
Towards a Precision Medicine Approach and In Situ Vaccination against Prostate Cancer by PSMA-Retargeted oHSV

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Antibodies
2.2. Viruses
2.3. Generation of Murine and Human Cancer Cells Expressing Human-PSMA
2.4. R-405 Tropism and Neutralization of Infection by MAbs to PSMA and HSV-1
2.5. Virus Growth and Cytotoxicity Assay
2.6. In Vivo Experiments
2.7. Determination of Tumor Microenvironment Infiltrating Cells
2.8. Determination of Intratumoral Cytokine and Immune Factors by RT-PCR
2.9. Detection of Splenocyte Reactivity to Cancer Cells
2.10. Detection of Serum Antibodies to Cancer Cells
3. Results
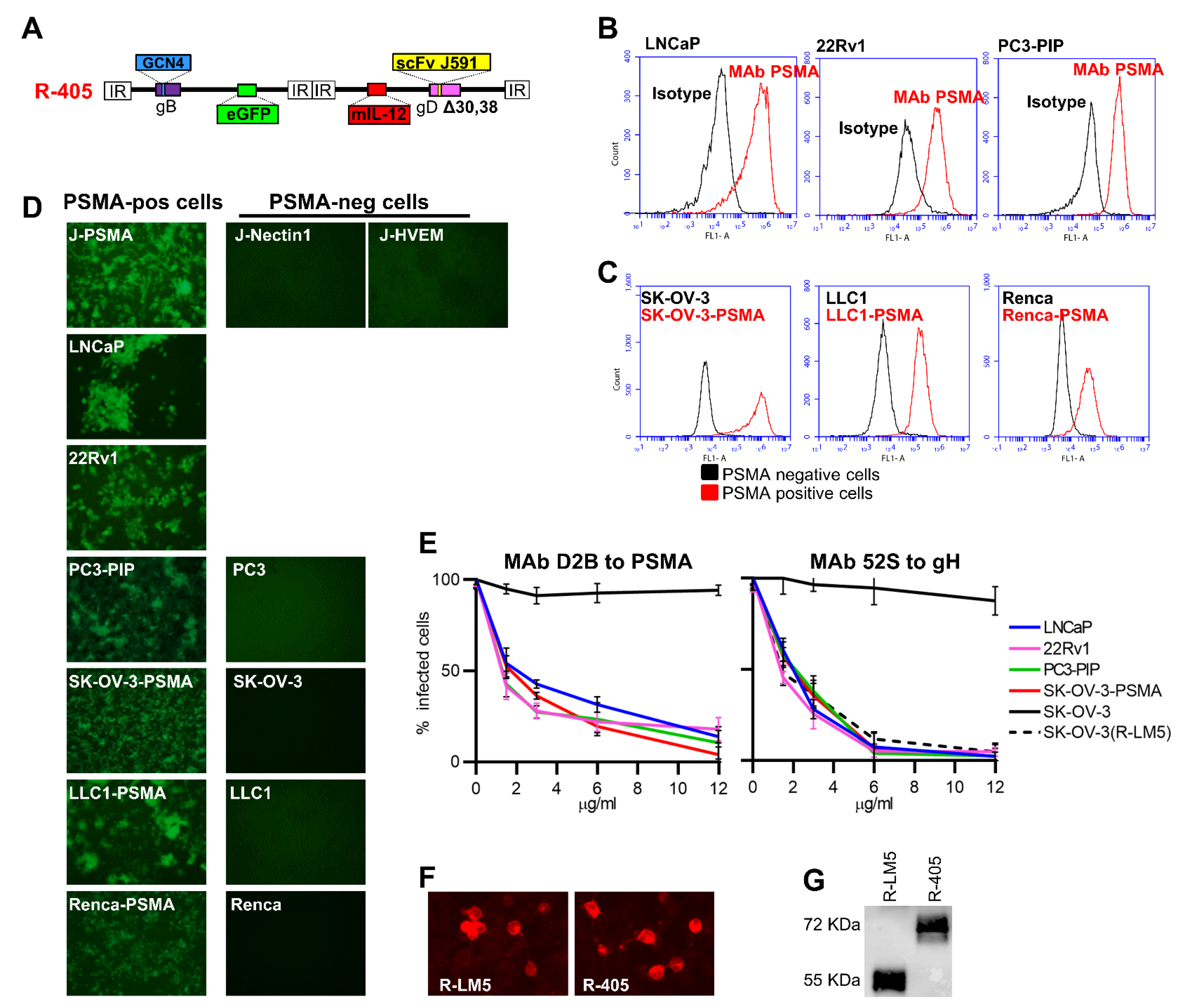
3.1. Genetic Engineering of R-405 and Generation of Human and Murine Cell Lines Expressing Human PSMA
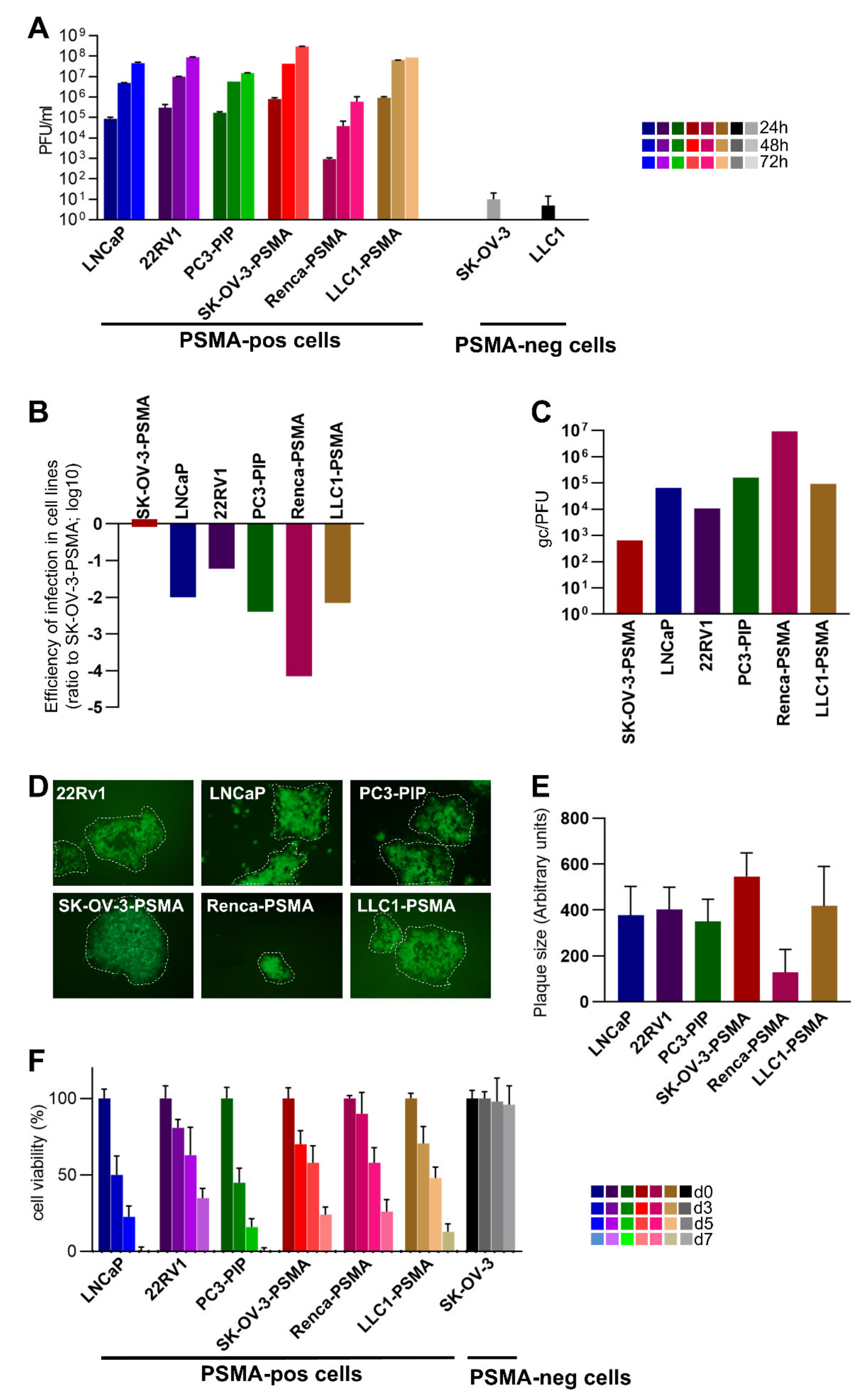
3.2. Infectivity, Cell-to-Cell Spread, and Cytotoxicity Caused by R-405 in Different Cell Lines
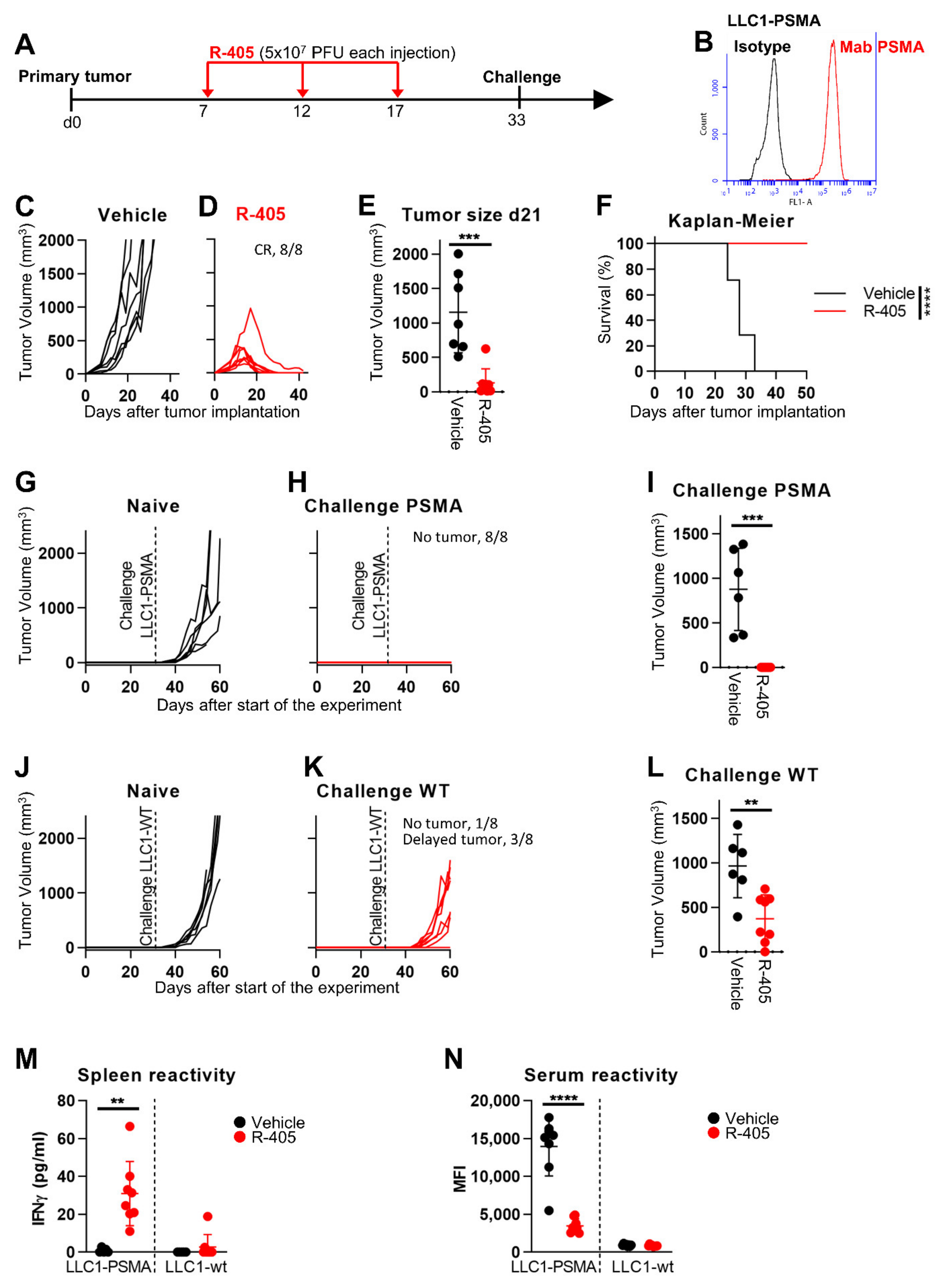
3.3. In Vivo Efficacy of R-405 Monotherapy against LLC1-PSMA Tumors
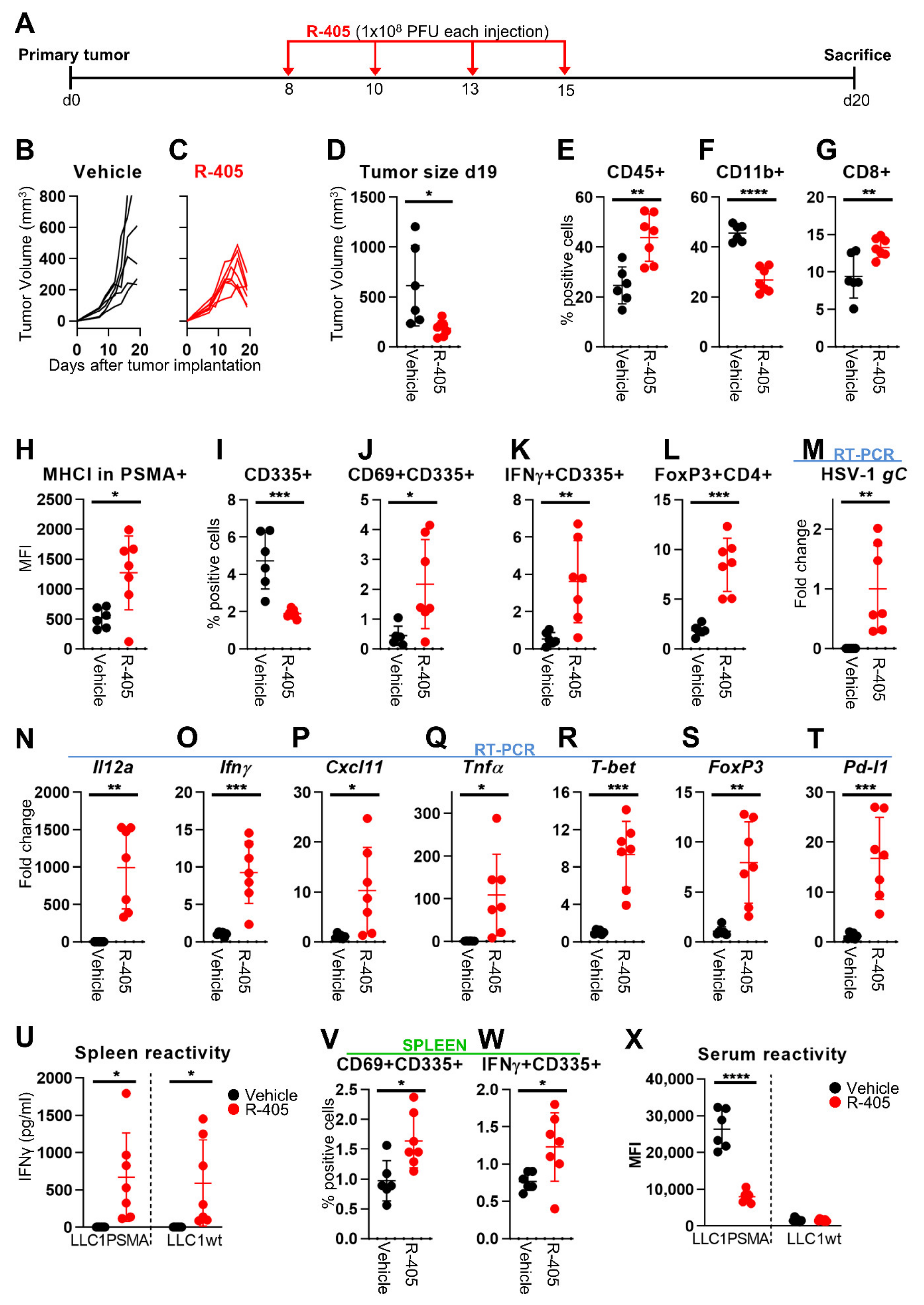
3.4. Changes to the LLC1-PSMA Tumor Microenvironment Induced by R-405
