
Transcriptional and Non-Transcriptional Activation, Posttranslational Modifications, and Antiviral Functions of Interferon Regulatory Factor 3 and Viral Antagonism by the SARS-Coronavirus

 , and
, and 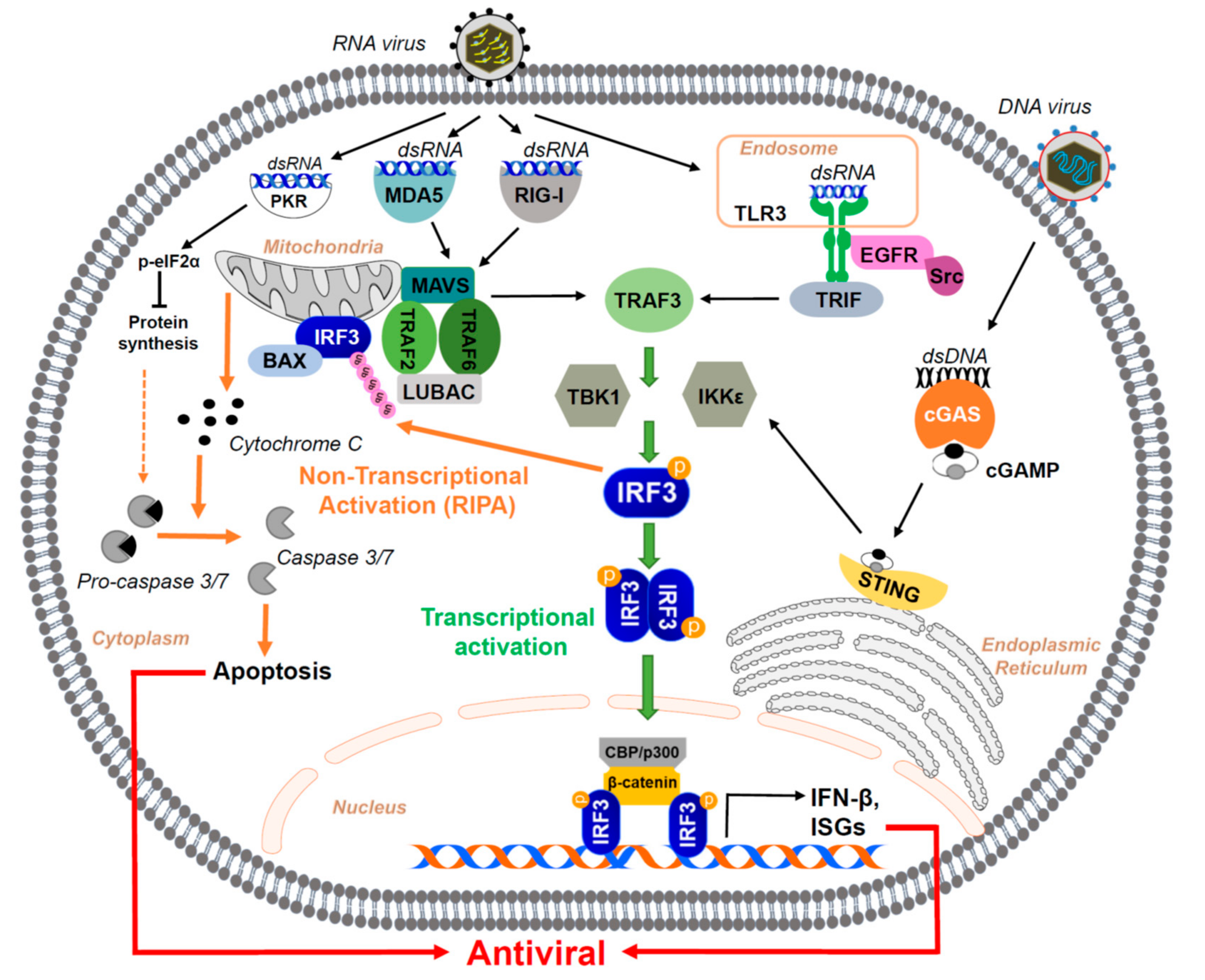{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. IRF3, a Member of the Family of Interferon Regulatory Factors (IRFs)
1.2. IRF3: Protein Expression and Structure
2. Role of IRF3 as a Mediator of Innate Immune Responses to Viral Infection
2.1. PRRs and Associated Pathways That Lead to IRF3 Activation
2.2. Transcriptional Activation of IRF3: Induction of IFN and Antiviral Genes
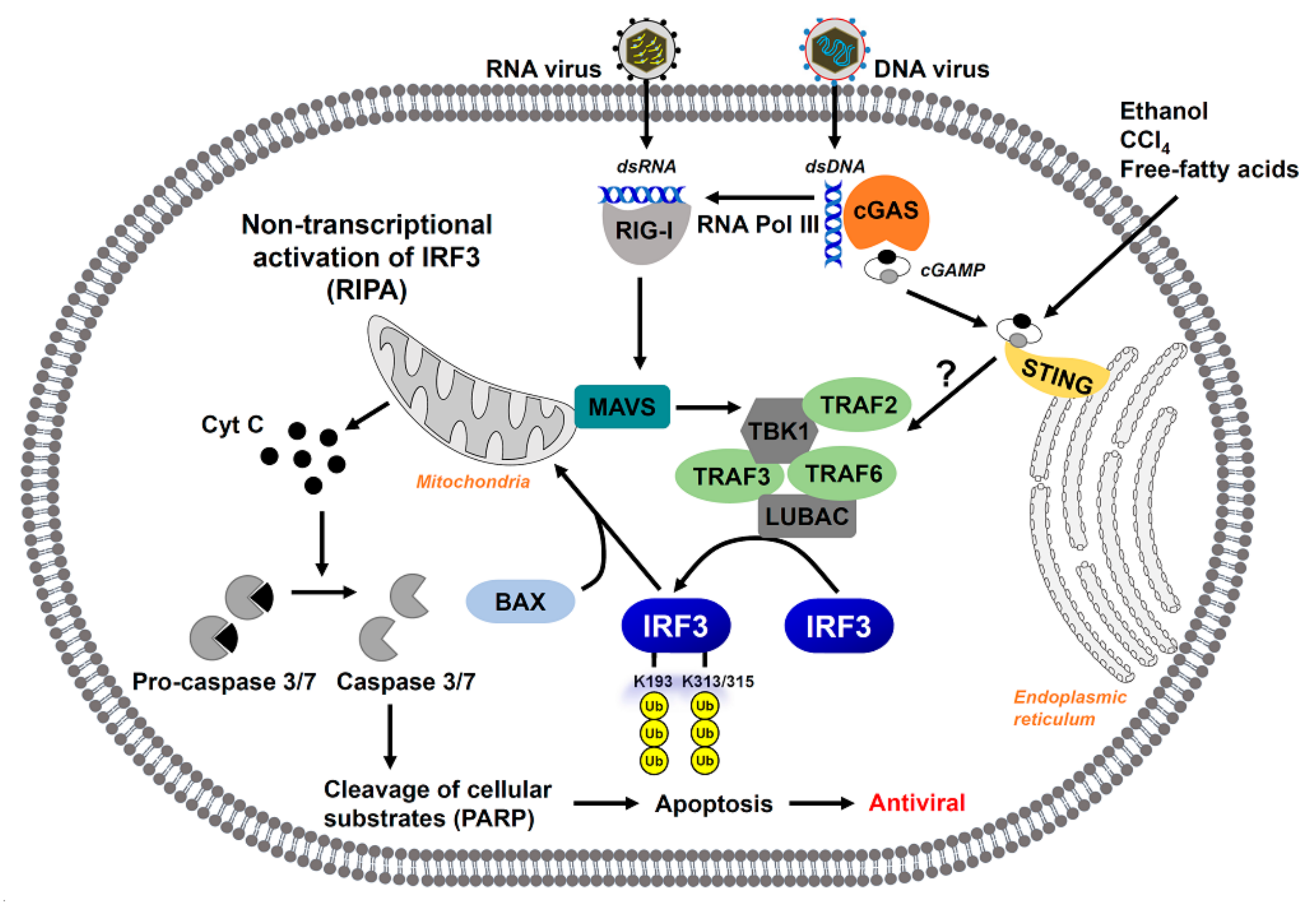
2.3. Non-Transcriptional Activation of IRF—The Pro-Apoptotic Pathway
2.4. Regulation of Non-Transcriptional Function of IRF3
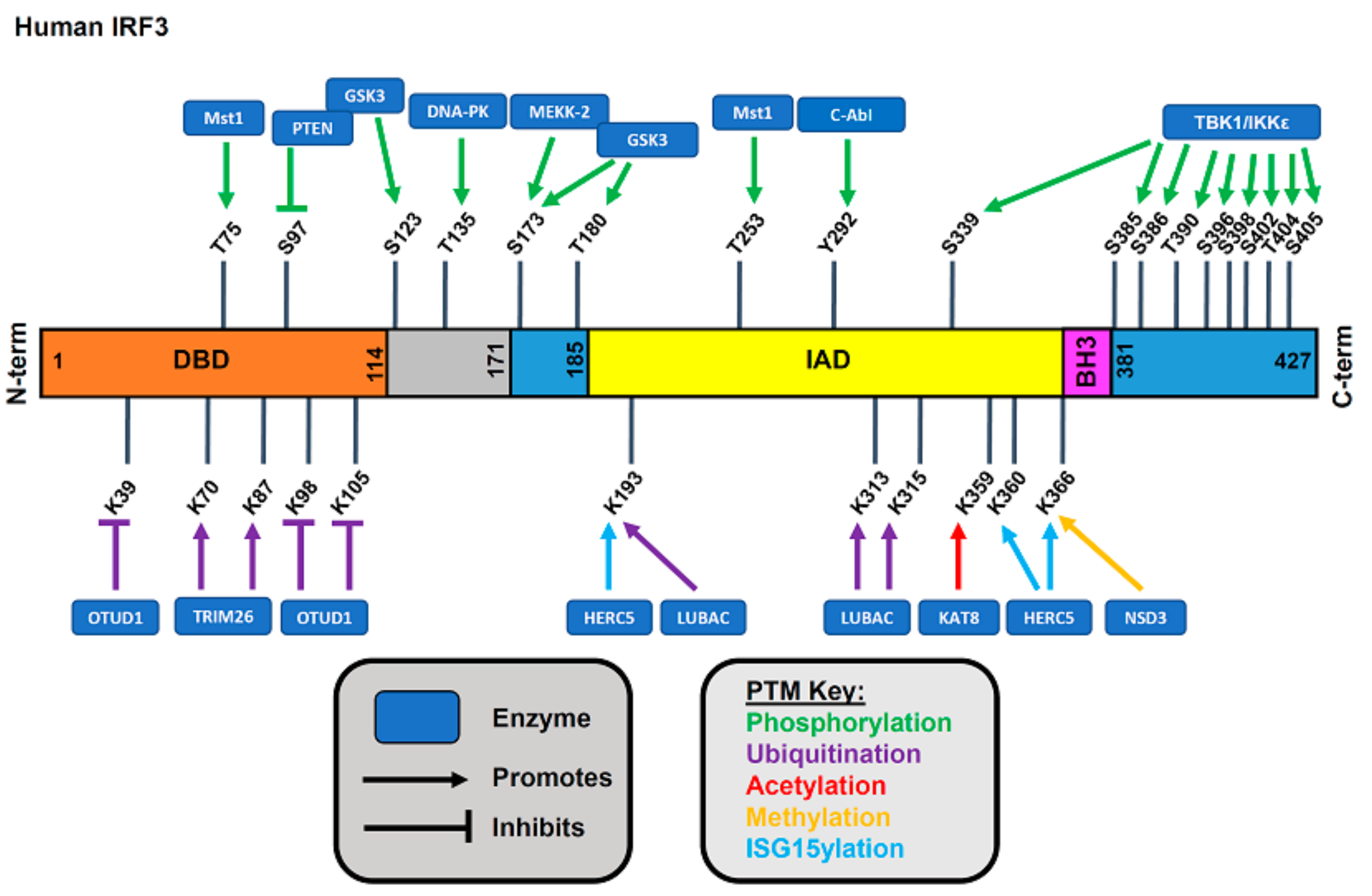
3. Regulation of IRF3 Functions by Posttranslational Modifications
3.1. IRF3 in Uninfected Cells
3.2. Phosphorylation of IRF3
3.2.1. Phosphorylation-Mediated Activation of IRF3
3.2.2. Phosphorylation-Mediated Downregulation of IRF3 Activity
3.3. Ubiquitination of IRF3
3.3.1. Protein Ubiquitination
3.3.2. Ubiquitin-Mediated Degradation of IRF3
3.3.3. Ubiquitin-Mediated Functional Change in IRF3 Activity
3.3.4. Deubiquitination of IRF3
3.4. SUMOylation
3.5. Glutathionylation
3.6. Methylation
3.7. ISG15ylation
3.8. Acetylation
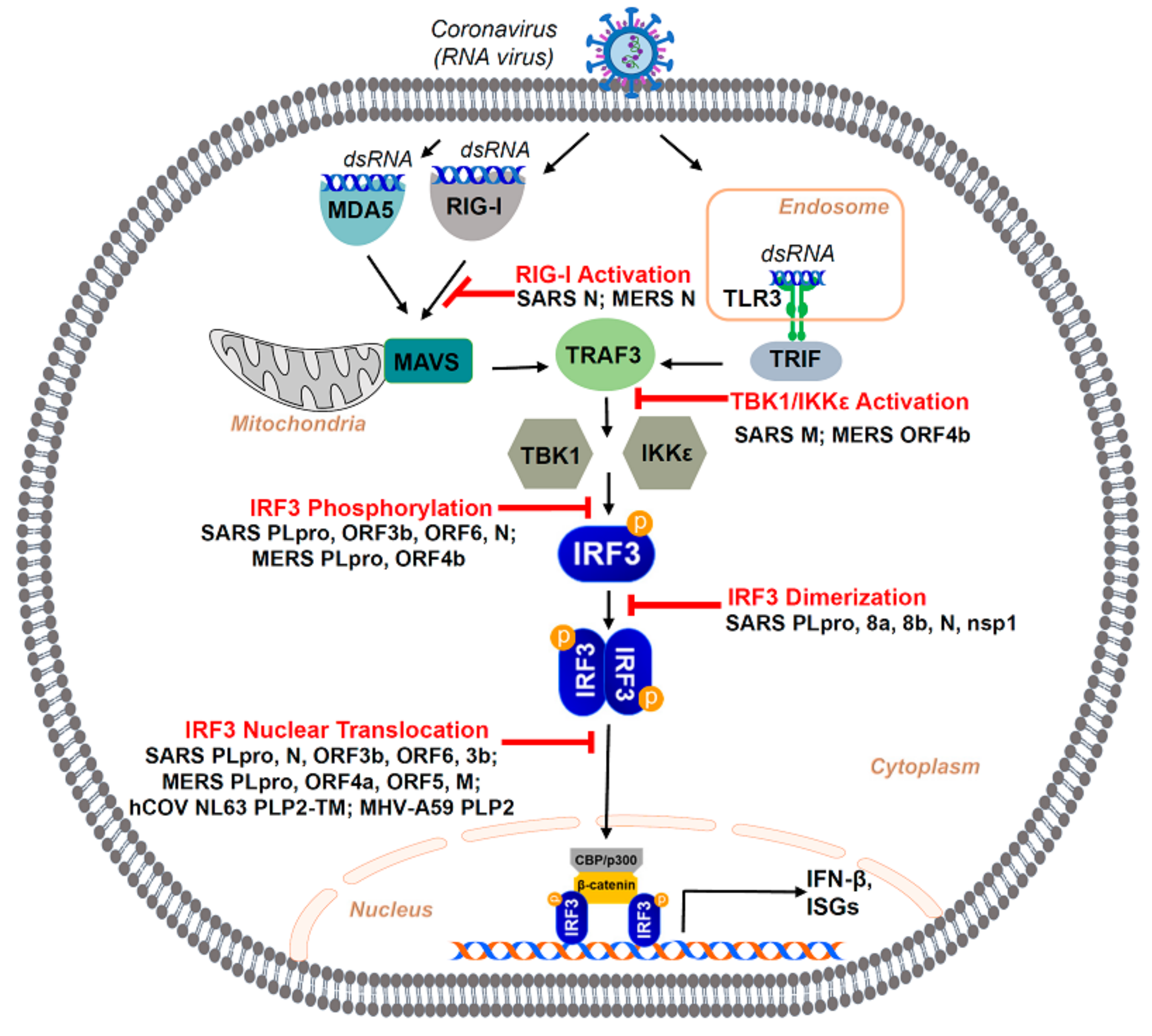
4. Viral Antagonism of IRF3: SARS-Coronavirus-Mediated Inhibition of IRF3 Activation
4.1. Antagonism of IRF3 by SARS-CoV Structural Proteins
4.2. Antagonism of IRF3 by SARS-CoV Non-Structural Proteins
4.3. Antagonism of IRF3 by SARS-CoV Accessory Proteins
5. Clinical Implications of IRF3 Activation and Therapeutic Applications
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Marsili, G.; Perrotti, E.; Remoli, A.L.; Acchioni, C.; Sgarbanti, M.; Battistini, A. IFN Regulatory Factors and Antiviral Innate Immunity: How Viruses Can Get Better. J. Interferon Cytokine Res. 2016, 36, 414–432. [Google Scholar] [CrossRef]
- Jefferies, C.A. Regulating IRFs in IFN Driven Disease. Front. Immunol. 2019, 10, 325. [Google Scholar] [CrossRef] [Green Version]
- Ozato, K.; Tailor, P.; Kubota, T. The interferon regulatory factor family in host defense: Mechanism of action. J. Biol. Chem. 2007, 282, 20065–20069. [Google Scholar] [CrossRef] [Green Version]
- Hiscott, J. Triggering the innate antiviral response through IRF-3 activation. J. Biol. Chem. 2007, 282, 15325–15329. [Google Scholar] [CrossRef] [Green Version]
- Petro, T.M. IFN Regulatory Factor 3 in Health and Disease. J. Immunol. 2020, 205, 1981–1989. [Google Scholar] [CrossRef]
- Ning, S.; Pagano, J.S.; Barber, G.N. IRF7: Activation, regulation, modification and function. Genes Immun. 2011, 12, 399–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern recognition receptors and the innate immune response to viral infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, K.; Bowie, A.G. Activation of host pattern recognition receptors by viruses. Curr. Opin. Microbiol. 2010, 13, 503–507. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Sen, G.C. RIG-I-like receptor-induced IRF3 mediated pathway of apoptosis (RIPA): A new antiviral pathway. Protein Cell 2017, 8, 165–168. [Google Scholar] [CrossRef] [Green Version]
- Escalante, C.R.; Yie, J.; Thanos, D.; Aggarwal, A.K. Structure of IRF-1 with bound DNA reveals determinants of interferon regulation. Nature 1998, 391, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Takahasi, K.; Suzuki, N.N.; Horiuchi, M.; Mori, M.; Suhara, W.; Okabe, Y.; Fukuhara, Y.; Terasawa, H.; Akira, S.; Fujita, T.; et al. X-ray crystal structure of IRF-3 and its functional implications. Nat. Struct. Biol. 2003, 10, 922–927. [Google Scholar] [CrossRef]
- Qin, B.Y.; Liu, C.; Lam, S.S.; Srinath, H.; Delston, R.; Correia, J.J.; Derynck, R.; Lin, K. Crystal structure of IRF-3 reveals mechanism of autoinhibition and virus-induced phosphoactivation. Nat. Struct. Biol. 2003, 10, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Karpova, A.Y.; Ronco, L.V.; Howley, P.M. Functional characterization of interferon regulatory factor 3a (IRF-3a), an alternative splice isoform of IRF-3. Mol. Cell Biol. 2001, 21, 4169–4176. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Ablasser, A.; Bauernfeind, F.; Hartmann, G.; Latz, E.; Fitzgerald, K.A.; Hornung, V. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol. 2009, 10, 1065–1072. [Google Scholar] [CrossRef] [Green Version]
- Ablasser, A.; Schmid-Burgk, J.L.; Hemmerling, I.; Horvath, G.L.; Schmidt, T.; Latz, E.; Hornung, V. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 2013, 503, 530–534. [Google Scholar] [CrossRef] [Green Version]
- Satoh, T.; Akira, S. Toll-Like Receptor Signaling and Its Inducible Proteins. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Bermejo-Jambrina, M.; Eder, J.; Helgers, L.C.; Hertoghs, N.; Nijmeijer, B.M.; Stunnenberg, M.; Geijtenbeek, T.B.H. C-Type Lectin Receptors in Antiviral Immunity and Viral Escape. Front. Immunol. 2018, 9, 590. [Google Scholar] [CrossRef] [PubMed]
