
Comparative Analysis of Within-Host Mutation Patterns and Diversity of Hepatitis C Virus Subtypes 1a, 1b, and 3a


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
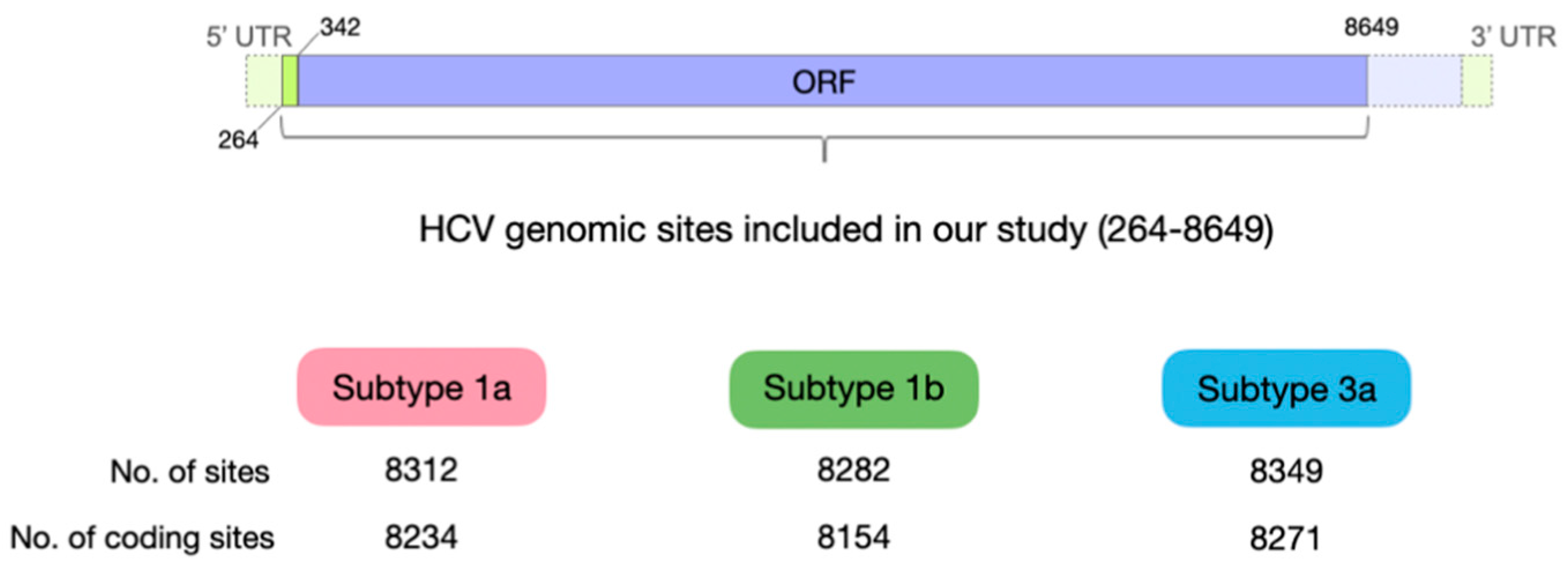
2.1. Viral Samples
2.2. Data Validation
2.3. Data Analysis
3. Results
3.1. Comparison of Within-Patient Evolutionary Patterns between HCV Subtypes
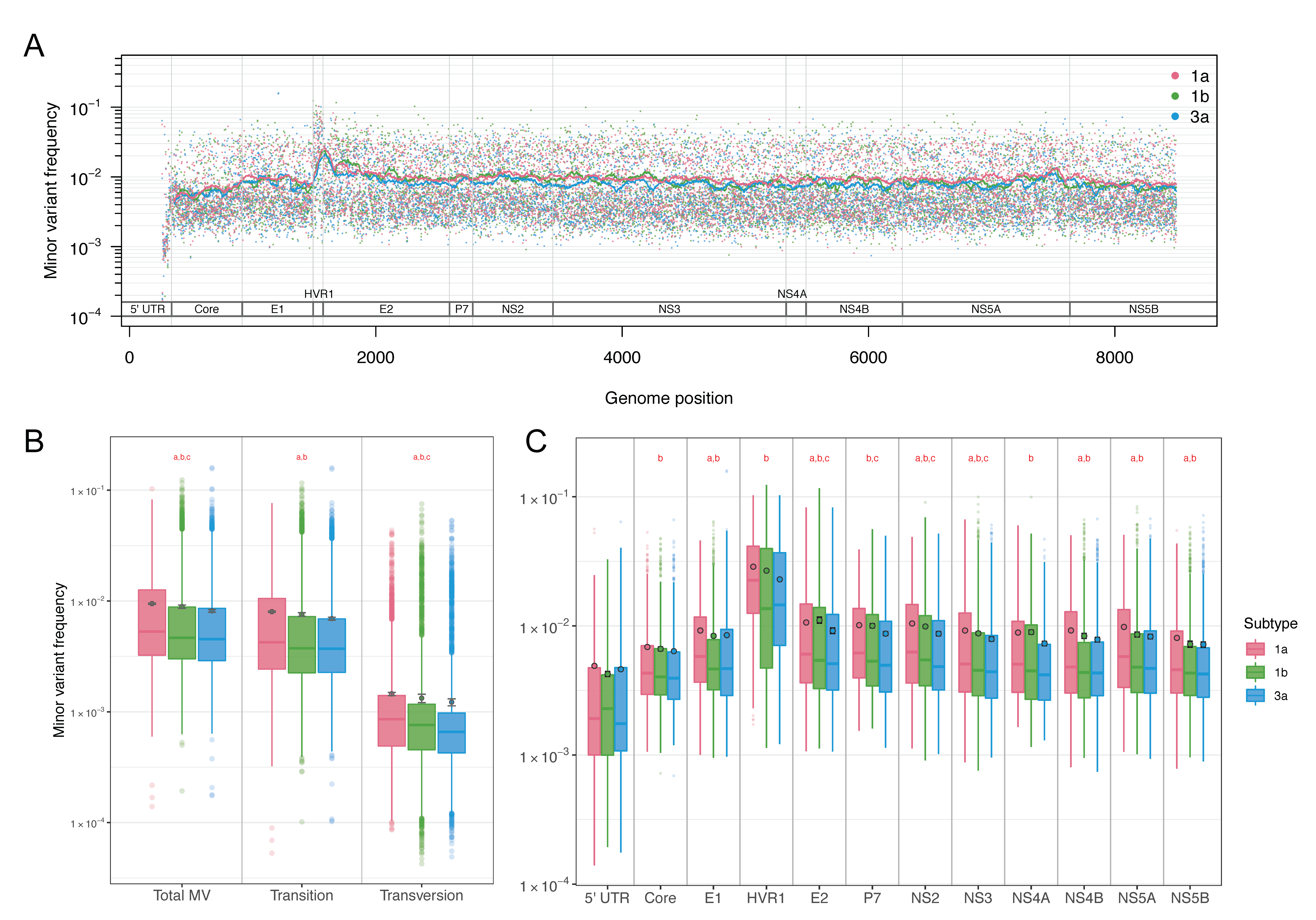