3.5. R-405 and Anti-PD-1 Combination Therapy against LLC1-PSMA Tumors
3.6. R-405 Monotherapy and Combination Therapy against Renca-PSMA Tumors
4. Discussion
- (i)
- Previous attempts to generate OVs specific for PC cells have included conditionally replicating transcriptionally retargeted adenoviruses (AdVs) carrying key viral genes under prostate specific antigen (PSA) or PSMA enhancer elements [62,63], an alphavirus encoding PSMA and acting as a vaccine [64], a vesicular stomatitis virus (VSV) pseudotyped with measles virus (MeV) glycoprotein, including a PSMA-retargeted MeV glycoprotein [65], and an MeV genetically retargeted to PSMA by insertion of scFv in the hemagglutinin gene [66]. The latter virus demonstrated in vivo efficacy against xenotransplants of LNCaP and 22Rv1 human PC cells [66]. Its properties against immunocompetent PSMA-positive murine models and the extent of in vivo off-tumor infections were not reported. The PSMA-MeV had not been additionally investigated in the past 10 years. Hence, how effective it is in eliciting anti-tumor in situ vaccination and in sensitizing to ICIs is not known. AdVs and Reolysin are being tested against PC in ongoing phase 1 or 2 clinical trials, without or in combination with ICIs.