- Dambuza, I.M.; Brown, G.D. C-type lectins in immunity: Recent developments. Curr. Opin. Immunol. 2015, 32, 21–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geijtenbeek, T.B.; Kwon, D.S.; Torensma, R.; van Vliet, S.J.; van Duijnhoven, G.C.; Middel, J.; Cornelissen, I.L.; Nottet, H.S.; KewalRamani, V.N.; Littman, D.R.; et al. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell 2000, 100, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Moris, A.; Nobile, C.; Buseyne, F.; Porrot, F.; Abastado, J.P.; Schwartz, O. DC-SIGN promotes exogenous MHC-I-restricted HIV-1 antigen presentation. Blood 2004, 103, 2648–2654. [Google Scholar] [CrossRef]
- Schlee, M. Master sensors of pathogenic RNA—RIG-I like receptors. Immunobiology 2013, 218, 1322–1335. [Google Scholar] [CrossRef] [PubMed]
- Fensterl, V.; Chattopadhyay, S.; Sen, G.C. No Love Lost Between Viruses and Interferons. Annu. Rev. Virol. 2015, 2, 549–572. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Hirai, R.; Loo, Y.M.; Owen, D.; Johnson, C.L.; Sinha, S.C.; Akira, S.; Fujita, T.; Gale, M., Jr. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc. Natl. Acad. Sci. USA 2007, 104, 582–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengoku, T.; Nureki, O.; Nakamura, A.; Kobayashi, S.; Yokoyama, S. Structural basis for RNA unwinding by the DEAD-box protein Drosophila Vasa. Cell 2006, 125, 287–300. [Google Scholar] [CrossRef] [Green Version]
- Brubaker, S.W.; Bonham, K.S.; Zanoni, I.; Kagan, J.C. Innate immune pattern recognition: A cell biological perspective. Annu. Rev. Immunol. 2015, 33, 257–290. [Google Scholar] [CrossRef] [Green Version]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzozka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef] [Green Version]
- Fujita, T. Virology. Sensing viral RNA amid your own. Science 2006, 314, 935–936. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337. [Google Scholar] [CrossRef] [Green Version]
- Broquet, A.H.; Hirata, Y.; McAllister, C.S.; Kagnoff, M.F. RIG-I/MDA5/MAVS are required to signal a protective IFN response in rotavirus-infected intestinal epithelium. J. Immunol. 2011, 186, 1618–1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horner, S.M.; Liu, H.M.; Park, H.S.; Briley, J.; Gale, M., Jr. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. USA 2011, 108, 14590–14595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, H.; Kawai, T.; Kato, H.; Sato, S.; Takahashi, K.; Coban, C.; Yamamoto, M.; Uematsu, S.; Ishii, K.J.; Takeuchi, O.; et al. Essential role of IPS-1 in innate immune responses against RNA viruses. J. Exp. Med. 2006, 203, 1795–1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Grandvaux, N.; Zhou, G.P.; Lin, R.; Hiscott, J. Triggering the interferon antiviral response through an IKK-related pathway. Science 2003, 300, 1148–1151. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.M.; Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef]
- Xia, P.; Wang, S.; Gao, P.; Gao, G.; Fan, Z. DNA sensor cGAS-mediated immune recognition. Protein Cell 2016, 7, 777–791. [Google Scholar] [CrossRef] [Green Version]
- Dalskov, L.; Narita, R.; Andersen, L.L.; Jensen, N.; Assil, S.; Kristensen, K.H.; Mikkelsen, J.G.; Fujita, T.; Mogensen, T.H.; Paludan, S.R.; et al. Characterization of distinct molecular interactions responsible for IRF3 and IRF7 phosphorylation and subsequent dimerization. Nucleic Acids Res. 2020, 48, 11421–11433. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef] [Green Version]
- Eckner, R.; Ewen, M.E.; Newsome, D.; Gerdes, M.; DeCaprio, J.A.; Lawrence, J.B.; Livingston, D.M. Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev. 1994, 8, 869–884. [Google Scholar] [CrossRef] [Green Version]
- Yoneyama, M.; Suhara, W.; Fukuhara, Y.; Fukuda, M.; Nishida, E.; Fujita, T. Direct triggering of the type I interferon system by virus infection: Activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 1998, 17, 1087–1095. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, S.; Fensterl, V.; Zhang, Y.; Veleeparambil, M.; Wetzel, J.L.; Sen, G.C. Inhibition of viral pathogenesis and promotion of the septic shock response to bacterial infection by IRF-3 are regulated by the acetylation and phosphorylation of its coactivators. mBio 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Coyne, C.B.; Sarkar, S.N. PKC alpha regulates Sendai virus-mediated interferon induction through HDAC6 and β-catenin. EMBO J. 2011, 30, 4838–4849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suhara, W.; Yoneyama, M.; Kitabayashi, I.; Fujita, T. Direct involvement of CREB-binding protein/p300 in sequence-specific DNA binding of virus-activated interferon regulatory factor-3 holocomplex. J. Biol. Chem. 2002, 277, 22304–22313. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, G.; Kuzmanovic, T.; Zhang, Y.; Peter, C.B.; Veleeparambil, M.; Chakravarti, R.; Sen, G.C.; Chattopadhyay, S. A new mechanism of interferon’s antiviral action: Induction of autophagy, essential for paramyxovirus replication, is inhibited by the interferon stimulated gene, TDRD7. PLoS Pathog. 