. 2021; 13(10):2041. https://0-doi-org.brum.beds.ac.uk/10.3390/v13102041
Chicago/Turabian StyleDo, Hai-Quynh, Van-Giap Nguyen, Chul-Un Chung, Yong-Shin Jeon, Sook Shin, Kuem-Chan Jang, Le Bich Hang Pham, Aeri Kong, Cheong-Ung Kim, Yong-Ho Park, and et al. 2021. "Genomic Characterization of a Novel Alphacoronavirus Isolated from Bats, Korea, 2020" Viruses 13, no. 10: 2041. https://0-doi-org.brum.beds.ac.uk/10.3390/v13102041