3.1.1. Within-Host Mutation Frequency Summary
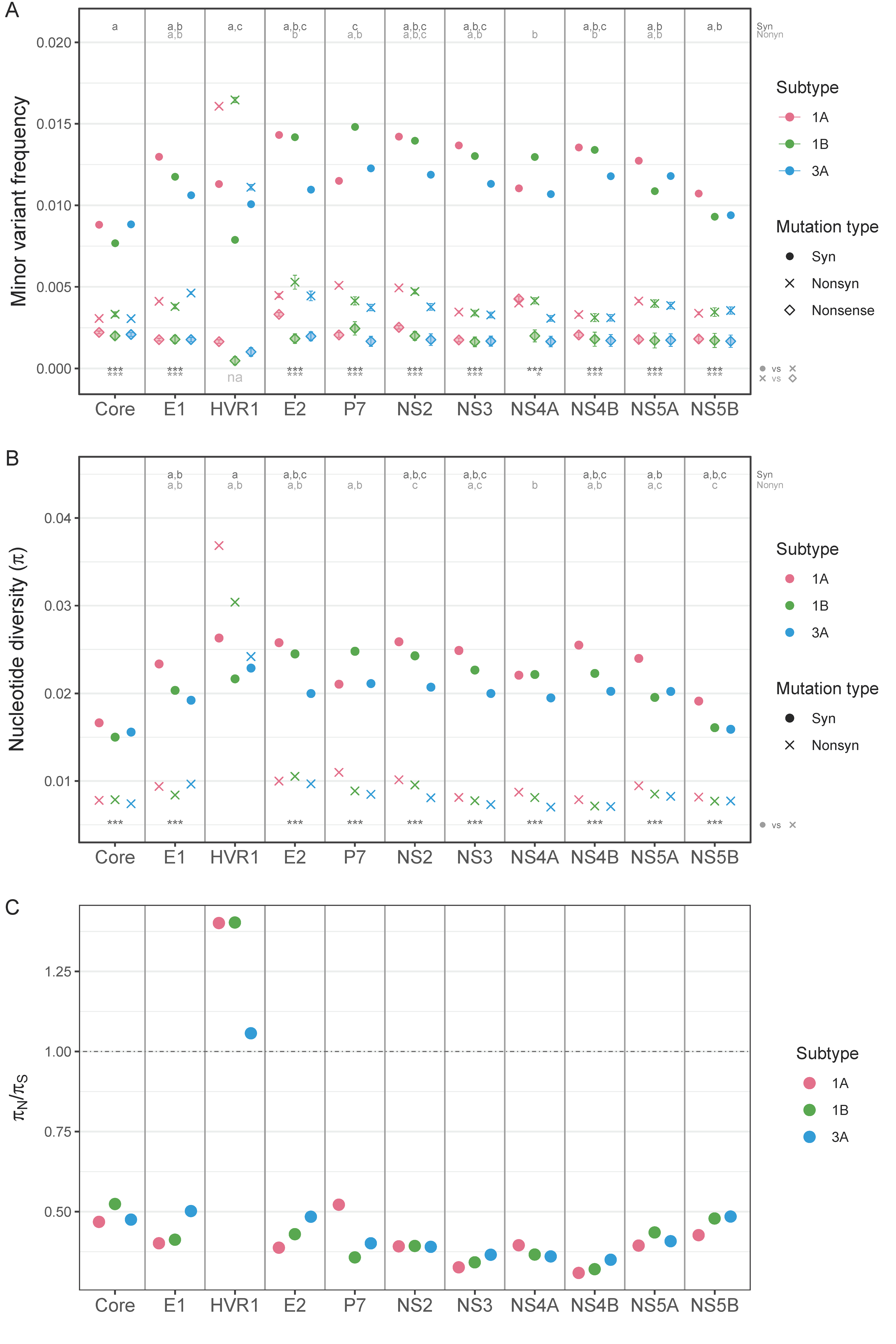
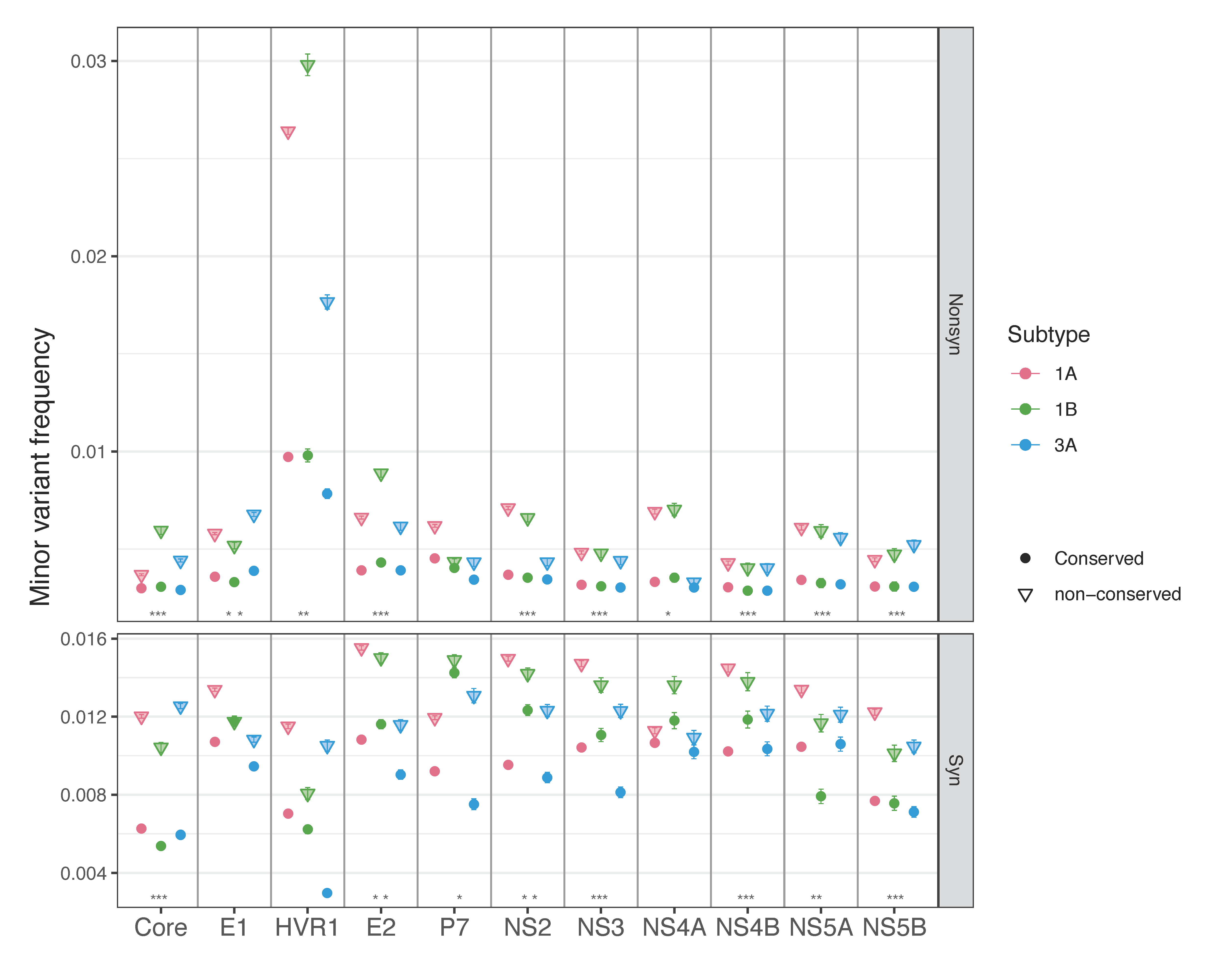
3.1.2. Nonsynonymous and Synonymous Transition Mutation Frequencies by Genomic Region
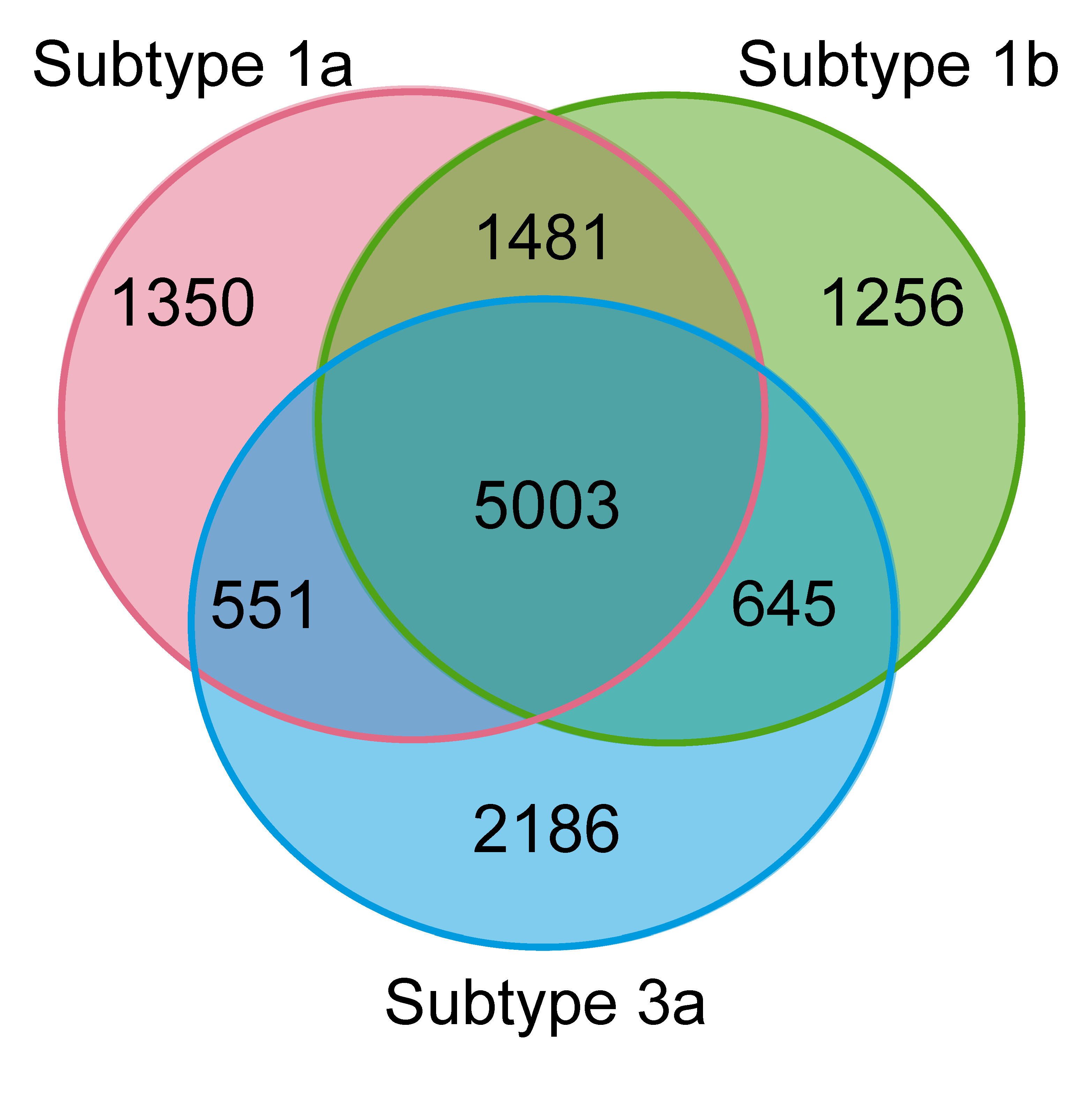
3.1.3. Sites Conserved across Different Subtypes Have Lower Mutation Frequencies
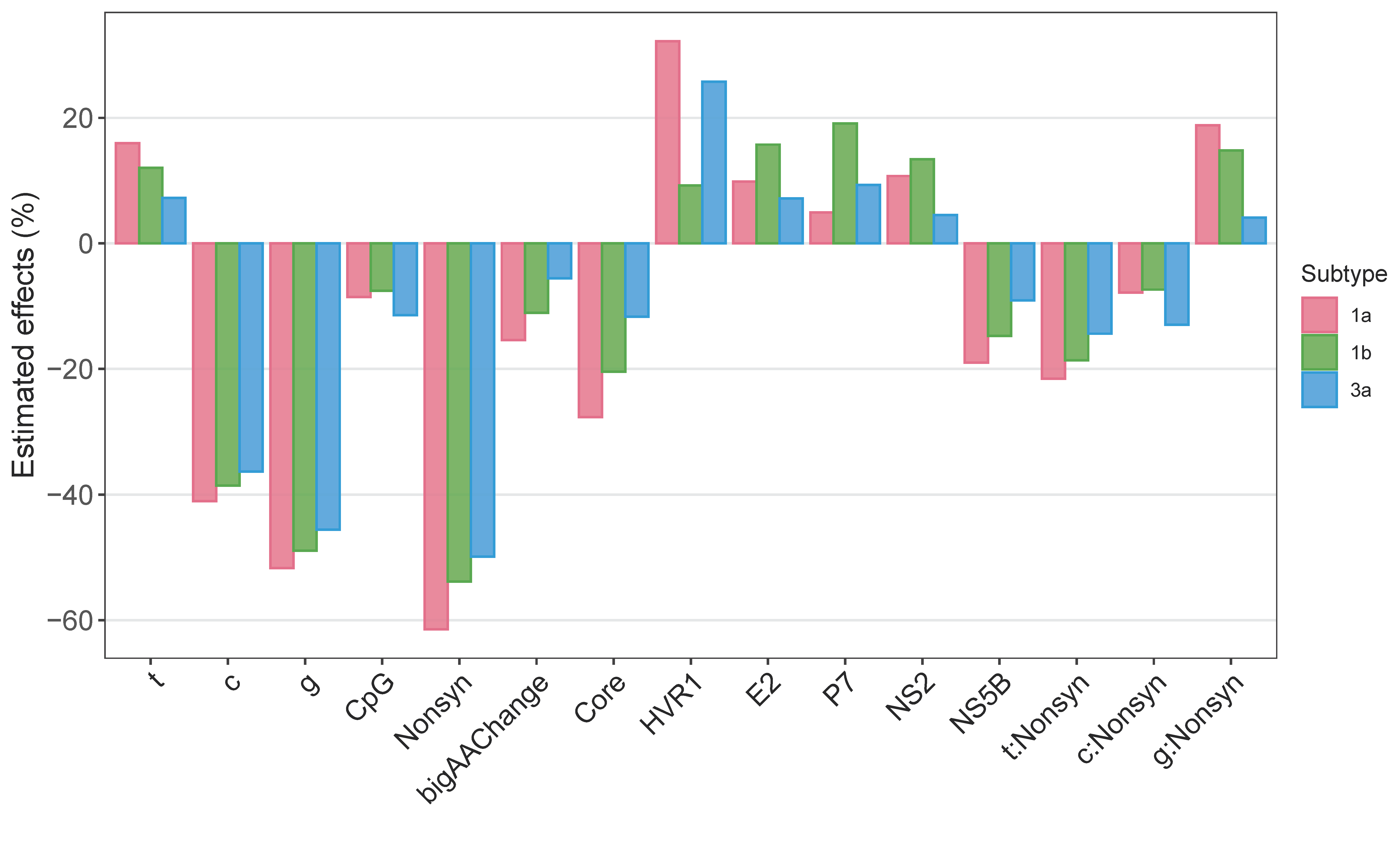
3.2. What Drives and Constrains Within-Host Mutations?