2018, 14, e1006877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, B.; Novick, D.; Barak, S.; Rubinstein, M. Ligand-induced association of the type I interferon receptor components. Mol. Cell Biol. 1995, 15, 4208–4214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandyopadhyay, S.K.; Leonard, G.T., Jr.; Bandyopadhyay, T.; Stark, G.R.; Sen, G.C. Transcriptional induction by double-stranded RNA is mediated by interferon-stimulated response elements without activation of interferon-stimulated gene factor 3. J. Biol. Chem. 1995, 270, 19624–19629. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, S.; Fensterl, V.; Zhang, Y.; Veleeparambil, M.; Yamashita, M.; Sen, G.C. Role of interferon regulatory factor 3-mediated apoptosis in the establishment and maintenance of persistent infection by Sendai virus. J. Virol. 2013, 87, 16–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chattopadhyay, S.; Kuzmanovic, T.; Zhang, Y.; Wetzel, J.L.; Sen, G.C. Ubiquitination of the Transcription Factor IRF-3 Activates RIPA, the Apoptotic Pathway that Protects Mice from Viral Pathogenesis. Immunity 2016, 44, 1151–1161. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, S.; Marques, J.T.; Yamashita, M.; Peters, K.L.; Smith, K.; Desai, A.; Williams, B.R.; Sen, G.C. Viral apoptosis is induced by IRF-3-mediated activation of Bax. EMBO J. 2010, 29, 1762–1773. [Google Scholar] [CrossRef] [Green Version]
- Peters, K.; Chattopadhyay, S.; Sen, G.C. IRF-3 activation by Sendai virus infection is required for cellular apoptosis and avoidance of persistence. J. Virol. 2008, 82, 3500–3508. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, S.; Yamashita, M.; Zhang, Y.; Sen, G.C. The IRF-3/Bax-mediated apoptotic pathway, activated by viral cytoplasmic RNA and DNA, inhibits virus replication. J. Virol. 2011, 85, 3708–3716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, C.L.; Chattopadhyay, S.; Sen, G.C. Phosphatidylinositol 3-kinase signaling delays sendai virus-induced apoptosis by preventing XIAP degradation. J. Virol. 2011, 85, 5224–5227. [Google Scholar] [CrossRef] [Green Version]
- Manzur, M.; Fleming, P.; Huang, D.C.; Degli-Esposti, M.A.; Andoniou, C.E. Virally mediated inhibition of Bax in leukocytes promotes dissemination of murine cytomegalovirus. Cell Death Differ. 2009, 16, 312–320. [Google Scholar] [CrossRef]
- Sze, A.; Belgnaoui, S.M.; Olagnier, D.; Lin, R.; Hiscott, J.; van Grevenynghe, J. Host restriction factor SAMHD1 limits human T cell leukemia virus type 1 infection of monocytes via STING-mediated apoptosis. Cell Host Microbe 2013, 14, 422–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zierhut, C.; Yamaguchi, N.; Paredes, M.; Luo, J.D.; Carroll, T.; Funabiki, H. The Cytoplasmic DNA Sensor cGAS Promotes Mitotic Cell Death. Cell 2019, 178, 302.e23–315.e23. [Google Scholar] [CrossRef]
- Petrasek, J.; Iracheta-Vellve, A.; Csak, T.; Satishchandran, A.; Kodys, K.; Kurt-Jones, E.A.; Fitzgerald, K.A.; Szabo, G. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc. Natl. Acad. Sci. USA 2013, 110, 16544–16549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iracheta-Vellve, A.; Petrasek, J.; Gyongyosi, B.; Satishchandran, A.; Lowe, P.; Kodys, K.; Catalano, D.; Calenda, C.D.; Kurt-Jones, E.A.; Fitzgerald, K.A.; et al. Endoplasmic Reticulum Stress-induced Hepatocellular Death Pathways Mediate Liver Injury and Fibrosis via Stimulator of Interferon Genes. J. Biol. Chem. 2016, 291, 26794–26805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, J.T.; Cui, C.; Qing, L.; Wang, L.S.; He, T.Y.; Yan, F.; Liu, F.Q.; Shen, Y.H.; Hou, X.G.; Chen, L. Activation of the STING-IRF3 pathway promotes hepatocyte inflammation, apoptosis and induces metabolic disorders in nonalcoholic fatty liver disease. Metabolism 2018, 81, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Garcia, C.; McMullen, M.R.; Chattopadhyay, S.; Roychowdhury, S.; Sen, G.; Nagy, L.E. Nontranscriptional Activity of Interferon Regulatory Factor 3 Protects Mice From High-Fat Diet-Induced Liver Injury. Hepatol. Commun. 2019, 3, 1626–1641. [Google Scholar] [CrossRef]
- Glanz, A.; Chawla, K.; Fabry, S.; Subramanian, G.; Garcia, J.; Jay, B.; Ciricillo, J.; Chakravarti, R.; Taylor, R.T.; Chattopadhyay, S. High Throughput Screening of FDA-Approved Drug Library Reveals the Compounds that Promote IRF3-Mediated Pro-Apoptotic Pathway Inhibit Virus Replication. Viruses 2020, 12, 442. [Google Scholar] [CrossRef] [Green Version]
- Vogel, O.A.; Han, J.; Liang, C.Y.; Manicassamy, S.; Perez, J.T.; Manicassamy, B. The p150 Isoform of ADAR1 Blocks Sustained RLR signaling and Apoptosis during Influenza Virus Infection. PLoS Pathog. 2020, 16, e1008842. [Google Scholar] [CrossRef]