- (ii)
- High PSMA expression is frequent (75.21%) in metastatic castration-resistant prostate cancer patients and portends a poor prognosis [31,32,33]. Anti-PSMA antibodies are utilized for diagnostic or combined diagnostic + therapeutic (theranostic) applications, including PSMA-SPECT and the delivery of radiometals. Most of the PSMA-based clinical trials utilize anti-PSMA as a diagnostic or theranostic tool. Of note, a recently closed phase 3 clinical trial regarding the effect of Lutetium-177–PSMA-617 prolonged progression-free survival and overall survival when added to standard care in patients with advanced PSMA-positive metastatic castration-resistant prostate cancer [32]. Notably, a wealth of data convincingly indicated that targeting PSMA results in low toxicity and suggested few off-tumor effects [34,35].
- (iii)
- The importance of PSMA as a target for precision medicine is strengthened by investigations regarding PSMA-targeted chemical antigen receptor T cells (CAR-Ts) [67]. Clinical trials are ongoing. Despite many efforts and rapid developments, the field of CAR-Ts regarding solid tumors still faces difficulties which need to be overcome. Oncolytic viruses, with their ability to attract lymphocytes to TME and to activate them, coupled with high tolerability, could be interesting partners for CAR-Ts.
- (iv)
- Very few trials address anti-PC vaccination. Sipuleucel-T is a dendritic cell (DC)-based vaccine recommended as first-line therapy for the treatment of metastatic castration-resistant prostate cancer [68]. It must be prepared individually for each patient, starting from the patient’s own blood [5,6].
5. Patents
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- WHO—Latest Global Cancer Data: Cancer Burden Rises to 18.1 Million New Cases and 9.6 Million Cancer Deaths in 2018. Available online: https://www.who.int/cancer/PRGlobocanFinal.pdf (accessed on 12 September 2018).
- The Global Cancer Observatory—All Cancers. Available online: https://gco.iarc.fr/today/data/factsheets/cancers/39-All-cancers-fact-sheet.pdf (accessed on 1 December 2020).