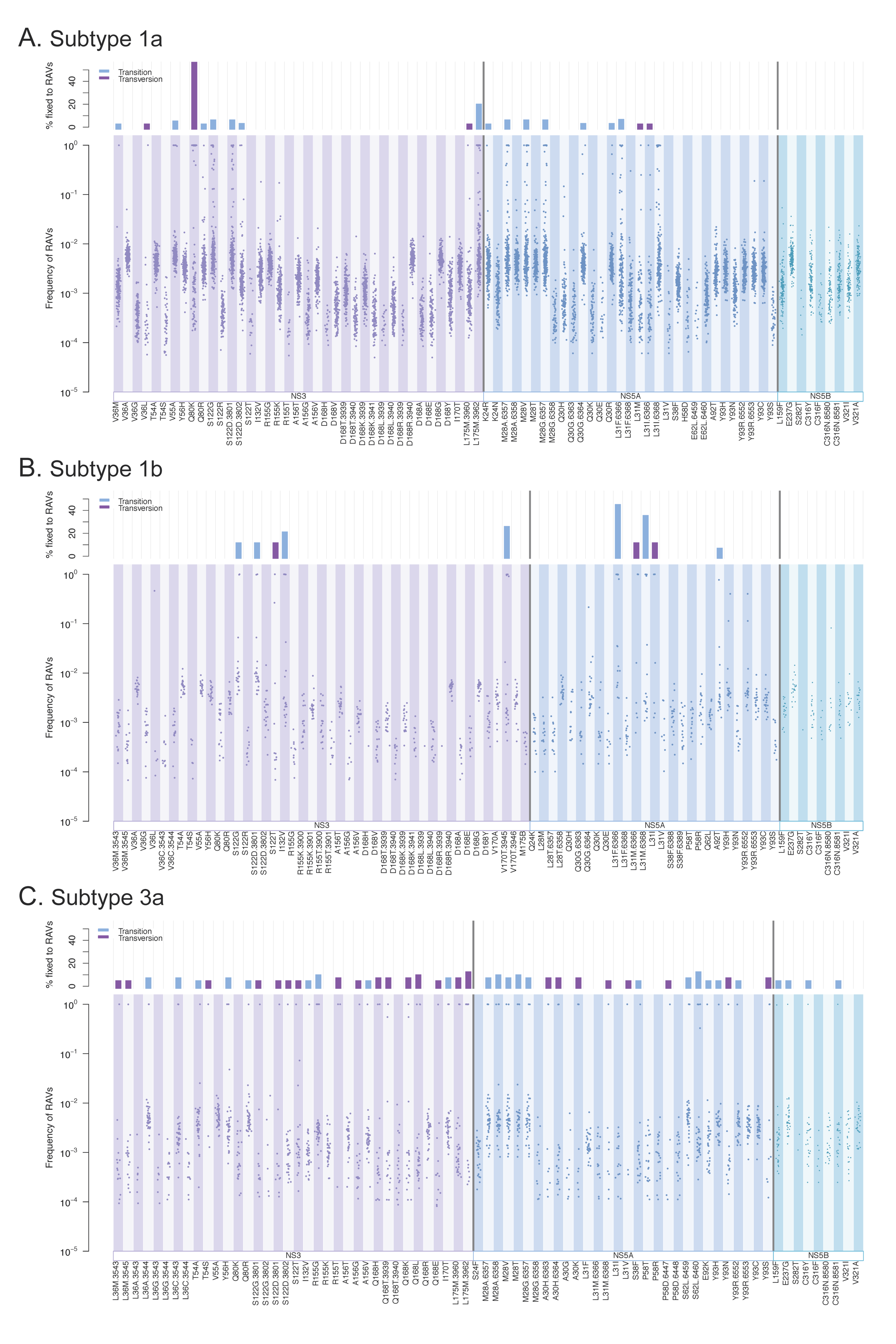
3.3. Resistance-Associated Variants (RAVs)
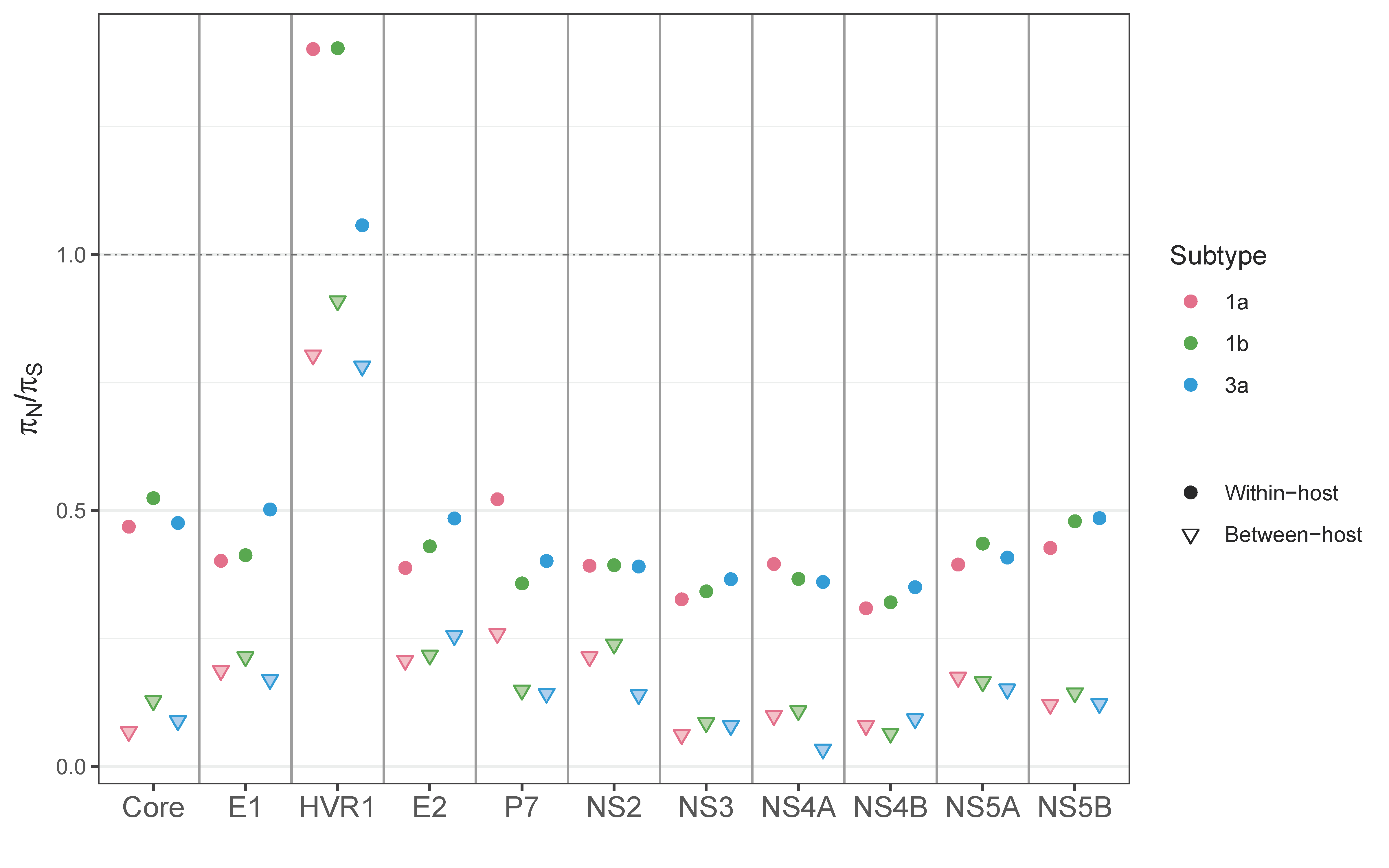
3.4. Between- vs. within-Host Genetic Variation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kato, N. Genome of Human Hepatitis C Virus (HCV): Gene organization, sequence diversity, and variation. Microb. Comp. Genom. 2000, 5, 129–151. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, M.; Rauff, B.; Rashid, H.; Lam, T.; Rafiq, S. Update on global epidemiology of viral hepatitis and preventive strategies. WJCC 2018, 6, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Coppola, N.; Alessio, L.; Onorato, L.; Sagnelli, C.; Macera, M.; Sagnelli, E.; Pisaturo, M. Epidemiology and management of hepatitis C virus infections in immigrant populations. Infect. Dis. Poverty 2019, 8, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauring, A.S.; Andino, R. Quasispecies Theory and the Behavior of RNA Viruses. PLoS Pathog. 2010, 6, e1001005. [Google Scholar] [CrossRef] [PubMed]
- Gallego, I.; Gregori, J.; Soria, M.E.; García-Crespo, C.; García-Álvarez, M.; Gómez-González, A.; Valiergue, R.; Gómez, J.; Esteban, J.I.; Quer, J.; et al. Resistance of high fitness hepatitis C virus to lethal mutagenesis. Virology 2018, 523, 100–109. [Google Scholar] [CrossRef]
- Sorbo, M.C.; Cento, V.; Di Maio, V.C.; Howe, A.Y.M.; Garcia, F.; Perno, C.F.; Ceccherini-Silberstein, F. Hepatitis C virus drug resistance associated substitutions and their clinical relevance: Update 2018. Drug Resist. Updates 2018, 37, 17–39. [Google Scholar] [CrossRef] [PubMed]
- WHO. Hepatitis C. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-c (accessed on 29 October 2020).