- Sanz-Garcia, C.; Poulsen, K.L.; Bellos, D.; Wang, H.; McMullen, M.R.; Li, X.; Chattopadhyay, S.; Sen, G.; Nagy, L.E. The non-transcriptional activity of IRF3 modulates hepatic immune cell populations in acute-on-chronic ethanol administration in mice. J. Hepatol. 2019, 70, 974–984. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.P.; McBride, K.M.; Weaver, B.K.; Dingwall, C.; Reich, N.C. Regulated nuclear-cytoplasmic localization of interferon regulatory factor 3, a subunit of double-stranded RNA-activated factor 1. Mol. Cell Biol. 2000, 20, 4159–4168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R.; Mamane, Y.; Hiscott, J. Structural and functional analysis of interferon regulatory factor 3: Localization of the transactivation and autoinhibitory domains. Mol. Cell Biol. 1999, 19, 2465–2474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Srinath, H.; Lam, S.S.; Schiffer, C.A.; Royer, W.E., Jr.; Lin, K. Contribution of Ser386 and Ser396 to activation of interferon regulatory factor 3. J. Mol. Biol. 2008, 379, 251–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panne, D.; McWhirter, S.M.; Maniatis, T.; Harrison, S.C. Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. J. Biol. Chem. 2007, 282, 22816–22822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergstroem, B.; Johnsen, I.B.; Nguyen, T.T.; Hagen, L.; Slupphaug, G.; Thommesen, L.; Anthonsen, M.W. Identification of a novel in vivo virus-targeted phosphorylation site in interferon regulatory factor-3 (IRF3). J. Biol. Chem. 2010, 285, 24904–24914. [Google Scholar] [CrossRef] [Green Version]
- Clément, J.F.; Bibeau-Poirier, A.; Gravel, S.P.; Grandvaux, N.; Bonneil, E.; Thibault, P.; Meloche, S.; Servant, M.J. Phosphorylation of IRF-3 on Ser 339 generates a hyperactive form of IRF-3 through regulation of dimerization and CBP association. J. Virol. 2008, 82, 3984–3996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpova, A.Y.; Trost, M.; Murray, J.M.; Cantley, L.C.; Howley, P.M. Interferon regulatory factor-3 is an in vivo target of DNA-PK. Proc. Natl. Acad. Sci. USA 2002, 99, 2818–2823. [Google Scholar] [CrossRef] [Green Version]
- Luo, F.; Liu, H.; Yang, S.; Fang, Y.; Zhao, Z.; Hu, Y.; Jin, Y.; Li, P.; Gao, T.; Cao, C.; et al. Nonreceptor Tyrosine Kinase c-Abl- and Arg-Mediated IRF3 Phosphorylation Regulates Innate Immune Responses by Promoting Type I IFN Production. J. Immunol. 2019, 202, 2254–2265. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Zhu, M.; Pan, R.; Fang, T.; Cao, Y.Y.; Chen, S.; Zhao, X.; Lei, C.Q.; Guo, L.; Chen, Y.; et al. The tumor suppressor PTEN has a critical role in antiviral innate immunity. Nat. Immunol. 2016, 17, 241–249. [Google Scholar] [CrossRef]
- Meng, F.; Zhou, R.; Wu, S.; Zhang, Q.; Jin, Q.; Zhou, Y.; Plouffe, S.W.; Liu, S.; Song, H.; Xia, Z.; et al. Mst1 shuts off cytosolic antiviral defense through IRF3 phosphorylation. Genes Dev. 2016, 30, 1086–1100. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.T.; Chang, L.S.; Chen, C.J.; Doong, S.L.; Chang, C.W.; Chen, M.R. Glycogen synthase kinase 3 negatively regulates IFN regulatory factor 3 transactivation through phosphorylation at its linker region. Innate Immun. 2014, 20, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.T.; Doong, S.L.; Teng, S.C.; Lee, C.P.; Tsai, C.H.; Chen, M.R. Epstein-Barr virus BGLF4 kinase suppresses the interferon regulatory factor 3 signaling pathway. J. Virol. 2009, 83, 1856–1869. [Google Scholar] [CrossRef] [Green Version]
- Saitoh, T.; Tun-Kyi, A.; Ryo, A.; Yamamoto, M.; Finn, G.; Fujita, T.; Akira, S.; Yamamoto, N.; Lu, K.P.; Yamaoka, S. Negative regulation of interferon-regulatory factor 3-dependent innate antiviral response by the prolyl isomerase Pin1. Nat. Immunol. 2006, 7, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Wang, L.; Dai, T.; Jin, K.; Zhang, Z.; Wang, S.; Xie, F.; Fang, P.; Yang, B.; Huang, H.; et al. Tumor-derived exosomes antagonize innate antiviral immunity. Nat. Immunol. 2018, 19, 233–245. [Google Scholar] [CrossRef]
- Bhoj, V.G.; Chen, Z.J. Ubiquitylation in innate and adaptive immunity. Nature 2009, 458, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Chen, Z.J. The role of ubiquitylation in immune defence and pathogen evasion. Nat. Rev. Immunol. 2011, 12, 35–48. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Wang, D.; Wang, P.; Zhao, Y.; You, F. OTUD1 Negatively Regulates Type I IFN Induction by Disrupting Noncanonical Ubiquitination of IRF3. J. Immunol. 2020, 204, 1904–1918. [Google Scholar] [CrossRef] [PubMed]
- Spit, M.; Rieser, E.; Walczak, H. Linear ubiquitination at a glance. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [Green Version]
- Inn, K.S.; Gack, M.U.; Tokunaga, F.; Shi, M.; Wong, L.Y.; Iwai, K.; Jung, J.U. Linear ubiquitin assembly complex negatively regulates RIG-I- and TRIM25-mediated type I interferon induction. Mol. Cell 2011, 41, 354–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieser, E.; Cordier, S.M.; Walczak, H. Linear ubiquitination: A newly discovered regulator of cell signalling. Trends Biochem. Sci. 2013, 38, 94–102. [Google Scholar] [CrossRef]
- Kayagaki, N.; Phung, Q.; Chan, S.; Chaudhari, R.; Quan, C.; O’Rourke, K.M.; Eby, M.; Pietras, E.; Cheng, G.; Bazan, J.F.; et al. DUBA: A deubiquitinase that regulates type I interferon production. Science 2007, 318, 1628–1632. [Google Scholar] [CrossRef] [PubMed]
- Sears, N.; Sen, G.C.; Stark, G.R.; Chattopadhyay, S. Caspase-8-mediated cleavage inhibits IRF-3 protein by facilitating its proteasome-mediated degradation. J. Biol. Chem. 2011, 286, 33037–33044. [Google Scholar] [CrossRef] [Green Version]
- Goswami, R.; Majumdar, T.; Dhar, J.; Chattopadhyay, S.; Bandyopadhyay, S.K.; Verbovetskaya, V.; Sen, G.C.; Barik, S. Viral degradasome hijacks mitochondria to suppress innate immunity. Cell Res. 2013, 23, 1025–1042. [Google Scholar] [CrossRef] [Green Version]