- Cooperberg, M.R.; Moul, J.W.; Carroll, P.R. The changing face of prostate cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 8146–8151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goswami, S.; Aparicio, A.; Subudhi, S.K. Immune checkpoint therapies in prostate cancer. Cancer J. 2016, 22, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheever, M.A.; Higano, C.S. PROVENGE (Sipuleucel-T) in prostate cancer: The first FDA-approved therapeutic cancer vaccine. Clin. Cancer Res. 2011, 17, 3520–3526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutherland, S.I.; Ju, X.; Horvath, L.; Clark, G.J. Moving on From Sipuleucel-T: New Dendritic Cell Vaccine Strategies for Prostate Cancer. Front. Immunol. 2021, 12, 562. [Google Scholar] [CrossRef]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichty, B.D.; Breitbach, C.J.; Stojdl, D.F.; Bell, J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 559–567. [Google Scholar] [CrossRef]
- Gujar, S.; Bell, J.; Diallo, J.-S. SnapShot: Cancer immunotherapy with oncolytic viruses. Cell 2019, 176, 1240–1240.e1241. [Google Scholar] [CrossRef] [PubMed]
- Hunter, W.D.; Martuza, R.L.; Feigenbaum, F.; Todo, T.; Mineta, T.; Yazaki, T.; Toda, M.; Newsome, J.T.; Platenberg, R.C.; Manz, H.J.; et al. Attenuated, replication-competent herpes simplex virus type 1 mutant G207: Safety evaluation of intracerebral injection in nonhuman primates. J. Virol. 1999, 73, 6319–6326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, J.; Kern, E.R.; Whitley, R.J.; Roizman, B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 1990, 250, 1262–1266. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Lee, P.; Gujar, S. Potentiating prostate cancer immunotherapy with oncolytic viruses. Nat. Rev. Urol. 2018, 15, 235–250. [Google Scholar] [CrossRef]
- Menotti, L.; Nicoletti, G.; Gatta, V.; Croci, S.; Landuzzi, L.; De Giovanni, C.; Nanni, P.; Lollini, P.L.; Campadelli-Fiume, G. Inhibition of human tumor growth in mice by an oncolytic herpes simplex virus designed to target solely HER-2-positive cells. Proc. Natl. Acad. Sci. USA 2009, 106, 9039–9044. [Google Scholar] [CrossRef] [Green Version]
- Menotti, L.; Cerretani, A.; Hengel, H.; Campadelli-Fiume, G. Construction of a fully retargeted herpes simplex virus 1 recombinant capable of entering cells solely via human epidermal growth factor receptor 2. J. Virol. 2008, 20, 10153–10161. [Google Scholar] [CrossRef] [Green Version]
- Nanni, P.; Nicoletti, G.; Landuzzi, L.; Croci, S.; Murgo, A.; Palladini, A.; Antognoli, A.; Ianzano, M.L.; Stivani, V.; Grosso, V.; et al. High metastatic efficiency of human sarcoma cells in Rag2/gammac double knockout mice provides a powerful test system for antimetastatic targeted therapy. Eur. J. Cancer 2010, 46, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Nanni, P.; Gatta, V.; Menotti, L.; De Giovanni, C.; Ianzano, M.; Palladini, A.; Grosso, V.; Dall’ora, M.; Croci, S.; Nicoletti, G.; et al. Preclinical Therapy of Disseminated HER-2(+) Ovarian and Breast Carcinomas with a HER-2-Retargeted Oncolytic Herpesvirus. PLoS Pathog. 2013, 9, e1003155. [Google Scholar] [CrossRef] [Green Version]
- Campadelli-Fiume, G.; De Giovanni, C.; Gatta, V.; Nanni, P.; Lollini, P.L.; Menotti, L. Rethinking herpes simplex virus: The way to oncolytic agents. Rev. Med. Virol. 2011, 21, 213–226. [Google Scholar] [CrossRef]
- Leoni, V.; Gatta, V.; Casiraghi, C.; Nicosia, A.; Petrovic, B.; Campadelli-Fiume, G. A Strategy for Cultivation of Retargeted Oncolytic Herpes Simplex Viruses in Non-cancer Cells. J. Virol. 2017, 91, e00067-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leoni, V.; Vannini, A.; Gatta, V.; Rambaldi, J.; Sanapo, M.; Barboni, C.; Zaghini, A.; Nanni, P.; Lollini, P.L.; Casiraghi, C.; et al. A fully-virulent retargeted oncolytic HSV armed with IL-12 elicits local immunity and vaccine therapy towards distant tumors. PLoS Pathog. 2018, 14, e1007209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrovic, B.; Gianni, T.; Gatta, V.; Campadelli-Fiume, G. Insertion of a ligand to HER2 in gB retargets HSV tropism and obviates the need for activation of the other entry glycoproteins. PLoS Pathog. 2017, 13, e1006352. [Google Scholar] [CrossRef]
- Gatta, V.; Petrovic, B.; Campadelli-Fiume, G. The Engineering of a Novel Ligand in gH Confers to HSV an Expanded Tropism Independent of gD Activation by Its Receptors. PLoS Pathog. 2015, 11, e1004907. [Google Scholar] [CrossRef] [Green Version]