- Echeverría, N. Hepatitis C virus genetic variability and evolution. WJH 2015, 7, 831–845. [Google Scholar] [CrossRef]
- Mauger, D.M.; Golden, M.; Yamane, D.; Williford, S.; Lemon, S.M.; Martin, D.P.; Weeks, K.M. Functionally conserved architecture of hepatitis C virus RNA genomes. Proc. Natl. Acad. Sci. USA 2015, 112, 3692–3697. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.B.; Pathirana, S.; Davidson, F.; Lawlor, E.; Power, J.; Yap, P.L.; Simmonds, P. The origin of hepatitis C virus genotypes. J. Gen. Virol. 1997, 78, 321–328. [Google Scholar] [CrossRef] [Green Version]
- Major, M.E.; Feinstone, S.M. The molecular virology of hepatitis C. Hepatology 1997, 25, 1527–1538. [Google Scholar] [CrossRef]
- Keck, Z.-Y.; Girard-Blanc, C.; Wang, W.; Lau, P.; Zuiani, A.; Rey, F.A.; Krey, T.; Diamond, M.S.; Foung, S.K.H. Antibody Response to Hypervariable Region 1 Interferes with Broadly Neutralizing Antibodies to Hepatitis C Virus. J. Virol. 2016, 90, 3112–3122. [Google Scholar] [CrossRef] [Green Version]
- Gray, R.R.; Parker, J.; Lemey, P.; Salemi, M.; Katzourakis, A.; Pybus, O.G. The mode and tempo of hepatitis C virus evolution within and among hosts. BMC Evol. Biol. 2011, 11, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Lello, F.A.; Culasso, A.C.A.; Campos, R.H. Inter and intrapatient evolution of hepatitis C virus. Ann. Hepatol. 2015, 14, 442–449. [Google Scholar] [CrossRef]
- Alizon, S.; Luciani, F.; Regoes, R.R. Epidemiological and clinical consequences of within-host evolution. Trends Microbiol. 2011, 19, 24–32. [Google Scholar] [CrossRef]
- Nelson, C.W.; Hughes, A.L. Within-host nucleotide diversity of virus populations: Insights from next-generation sequencing. Infect. Genet. Evol. 2015, 30, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Caporossi, A.; Kulkarni, O.; Blum, M.G.B.; Leroy, V.; Morand, P.; Larrat, S.; François, O. Using high-throughput sequencing for investigating intra-host hepatitis C evolution over long retrospective periods. Infect. Genet. Evol. 2019, 67, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Raghwani, J.; Wu, C.-H.; Ho, C.K.Y.; de Jong, M.; Molenkamp, R.; Schinkel, J.; Pybus, O.G.; Lythgoe, K.A. High-Resolution Evolutionary Analysis of Within-Host Hepatitis C Virus Infection. J. Infect. Dis. 2019, 219, 1722–1729. [Google Scholar] [CrossRef] [Green Version]
- Tisthammer, K.H.; Dong, W.; Joy, J.B.; Pennings, P.S. Assessing in vivo mutation frequencies and creating a high-resolution genome-wide map of fitness costs of Hepatitis C virus. bioRxiv 2020. in review. Available online: https://www.biorxiv.org/content/10.1101/2020.10.01.323253v1 (accessed on 1 October 2020).
- Lindsay, K.L. Introduction to therapy of hepatitis C. J. Med. Virol. 2003, 36, s114–s120. [Google Scholar]
- Huang, J.-F.; Huang, C.-F.; Yeh, M.-L.; Dai, C.-Y.; Yu, M.-L.; Chuang, W.-L. Updates in the management and treatment of HCV genotype 3, what are the remaining challenges? Expert Rev. Anti-Infect. Ther. 2018, 16, 907–912. [Google Scholar] [CrossRef]
- Klein, M.B.; Saeed, S.; Yang, H.; Cohen, J.; Conway, B.; Cooper, C.; Côté, P.; Cox, J.; Gill, J.; Haase, D. Cohort profile: The Canadian HIV–hepatitis C co-infection cohort study. Int. J. Epidemiol. 2010, 39, 1162–1169. [Google Scholar] [CrossRef]
- Bushnell, B.; Rood, J.; Singer, E. BBMerge—Accurate paired shotgun read merging via overlap. PLoS ONE 2017, 12, e0185056. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanini, F.; Brodin, J.; Thebo, L.; Lanz, C.; Bratt, G.; Elife, J.A.; Neher, R.A. Population genomics of intrapatient HIV-1 evolution. eLife 2015, 4, e11282. [Google Scholar] [CrossRef]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 15 December 2020).
- Chui, C.K.S.; Dong, W.W.Y.; Joy, J.B.; Poon, A.F.Y.; Dong, W.Y.; Mo, T.; Woods, C.K.; Beatty, C.; Hew, H.; Harrigan, P.R.; et al. Development and Validation of Two Screening Assays for the Hepatitis C Virus NS3 Q80K Polymorphism Associated with Reduced Response to Combination Treatment Regimens Containing Simeprevir. J. Clin. Microbiol. 2015, 53, 2942–2950. [Google Scholar] [CrossRef] [Green Version]
- Fox, E.J.; Reid-Bayliss, K.S.; Emond, M.J.; Loeb, L.A. Accuracy of Next Generation Sequencing Platforms. Next Gener. Seq. Appl. 2014, 1, 1000106. [Google Scholar] [CrossRef] [Green Version]
- Caribari-Neto, F.; Zeileis, A. Beta Regression in R. J. Stat. Softw. 2019, 34, 1–24. [Google Scholar]
- Paradis, E.; Jombart, T.; Kamvar, Z.; Knaus, B.; Sochliep, K.; Potts, A.; Winter, D. Package “pegas”: An R package for population genetics with an integrated–modular approach. Bioinformatics. 2010, 26, 419–420. [Google Scholar] [CrossRef] [Green Version]
- Poveda, E.; Wyles, D.L.; Mena, Á.; Pedreira, J.D.; Castro-Iglesias, Á.; Cachay, E. Update on hepatitis C virus resistance to direct-acting antiviral agents. Antiviral Res. 2014, 108, 181–191. [Google Scholar] [CrossRef] [Green Version]