- Bibeau-Poirier, A.; Gravel, S.P.; Clement, J.F.; Rolland, S.; Rodier, G.; Coulombe, P.; Hiscott, J.; Grandvaux, N.; Meloche, S.; Servant, M.J. Involvement of the IkappaB kinase (IKK)-related kinases tank-binding kinase 1/IKKi and cullin-based ubiquitin ligases in IFN regulatory factor-3 degradation. J. Immunol. 2006, 177, 5059–5067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Hayward, G.S. The ubiquitin E3 ligase RAUL negatively regulates type i interferon through ubiquitination of the transcription factors IRF7 and IRF3. Immunity 2010, 33, 863–877. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Tian, Y.; Wang, R.P.; Gao, D.; Zhang, Y.; Diao, F.C.; Chen, D.Y.; Zhai, Z.H.; Shu, H.B. Negative feedback regulation of cellular antiviral signaling by RBCK1-mediated degradation of IRF3. Cell Res. 2008, 18, 1096–1104. [Google Scholar] [CrossRef]
- Higgs, R.; Ni Gabhann, J.; Ben Larbi, N.; Breen, E.P.; Fitzgerald, K.A.; Jefferies, C.A. The E3 ubiquitin ligase Ro52 negatively regulates IFN-beta production post-pathogen recognition by polyubiquitin-mediated degradation of IRF3. J. Immunol. 2008, 181, 1780–1786. [Google Scholar] [CrossRef]
- Rhodes, D.A.; Trowsdale, J. TRIM21 is a trimeric protein that binds IgG Fc via the B30.2 domain. Mol. Immunol. 2007, 44, 2406–2414. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.S.; Imm, J.H.; Min, C.K.; Kim, K.J.; Cha, S.S.; Oh, B.H. Structural and functional insights into the B30.2/SPRY domain. EMBO J. 2006, 25, 1353–1363. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Zhao, W.; Zhao, K.; Zhang, L.; Gao, C. TRIM26 negatively regulates interferon-beta production and antiviral response through polyubiquitination and degradation of nuclear IRF3. PLoS Pathog. 2015, 11, e1004726. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Zhou, M.T.; Hu, M.M.; Hu, Y.H.; Zhang, J.; Guo, L.; Zhong, B.; Shu, H.B. RNF26 temporally regulates virus-triggered type I interferon induction by two distinct mechanisms. PLoS Pathog. 2014, 10, e1004358. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.Q.; Zhang, Y.; Xia, T.; Jiang, L.Q.; Zhong, B.; Shu, H.B. FoxO1 negatively regulates cellular antiviral response by promoting degradation of IRF3. J. Biol. Chem. 2013, 288, 12596–12604. [Google Scholar] [CrossRef] [Green Version]
- Litvak, V.; Ratushny, A.V.; Lampano, A.E.; Schmitz, F.; Huang, A.C.; Raman, A.; Rust, A.G.; Bergthaler, A.; Aitchison, J.D.; Aderem, A. A FOXO3-IRF7 gene regulatory circuit limits inflammatory sequelae of antiviral responses. Nature 2012, 490, 421–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Zhu, H.; Yu, J.; Li, H.; Ge, J.; Chen, W. c-Cbl-mediated ubiquitination of IRF3 negatively regulates IFN-beta production and cellular antiviral response. Cell. Signal. 2016, 28, 1683–1693. [Google Scholar] [CrossRef]
- Xiong, H.; Li, H.; Kong, H.J.; Chen, Y.; Zhao, J.; Xiong, S.; Huang, B.; Gu, H.; Mayer, L.; Ozato, K.; et al. Ubiquitin-dependent degradation of interferon regulatory factor-8 mediated by Cbl down-regulates interleukin-12 expression. J. Biol. Chem. 2005, 280, 23531–23539. [Google Scholar] [CrossRef] [Green Version]
- Zeng, W.; Xu, M.; Liu, S.; Sun, L.; Chen, Z.J. Key role of Ubc5 and lysine-63 polyubiquitination in viral activation of IRF3. Mol. Cell 2009, 36, 315–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, D.; Song, J.; Sun, Y.; Qi, F.; Liu, L.; Jin, Y.; McNutt, M.A.; Yin, Y. Mutations of deubiquitinase OTUD1 are associated with autoimmune disorders. J. Autoimmun. 2018, 94, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Jayappa, K.D.; Ao, Z.; Qiu, X.; Su, R.C.; Yao, X. Noncovalent SUMO-interaction motifs in HIV integrase play important roles in SUMOylation, cofactor binding, and virus replication. Virol. J. 2019, 16, 42. [Google Scholar] [CrossRef]
- Hu, M.M.; Yang, Q.; Xie, X.Q.; Liao, C.Y.; Lin, H.; Liu, T.T.; Yin, L.; Shu, H.B. Sumoylation Promotes the Stability of the DNA Sensor cGAS and the Adaptor STING to Regulate the Kinetics of Response to DNA Virus. Immunity 2016, 45, 555–569. [Google Scholar] [CrossRef] [Green Version]
- Kubota, T.; Matsuoka, M.; Chang, T.H.; Tailor, P.; Sasaki, T.; Tashiro, M.; Kato, A.; Ozato, K. Virus infection triggers SUMOylation of IRF3 and IRF7, leading to the negative regulation of type I interferon gene expression. J. Biol. Chem. 2008, 283, 25660–25670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, Y.; Liu, T.T.; Zhou, Q.; Li, S.; Mao, A.P.; Li, Y.; Liu, L.J.; Cheng, J.K.; Shu, H.B. SENP2 negatively regulates cellular antiviral response by deSUMOylating IRF3 and conditioning it for ubiquitination and degradation. J. Mol. Cell Biol. 2011, 3, 283–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maarifi, G.; Hannoun, Z.; Geoffroy, M.C.; El Asmi, F.; Zarrouk, K.; Nisole, S.; Blondel, D.; Chelbi-Alix, M.K. MxA Mediates SUMO-Induced Resistance to Vesicular Stomatitis Virus. J. Virol. 2016, 90, 6598–6610. [Google Scholar] [CrossRef] [Green Version]
- Dalle-Donne, I.; Rossi, R.; Colombo, G.; Giustarini, D.; Milzani, A. Protein S-glutathionylation: A regulatory device from bacteria to humans. Trends Biochem. Sci. 2009, 34, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ye, Z.W.; Singh, S.; Townsend, D.M.; Tew, K.D. An evolving understanding of the S-glutathionylation cycle in pathways of redox regulation. Free Radic. Biol. Med. 2018, 120, 204–216. [Google Scholar] [CrossRef]