- De Lucia, M.; Cotugno, G.; Bignone, V.; Garzia, I.; Nocchi, L.; Langone, F.; Petrovic, B.; Sasso, E.; Pepe, S.; Froechlich, G.; et al. Retargeted and multi-cytokine armed herpes virus is a potent cancer endovaccine for local and systemic anti-tumor treatment in combination with anti-PD1. Mol. Ther.-Oncolytics 2020, 19, 253–264. [Google Scholar] [CrossRef]
- Vannini, A.; Leoni, V.; Sanapo, M.; Gianni, T.; Giordani, G.; Gatta, V.; Barboni, C.; Zaghini, A.; Campadelli-Fiume, G. Immunotherapeutic Efficacy of Retargeted oHSVs Designed for Propagation in an Ad Hoc Cell Line. Cancers 2021, 13, 266. [Google Scholar] [CrossRef]
- Menotti, L.; Avitabile, E. Herpes simplex virus oncolytic immunovirotherapy: The blossoming branch of multimodal therapy. Int. J. Mol. Sci. 2020, 21, 8310. [Google Scholar] [CrossRef]
- Leoni, V.; Gatta, V.; Palladini, A.; Nicoletti, G.; Ranieri, D.; Dall’Ora, M.; Grosso, V.; Rossi, M.; Alviano, F.; Bonsi, L.; et al. Systemic delivery of HER2-retargeted oncolytic-HSV by mesenchymal stromal cells protects from lung and brain metastases. Oncotarget 2015, 6, 34774. [Google Scholar] [CrossRef] [Green Version]
- Gambini, E.; Reisoli, E.; Appolloni, I.; Gatta, V.; Campadelli-Fiume, G.; Menotti, L.; Malatesta, P. Replication-competent herpes simplex virus retargeted to HER2 as therapy for high-grade glioma. Mol. Ther. 2012, 20, 994–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannini, A.; Leoni, V.; Campadelli-Fiume, G. Targeted Delivery of IL-12 adjuvants immunotherapy by oncolytic viruses. Adv. Exp. Med. Biol. 2021, 1290, 67–80. [Google Scholar] [PubMed]
- Kiess, A.P.; Banerjee, S.R.; Mease, R.C.; Rowe, S.P.; Rao, A.; Foss, C.A.; Chen, Y.; Yang, X.; Cho, S.Y.; Nimmagadda, S.; et al. Prostate-specific membrane antigen as a target for cancer imaging and therapy. Q. J. Nucl. Med. Mol. Imaging 2015, 59, 241–268. [Google Scholar] [PubMed]
- Van de Wiele, C.; Sathekge, M.; De Spiegeleer, B.; De Jonghe, P.J.; Debruyne, P.R.; Borms, M.; Beels, L.; Maes, A. PSMA expression on neovasculature of solid tumors. Histol. Histopathol. 2020, 35, 919–927. [Google Scholar]
- Vlachostergios, P.J.; Niaz, M.J.; Sun, M.; Mosallaie, S.A.; Thomas, C.; Christos, P.J.; Osborne, J.R.; Molina, A.M.; Nanus, D.M.; Bander, N.H. Prostate-Specific Membrane Antigen Uptake and Survival in Metastatic Castration-Resistant Prostate Cancer. Front. Oncol. 2021, 11, 4. [Google Scholar] [CrossRef]
- Sartor, O.; de Bono, J.; Chi, K.N.; Fizazi, K.; Herrmann, K.; Rahbar, K.; Tagawa, S.T.; Nordquist, L.T.; Vaishampayan, N.; El-Haddad, G. Lutetium-177–PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2021, 385, 1091–1103. [Google Scholar] [CrossRef]
- Seifert, R.; Seitzer, K.; Herrmann, K.; Kessel, K.; Schäfers, M.; Kleesiek, J.; Weckesser, M.; Boegemann, M.; Rahbar, K. Analysis of PSMA expression and outcome in patients with advanced Prostate Cancer receiving 177Lu-PSMA-617 Radioligand Therapy. Theranostics 2020, 10, 7812. [Google Scholar] [CrossRef] [PubMed]
- Donin, N.M.; Reiter, R.E. Why targeting PSMA is a game changer in the management of prostate cancer. J. Nucl. Med. 2018, 59, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Heidegger, I.; Brandt, M.P.; Heck, M.M. Treatment of non-metastatic castration resistant prostate cancer in 2020: What is the best? Urol. Oncol. 2020, 38, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Cocchi, F.; Menotti, L.; Mirandola, P.; Lopez, M.; Campadelli-Fiume, G. The ectodomain of a novel member of the immunoglobulin subfamily related to the poliovirus receptor has the attributes of a bona fide receptor for herpes simplex virus types 1 and 2 in human cells. J. Virol. 1998, 72, 9992–10002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menotti, L.; Avitabile, E.; Gatta, V.; Petrovic, B.; Campadelli-Fiume, G. HSV as a platform for the generation of retargeted, armed, and reporter-expressing oncolytic viruses. Viruses 2018, 10, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.; Wang, X.; Klein, E.; Heston, W.D. Novel role of prostate-specific membrane antigen in suppressing prostate cancer invasiveness. Cancer Res. 2005, 65, 727–731. [Google Scholar]