- Lontok, E.; Harrington, P.; Howe, A.; Kieffer, T.; Lennerstrand, J.; Lenz, O.; McPhee, F.; Mo, H.; Parkin, N.; Pilot-Matias, T.; et al. Hepatitis C virus drug resistance-associated substitutions: State of the art summary. Hepatology 2015, 62, 1623–1632. [Google Scholar] [CrossRef]
- Sarrazin, C. The importance of resistance to direct antiviral drugs in HCV infection in clinical practice. J. Hepatol. 2016, 64, 486–504. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Han, K.-H.; Ahn, S.H. Hepatitis C Virus and Antiviral Drug Resistance. Gut Liver 2016, 10, 890–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyles, D.L.; Luetkemeyer, A.F. Understanding Hepatitis C Virus Drug Resistance: Clinical Implications for Current and Future Regimens. Top. Antivir. Med. 2017, 25, 103–109. [Google Scholar]
- Petruzziello, A.; Marigliano, S.; Loquercio, G.; Cozzolino, A.; Cacciapuoti, C. Global epidemiology of hepatitis C virus infection: An up-date of the distribution and circulation of hepatitis C virus genotypes. World J. Gastroenterol. 2016, 22, 7824. [Google Scholar] [CrossRef] [PubMed]
- Abayasingam, A.; Leung, P.; Eltahla, A.; Bull, R.A.; Luciani, F.; Grebely, J.; Dore, G.J.; Applegate, T.; Page, K.; Bruneau, J.; et al. Genomic characterization of hepatitis C virus transmitted founder variants with deep sequencing. Infect. Genet. Evol. 2019, 71, 36–41. [Google Scholar] [CrossRef]
- Karlin, S.; Doerfler, W.; Cardon, L.R. Why is CpG suppressed in the genomes of virtually all small eukaryotic viruses but not in those of large eukaryotic viruses? J. Virol. 1994, 68, 2889–2897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.; Virk, N.; Chen, W.; Ji, S.; Ji, S.; Sun, Y.; Wu, X. CpG usage in RNA viruses: Data and hypotheses. PLoS ONE 2013, 8, e74109. [Google Scholar] [CrossRef] [Green Version]
- Caudill, V.R.; Qin, S.; Winstead, R.; Kaur, J.; Tisthammer, K.; Pineda, E.G.; Solis, C.; Cobey, S.; Bedford, T.; Carja, O.; et al. CpG-creating mutations are costly in many human viruses. Evol. Ecol. 2020, 34, 339–359. [Google Scholar] [CrossRef] [Green Version]
- Witteveldt, J.; Martin-Gans, M.; Simmonds, P. Enhancement of the Replication of Hepatitis C Virus Replicons of Genotypes 1 to 4 by Manipulation of CpG and UpA Dinucleotide Frequencies and Use of Cell Lines Expressing SECL14L2 for Antiviral Resistance Testing. Antimicrob. Agents Chemother. 2016, 60, 2981–2992. [Google Scholar] [CrossRef] [Green Version]
- Berger, A.; Prondzinski, M.D.; Doerr, H.W.; Rabenau, H.; Weber, B. Hepatitis C plasma viral load is associated with HCV genotype but not with HIV coinfection. Conserv. Lett. 1996, 48, 339–343. [Google Scholar] [CrossRef]
- Chakravarti, A.; Dogra, G.; Verma, V.; Srivastava, A.P. Distribution pattern of HCV genotypes & its association with viral load. Indian J. Med. Res. 2011, 133, 326–331. [Google Scholar]
- Petruzziello, A.; Coppola, N.; Loquercio, G.; Marigliano, S.; Giordano, M.; Azzaro, R.; Diodato, A.M.; Iervolino, V.; Di Costanzo, G.; Di Macchia, C.A.; et al. Distribution Pattern of Hepatitis C Virus Genotypes and Correlation with Viral Load and Risk Factors in Chronic Positive Patients. Intervirology 2014, 57, 311–318. [Google Scholar] [CrossRef]
- Ansari, M.A.; Pedergnana, V.; Ip, C.L.C.; Magri, A.; Von Delft, A.; Bonsall, D.; Chaturvedi, N.; Bartha, I.; Smith, D.; Nicholson, G.; et al. Genome-to-genome analysis highlights the effect of the human innate and adaptive immune systems on the hepatitis C virus. Nat. Genet. 2017, 49, 666–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, K.S.; Bloom, J.D. Linking influenza virus evolution within and between human hosts. Virus Evol. 2020, 6, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ampuero, J.; Romero-Gómez, M.; Reddy, K.R. Review article: HCV genotype 3—The new treatment challenge. Aliment. Pharmacol. Ther. 2014, 39, 686–698. [Google Scholar] [CrossRef]
- Liu, R.; Curry, S.; McMonagle, P.; Yeh, W.W.; Ludmerer, S.W.; Jumes, P.A.; Marshall, W.L.; Kong, S.; Ingravallo, P.; Black, S.; et al. Susceptibilities of Genotype 1a, 1b, and 3 Hepatitis C Virus Variants to the NS5A Inhibitor Elbasvir. Antimicrob. Agents Chemother. 2015, 59, 6922–6929. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tisthammer, K.H.; Dong, W.; Joy, J.B.; Pennings, P.S. Comparative Analysis of Within-Host Mutation Patterns and Diversity of Hepatitis C Virus Subtypes 1a, 1b, and 3a. Viruses 2021, 13, 511. https://0-doi-org.brum.beds.ac.uk/10.3390/v13030511
Tisthammer KH, Dong W, Joy JB, Pennings PS. Comparative Analysis of Within-Host Mutation Patterns and Diversity of Hepatitis C Virus Subtypes 1a, 1b, and 3a. Viruses. 2021; 13(3):511. https://0-doi-org.brum.beds.ac.uk/10.3390/v13030511
Chicago/Turabian StyleTisthammer, Kaho H., Weiyan Dong, Jeffrey B. Joy, and Pleuni S. Pennings. 2021. "Comparative Analysis of Within-Host Mutation Patterns and Diversity of Hepatitis C Virus Subtypes 1a, 1b, and 3a" Viruses 13, no. 3: 511. https://0-doi-org.brum.beds.ac.uk/10.3390/v13030511