- Shelton, M.D.; Mieyal, J.J. Regulation by reversible S-glutathionylation: Molecular targets implicated in inflammatory diseases. Mol. Cells 2008, 25, 332–346. [Google Scholar]
- Prinarakis, E.; Chantzoura, E.; Thanos, D.; Spyrou, G. S-glutathionylation of IRF3 regulates IRF3-CBP interaction and activation of the IFN beta pathway. EMBO J. 2008, 27, 865–875. [Google Scholar] [CrossRef]
- Mowen, K.A.; David, M. Unconventional post-translational modifications in immunological signaling. Nat. Immunol. 2014, 15, 512–520. [Google Scholar] [CrossRef]
- Biggar, K.K.; Li, S.S. Non-histone protein methylation as a regulator of cellular signalling and function. Nat. Rev. Mol. Cell Biol. 2015, 16, 5–17. [Google Scholar] [CrossRef]
- Wang, C.; Wang, Q.; Xu, X.; Xie, B.; Zhao, Y.; Li, N.; Cao, X. The methyltransferase NSD3 promotes antiviral innate immunity via direct lysine methylation of IRF3. J. Exp. Med. 2017, 214, 3597–3610. [Google Scholar] [CrossRef] [Green Version]
- Morales, D.J.; Lenschow, D.J. The antiviral activities of ISG15. J. Mol. Biol 2013, 425, 4995–5008. [Google Scholar] [CrossRef]
- Haas, A.L.; Ahrens, P.; Bright, P.M.; Ankel, H. Interferon induces a 15-kilodalton protein exhibiting marked homology to ubiquitin. J. Biol. Chem. 1987, 262, 11315–11323. [Google Scholar] [CrossRef]
- Dao, C.T.; Zhang, D.E. ISG15: A ubiquitin-like enigma. Front. Biosci. 2005, 10, 2701–2722. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, J.; Wang, M.; Fu, Z.; Klein, J.M.; Haas, A.L.; Kim, J.J. Crystal structure of the interferon-induced ubiquitin-like protein ISG15. J. Biol. Chem. 2005, 280, 27356–27365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Denison, C.; Huibregtse, J.M.; Gygi, S.; Krug, R.M. Human ISG15 conjugation targets both IFN-induced and constitutively expressed proteins functioning in diverse cellular pathways. Proc. Natl. Acad. Sci. USA 2005, 102, 10200–10205. [Google Scholar] [CrossRef] [Green Version]
- Arimoto, K.; Konishi, H.; Shimotohno, K. UbcH8 regulates ubiquitin and ISG15 conjugation to RIG-I. Mol. Immunol. 2008, 45, 1078–1084. [Google Scholar] [CrossRef]
- Giannakopoulos, N.V.; Luo, J.K.; Papov, V.; Zou, W.; Lenschow, D.J.; Jacobs, B.S.; Borden, E.C.; Li, J.; Virgin, H.W.; Zhang, D.E. Proteomic identification of proteins conjugated to ISG15 in mouse and human cells. Biochem. Biophys. Res. Commun. 2005, 336, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Dastur, A.; Beaudenon, S.; Kelley, M.; Krug, R.M.; Huibregtse, J.M. Herc5, an interferon-induced HECT E3 enzyme, is required for conjugation of ISG15 in human cells. J. Biol. Chem. 2006, 281, 4334–4338. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.J.; Pung, Y.F.; Sze, N.S.; Chin, K.C. HERC5 is an IFN-induced HECT-type E3 protein ligase that mediates type I IFN-induced ISGylation of protein targets. Proc. Natl. Acad. Sci. USA 2006, 103, 10735–10740. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.X.; Yang, K.; Liu, X.; Liu, X.Y.; Wei, B.; Shan, Y.F.; Zhu, L.H.; Wang, C. Positive regulation of interferon regulatory factor 3 activation by Herc5 via ISG15 modification. Mol. Cell Biol. 2010, 30, 2424–2436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, D.M. The presence of acetyl groups of histones. Biochem. J. 1963, 87, 258–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef] [Green Version]
- Caillaud, A.; Prakash, A.; Smith, E.; Masumi, A.; Hovanessian, A.G.; Levy, D.E.; Marie, I. Acetylation of interferon regulatory factor-7 by p300/CREB-binding protein (CBP)-associated factor (PCAF) impairs its DNA binding. J. Biol. Chem. 2002, 277, 49417–49421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Zang, C.; Cui, K.; Schones, D.E.; Barski, A.; Peng, W.; Zhao, K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 2009, 138, 1019–1031. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Gao, J.S.; Guan, Y.J.; McLane, K.E.; Yuan, Z.L.; Ramratnam, B.; Chin, Y.E. Acetylation-dependent signal transduction for type I interferon receptor. Cell 2007, 131, 93–105. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Yu, Y.; Eugene Chin, Y.; Xia, Q. The role of acetylation in TLR4-mediated innate immune responses. Immunol. Cell Biol. 2013, 91, 611–614. [Google Scholar] [CrossRef]
- Masumi, A.; Ozato, K. Coactivator p300 acetylates the interferon regulatory factor-2 in U937 cells following phorbol ester treatment. J. Biol. Chem. 2001, 276, 20973–20980. [Google Scholar] [CrossRef] [Green Version]
- Huai, W.; Liu, X.; Wang, C.; Zhang, Y.; Chen, X.; Chen, X.; Xu, S.; Thomas, T.; Li, N.; Cao, X. KAT8 selectively inhibits antiviral immunity by acetylating IRF3. J. Exp. Med. 2019, 216, 772–785. [Google Scholar] [CrossRef] [Green Version]
- Perlman, S.; Netland, J. Coronaviruses post-SARS: Update on replication and pathogenesis. Nat. Rev. Microbiol. 2009, 7, 439–450. [Google Scholar] [CrossRef] [Green Version]