- Lütje, S.; van Rij, C.M.; Franssen, G.M.; Fracasso, G.; Helfrich, W.; Eek, A.; Oyen, W.J.; Colombatti, M.; Boerman, O.C. Targeting human prostate cancer with 111In-labeled D2B IgG, F (ab′) 2 and Fab fragments in nude mice with PSMA-expressing xenografts. Contrast Media Mol. Imaging 2015, 10, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Showalter, S.D.; Zweig, M.; Hampar, B. Monoclonal antibodies to herpes simplex virus type 1 proteins, including the immediate-early protein ICP 4. Infect. Immun. 1981, 34, 684–692. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Moy, P.; Kim, S.; Xia, Y.; Rajasekaran, A.; Navarro, V.; Knudsen, B.; Bander, N.H. Monoclonal antibodies to the extracellular domain of prostate-specific membrane antigen also react with tumor vascular endothelium. Cancer Res. 1997, 57, 3629–3634. [Google Scholar]
- Vannini, A.; Petrovic, B.; Gatta, V.; Leoni, V.; Pepe, S.; Menotti, L.; Campadelli-Fiume, G.; Gianni, T. Rescue, Purification, and Characterization of a Recombinant HSV Expressing a Transgenic Protein. In Herpes Simplex Virus; Springer: Berlin/Heidelberg, Germany, 2020; pp. 153–168. [Google Scholar]
- Menotti, L.; Leoni, V.; Gatta, V.; Petrovic, B.; Vannini, A.; Pepe, S.; Gianni, T.; Campadelli-Fiume, G. oHSV Genome Editing by Means of galK Recombineering. In Herpes Simplex Virus; Springer: Berlin/Heidelberg, Germany, 2020; pp. 131–151. [Google Scholar]
- Gianni, T.; Leoni, V.; Chesnokova, L.S.; Hutt-Fletcher, L.M.; Campadelli-Fiume, G. alphavbeta3-integrin is a major sensor and activator of innate immunity to herpes simplex virus-1. Proc. Natl. Acad. Sci. USA 2012, 109, 19792–19797. [Google Scholar] [CrossRef] [Green Version]
- Petrovic, B.; Leoni, V.; Gatta, V.; Zaghini, A.; Vannini, A.; Campadelli-Fiume, G. Dual Ligand Insertion in gB and gD of Oncolytic Herpes Simplex Viruses for Retargeting to a Producer Vero Cell Line and to Cancer Cells. J. Virol. 2018, 92, e02122-17. [Google Scholar] [CrossRef] [Green Version]
- Lieschke, G.J.; Rao, P.K.; Gately, M.K.; Mulligan, R.C. Bioactive murine and human interleukin-12 fusion proteins which retain antitumor activity in vivo. Nat. Biotechnol. 1997, 15, 35–40. [Google Scholar] [CrossRef]
- Veinalde, R.; Grossardt, C.; Hartmann, L.; Bourgeois-Daigneault, M.-C.; Bell, J.C.; Jäger, D.; von Kalle, C.; Ungerechts, G.; Engeland, C.E. Oncolytic measles virus encoding interleukin-12 mediates potent antitumor effects through T cell activation. Oncoimmunology 2017, 6, e1285992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horoszewicz, J.S.; Leong, S.S.; Kawinski, E.; Karr, J.P.; Rosenthal, H.; Chu, T.M.; Mirand, E.A.; Murphy, G.P. LNCaP model of human prostatic carcinoma. Cancer Res. 1983, 43, 1809–1818. [Google Scholar] [PubMed]
- Korenchuk, S.; Lehr, J.; MClean, L.; Lee, Y.; Whitney, S.; Vessella, R.; Lin, D.; Pienta, K. VCaP, a cell-based model system of human prostate cancer. Vivo 2001, 15, 163–168. [Google Scholar]
- Kaighn, M.; Narayan, K.S.; Ohnuki, Y.; Lechner, J.F.; Jones, L. Establishment and characterization of a human prostatic carcinoma cell line (PC-3). Investig. Urol. 1979, 17, 16–23. [Google Scholar]
- Lechner, M.G.; Karimi, S.S.; Barry-Holson, K.; Angell, T.E.; Murphy, K.A.; Church, C.H.; Ohlfest, J.R.; Hu, P.; Epstein, A.L. Immunogenicity of murine solid tumor models as a defining feature of in vivo behavior and response to immunotherapy. J. Immunother. 2013, 36, 477–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.W.; Bhattacharya, S.; Yanamandra, N.; Kilian, D.; Shi, H.; Yadavilli, S.; Katlinskaya, Y.; Kaczynski, H.; Conner, M.; Benson, W. Tumor-immune profiling of murine syngeneic tumor models as a framework to guide mechanistic studies and predict therapy response in distinct tumor microenvironments. PLoS ONE 2018, 13, e0206223. [Google Scholar] [CrossRef] [PubMed]
- Mosely, S.I.; Prime, J.E.; Sainson, R.C.; Koopmann, J.-O.; Wang, D.Y.; Greenawalt, D.M.; Ahdesmaki, M.J.; Leyland, R.; Mullins, S.; Pacelli, L. Rational selection of syngeneic preclinical tumor models for immunotherapeutic drug discovery. Cancer Immunol. Res. 2017, 5, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Gianni, T.; Leoni, V.; Sanapo, M.; Parenti, F.; Bressanin, D.; Barboni, C.; Zaghini, A.; Campadelli-Fiume, G.; Vannini, A. Genotype of Immunologically Hot or Cold Tumors Determines the Antitumor Immune Response and Efficacy by Fully Virulent Retargeted oHSV. Viruses 2021, 13, 1747. [Google Scholar] [CrossRef]