- Zhong, N.S.; Zheng, B.J.; Li, Y.M.; Poon, L.L.M.; Xie, Z.H.; Chan, K.H.; Li, P.H.; Tan, S.Y.; Chang, Q.; Xie, J.P.; et al. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People’s Republic of China, in February, 2003. Lancet 2003, 362, 1353–1358. [Google Scholar] [CrossRef] [Green Version]
- Golonka, R.M.; Saha, P.; Yeoh, B.S.; Chattopadhyay, S.; Gewirtz, A.T.; Joe, B.; Vijay-Kumar, M. Harnessing innate immunity to eliminate SARS-CoV-2 and ameliorate COVID-19 disease. Physiol. Genom. 2020, 52, 217–221. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Liu, Y.; Zhang, X. Murine coronavirus induces type I interferon in oligodendrocytes through recognition by RIG-I and MDA5. J. Virol. 2010, 84, 6472–6482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kell, A.M.; Gale, M., Jr. RIG-I in RNA virus recognition. Virology 2015, 479–480, 110–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Totura, A.L.; Whitmore, A.; Agnihothram, S.; Schäfer, A.; Katze, M.G.; Heise, M.T.; Baric, R.S. Toll-Like Receptor 3 Signaling via TRIF Contributes to a Protective Innate Immune Response to Severe Acute Respiratory Syndrome Coronavirus Infection. mBio 2015, 6, e00638-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegel, M.; Pichlmair, A.; Martínez-Sobrido, L.; Cros, J.; García-Sastre, A.; Haller, O.; Weber, F. Inhibition of Beta interferon induction by severe acute respiratory syndrome coronavirus suggests a two-step model for activation of interferon regulatory factor 3. J. Virol. 2005, 79, 2079–2086. [Google Scholar] [CrossRef] [Green Version]
- Totura, A.L.; Baric, R.S. SARS coronavirus pathogenesis: Host innate immune responses and viral antagonism of interferon. Curr. Opin. Virol. 2012, 2, 264–275. [Google Scholar] [CrossRef]
- Clementz, M.A.; Chen, Z.; Banach, B.S.; Wang, Y.; Sun, L.; Ratia, K.; Baez-Santos, Y.M.; Wang, J.; Takayama, J.; Ghosh, A.K.; et al. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J. Virol. 2010, 84, 4619–4629. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.H.; Fung, T.S.; Fang, S.; Huang, M.; Le, M.T.; Liu, D.X. Accessory proteins 8b and 8ab of severe acute respiratory syndrome coronavirus suppress the interferon signaling pathway by mediating ubiquitin-dependent rapid degradation of interferon regulatory factor 3. Virology 2018, 515, 165–175. [Google Scholar] [CrossRef]
- Devaraj, S.G.; Wang, N.; Chen, Z.; Chen, Z.; Tseng, M.; Barretto, N.; Lin, R.; Peters, C.J.; Tseng, C.T.; Baker, S.C.; et al. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J. Biol. Chem. 2007, 282, 32208–32221. [Google Scholar] [CrossRef] [Green Version]
- Rota, P.A.; Oberste, M.S.; Monroe, S.S.; Nix, W.A.; Campagnoli, R.; Icenogle, J.P.; Peñaranda, S.; Bankamp, B.; Maher, K.; Chen, M.H.; et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 2003, 300, 1394–1399. [Google Scholar] [CrossRef] [Green Version]
- Schoeman, D.; Fielding, B.C. Coronavirus envelope protein: Current knowledge. Virol. J. 2019, 16, 69. [Google Scholar] [CrossRef] [Green Version]
- Siu, K.L.; Kok, K.H.; Ng, M.H.; Poon, V.K.; Yuen, K.Y.; Zheng, B.J.; Jin, D.Y. Severe acute respiratory syndrome coronavirus M protein inhibits type I interferon production by impeding the formation of TRAF3.TANK.TBK1/IKKepsilon complex. J. Biol. Chem. 2009, 284, 16202–16209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Zhuang, M.-W.; Han, L.; Zhang, J.; Nan, M.-L.; Gao, C.; Wang, P.-H. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Membrane (M) Protein Inhibits Type I and III Interferon Production by Targeting RIG-I/MDA-5 Signaling. bioRxiv 2020, 5, 299. [Google Scholar] [CrossRef]
- Lui, P.Y.; Wong, L.Y.; Fung, C.L.; Siu, K.L.; Yeung, M.L.; Yuen, K.S.; Chan, C.P.; Woo, P.C.; Yuen, K.Y.; Jin, D.Y. Middle East respiratory syndrome coronavirus M protein suppresses type I interferon expression through the inhibition of TBK1-dependent phosphorylation of IRF3. Emerg. Microbes Infect. 2016, 5, e39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Zhang, L.; Geng, H.; Deng, Y.; Huang, B.; Guo, Y.; Zhao, Z.; Tan, W. The structural and accessory proteins M, ORF 4a, ORF 4b, and ORF 5 of Middle East respiratory syndrome coronavirus (MERS-CoV) are potent interferon antagonists. Protein Cell 2013, 4, 951–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Pan, J.; Tao, J.; Guo, D. SARS-CoV nucleocapsid protein antagonizes IFN-β response by targeting initial step of IFN-β induction pathway, and its C-terminal region is critical for the antagonism. Virus Genes 2011, 42, 37–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Li, W.; Gao, T.; Cui, Y.; Jin, Y.; Li, P.; Ma, Q.; Liu, X.; Cao, C. The Severe Acute Respiratory Syndrome Coronavirus Nucleocapsid Inhibits Type I Interferon Production by Interfering with TRIM25-Mediated RIG-I Ubiquitination. J. Virol. 2017, 91, e02143-16. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.Y.; Liu, H.M.; Chang, M.F.; Chang, S.C. Middle East respiratory syndrome coronavirus nucleocapsid protein suppresses type I and type III interferon induction by targeting RIG-I signaling. J. Virol. 2020, 94, e00099-20. [Google Scholar] [CrossRef]