- Hammerich, L.; Binder, A.; Brody, J.D. In situ vaccination: Cancer immunotherapy both personalized and off-the-shelf. Mol. Oncol. 2015, 9, 1966–1981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, H.L.; Kim, D.W.; DeRaffele, G.; Mitcham, J.; Coffin, R.S.; Kim-Schulze, S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann. Surg. Oncol. 2010, 17, 718–730. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, D.L.; Liu, Z.; Sathaiah, M.; Ravindranathan, R.; Guo, Z.; He, Y.; Guo, Z.S. Oncolytic viruses as therapeutic cancer vaccines. Mol. Cancer 2013, 12, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamarin, D.; Holmgaard, R.B.; Ricca, J.; Plitt, T.; Palese, P.; Sharma, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Intratumoral modulation of the inducible co-stimulator ICOS by recombinant oncolytic virus promotes systemic anti-tumour immunity. Nat. Commun. 2017, 8, 14340. [Google Scholar] [CrossRef] [PubMed]
- Motoshima, T.; Komohara, Y.; Horlad, H.; Takeuchi, A.; Maeda, Y.; Tanoue, K.; Kawano, Y.; Harada, M.; Takeya, M.; Eto, M. Sorafenib enhances the antitumor effects of anti-CTLA-4 antibody in a murine cancer model by inhibiting myeloid-derived suppressor cells. Oncol. Rep. 2015, 33, 2947–2953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, J.J.; Sampath, P.; Hou, W.; Thorne, S.H. Defining effective combinations of immune checkpoint blockade and oncolytic virotherapy. Clin. Cancer Res. 2015, 21, 5543–5551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannini, A.; Leoni, V.; Barboni, C.; Sanapo, M.; Zaghini, A.; Malatesta, P.; Campadelli-Fiume, G.; Gianni, T. αvβ3-integrin regulates PD-L1 expression and is involved in cancer immune evasion. Proc. Natl. Acad. Sci. USA 2019, 116, 20141–20150. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S.; Lee, S.D.; Lee, S.J.; Chung, M.K. Development of an immunotherapeutic adenovirus targeting hormone-independent prostate cancer. OncoTargets Ther. 2013, 6, 1635. [Google Scholar] [CrossRef] [Green Version]
- Cheng, W.-S.; Dzojic, H.; Nilsson, B.; Tötterman, T.H.; Essand, M. An oncolytic conditionally replicating adenovirus for hormone-dependent and hormone-independent prostate cancer. Cancer Gene Ther. 2006, 13, 13–20. [Google Scholar] [CrossRef]
- Durso, R.J.; Andjelic, S.; Gardner, J.P.; Margitich, D.J.; Donovan, G.P.; Arrigale, R.R.; Wang, X.; Maughan, M.F.; Talarico, T.L.; Olmsted, R.A. A novel alphavirus vaccine encoding prostate-specific membrane antigen elicits potent cellular and humoral immune responses. Clin. Cancer Res. 2007, 13, 3999–4008. [Google Scholar] [CrossRef] [Green Version]
- Ayala-Breton, C.; Barber, G.N.; Russell, S.J.; Peng, K.-W. Retargeting vesicular stomatitis virus using measles virus envelope glycoproteins. Hum. Gene Ther. 2012, 23, 484–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Hasegawa, K.; Russell, S.J.; Sadelain, M.; Peng, K.W. Prostate-specific membrane antigen retargeted measles virotherapy for the treatment of prostate cancer. Prostate 2009, 69, 1128–1141. [Google Scholar] [CrossRef] [Green Version]
- Narayan, V.; Gladney, W.; Plesa, G.; Vapiwala, N.; Carpenter, E.; Maude, S.L.; Lal, P.; Lacey, S.F.; Melenhorst, J.J.; Sebro, R. A phase I clinical trial of PSMA-directed/TGFβ-insensitive CAR-T cells in metastatic castration-resistant prostate cancer. Am. Soc. Clin. Oncol. 2019, 37, TPS269. [Google Scholar] [CrossRef]
- Madan, R.A.; Antonarakis, E.S.; Drake, C.G.; Fong, L.; Yu, E.Y.; McNeel, D.G.; Lin, D.W.; Chang, N.N.; Sheikh, N.A.; Gulley, J.L. Putting the pieces together: Completing the mechanism of action jigsaw for sipuleucel-T. JNCI J. Natl. Cancer Inst. 2020, 112, 562–573. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vannini, A.; Parenti, F.; Bressanin, D.; Barboni, C.; Zaghini, A.; Campadelli-Fiume, G.; Gianni, T. Towards a Precision Medicine Approach and In Situ Vaccination against Prostate Cancer by PSMA-Retargeted oHSV. Viruses 2021, 13, 2085. https://0-doi-org.brum.beds.ac.uk/10.3390/v13102085
Vannini A, Parenti F, Bressanin D, Barboni C, Zaghini A, Campadelli-Fiume G, Gianni T. Towards a Precision Medicine Approach and In Situ Vaccination against Prostate Cancer by PSMA-Retargeted oHSV. Viruses. 2021; 13(10):2085. https://0-doi-org.brum.beds.ac.uk/10.3390/v13102085
Chicago/Turabian StyleVannini, Andrea, Federico Parenti, Daniela Bressanin, Catia Barboni, Anna Zaghini, Gabriella Campadelli-Fiume, and Tatiana Gianni. 2021. "Towards a Precision Medicine Approach and In Situ Vaccination against Prostate Cancer by PSMA-Retargeted oHSV" Viruses 13, no. 10: 2085. https://0-doi-org.brum.beds.ac.uk/10.3390/v13102085