- Chen, X.; Yang, X.; Zheng, Y.; Yang, Y.; Xing, Y.; Chen, Z. SARS coronavirus papain-like protease inhibits the type I interferon signaling pathway through interaction with the STING-TRAF3-TBK1 complex. Protein Cell 2014, 5, 369–381. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chen, X.; Bian, G.; Tu, J.; Xing, Y.; Wang, Y.; Chen, Z. Proteolytic processing, deubiquitinase and interferon antagonist activities of Middle East respiratory syndrome coronavirus papain-like protease. J. Gen. Virol. 2014, 95, 614–626. [Google Scholar] [CrossRef]
- Wathelet, M.G.; Orr, M.; Frieman, M.B.; Baric, R.S. Severe acute respiratory syndrome coronavirus evades antiviral signaling: Role of nsp1 and rational design of an attenuated strain. J. Virol. 2007, 81, 11620–11633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuen, C.K.; Lam, J.Y.; Wong, W.M.; Mak, L.F.; Wang, X.; Chu, H.; Cai, J.P.; Jin, D.Y.; To, K.K.; Chan, J.F.; et al. SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerg. Microbes Infect. 2020, 9, 1418–1428. [Google Scholar] [CrossRef] [PubMed]
- Michel, C.J.; Mayer, C.; Poch, O.; Thompson, J.D. Characterization of accessory genes in coronavirus genomes. Virol. J. 2020, 17, 131. [Google Scholar] [CrossRef]
- Narayanan, K.; Huang, C.; Makino, S. SARS coronavirus accessory proteins. Virus Res. 2008, 133, 113–121. [Google Scholar] [CrossRef]
- Kopecky-Bromberg, S.A.; Martínez-Sobrido, L.; Frieman, M.; Baric, R.A.; Palese, P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J. Virol. 2007, 81, 548–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, X.; Dong, X.; Ma, R.; Wang, W.; Xiao, X.; Tian, Z.; Wang, C.; Wang, Y.; Li, L.; Ren, L.; et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 2020, 11, 3810. [Google Scholar] [CrossRef]
- Sato, M.; Suemori, H.; Hata, N.; Asagiri, M.; Ogasawara, K.; Nakao, K.; Nakaya, T.; Katsuki, M.; Noguchi, S.; Tanaka, N.; et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 2000, 13, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.W.; King, K.; Tu, J.; Sanchez, M.; Luster, A.D.; Shresta, S. The roles of IRF-3 and IRF-7 in innate antiviral immunity against dengue virus. J. Immunol. 2013, 191, 4194–4201. [Google Scholar] [CrossRef] [Green Version]
- Lazear, H.M.; Lancaster, A.; Wilkins, C.; Suthar, M.S.; Huang, A.; Vick, S.C.; Clepper, L.; Thackray, L.; Brassil, M.M.; Virgin, H.W.; et al. IRF-3, IRF-5, and IRF-7 coordinately regulate the type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PLoS Pathog. 2013, 9, e1003118. [Google Scholar] [CrossRef]
- Mogensen, T.H. IRF and STAT Transcription Factors—From Basic Biology to Roles in Infection, Protective Immunity, and Primary Immunodeficiencies. Front. Immunol. 2018, 9, 3047. [Google Scholar] [CrossRef]
- Jorgensen, S.E.; Al-Mousawi, A.; Assing, K.; Hartling, U.; Grosen, D.; Fisker, N.; Nielsen, C.; Jakobsen, M.A.; Mogensen, T.H. STK4 Deficiency Impairs Innate Immunity and Interferon Production Through Negative Regulation of TBK1-IRF3 Signaling. J. Clin. Immunol. 2021, 41, 109–124. [Google Scholar] [CrossRef]
- Zhang, S.Y.; Casanova, J.L. Inborn errors underlying herpes simplex encephalitis: From TLR3 to IRF3. J. Exp. Med. 2015, 212, 1342–1343. [Google Scholar] [CrossRef] [Green Version]
- Andersen, L.L.; Mork, N.; Reinert, L.S.; Kofod-Olsen, E.; Narita, R.; Jorgensen, S.E.; Skipper, K.A.; Honing, K.; Gad, H.H.; Ostergaard, L.; et al. Functional IRF3 deficiency in a patient with herpes simplex encephalitis. J. Exp. Med. 2015, 212, 1371–1379. [Google Scholar] [CrossRef]
- Levy, R.; Bastard, P.; Lanternier, F.; Lecuit, M.; Zhang, S.Y.; Casanova, J.L. IFN-alpha2a Therapy in Two Patients with Inborn Errors of TLR3 and IRF3 Infected with SARS-CoV-2. J. Clin. Immunol. 2021, 41, 26–27. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.K.; Huang, S.X.L.; Chen, J.; Kerner, G.; Gilliaux, O.; Bastard, P.; Dobbs, K.; Hernandez, N.; Goudin, N.; Hasek, M.L.; et al. Severe influenza pneumonitis in children with inherited TLR3 deficiency. J. Exp. Med. 2019, 216, 2038–2056. [Google Scholar] [CrossRef]
- Ogunjimi, B.; Zhang, S.Y.; Sorensen, K.B.; Skipper, K.A.; Carter-Timofte, M.; Kerner, G.; Luecke, S.; Prabakaran, T.; Cai, Y.; Meester, J.; et al. Inborn errors in RNA polymerase III underlie severe varicella zoster virus infections. J. Clin. Investig. 2017, 127, 3543–3556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, S.E.; Christiansen, M.; Ryo, L.B.; Gad, H.H.; Gjedsted, J.; Staeheli, P.; Mikkelsen, J.G.; Storgaard, M.; Hartmann, R.; Mogensen, T.H. Defective RNA sensing by RIG-I in severe influenza virus infection. Clin. Exp. Immunol. 2018, 192, 366–376. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, M.M.; Jorgensen, S.E.; Gad, H.H.; Storgaard, M.; Gjedsted, J.; Christiansen, M.; Hartmann, R.; Mogensen, T.H. Defective interferon priming and impaired antiviral responses in a patient with an IRF7 variant and severe influenza. Med. Microbiol. Immunol. 2019, 208, 869–876. [Google Scholar] [CrossRef]
- Kim, T.; Kim, T.Y.; Lee, W.G.; Yim, J.; Kim, T.K. Signaling pathways to the assembly of an interferon-beta enhanceosome. Chemical genetic studies with a small molecule. J. Biol. Chem. 2000, 275, 16910–16917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattabhi, S.; Wilkins, C.R.; Dong, R.; Knoll, M.L.; Posakony, J.; Kaiser, S.; Mire, C.E.; Wang, M.L.; Ireton, R.C.; Geisbert, T.W.; et al. Targeting Innate Immunity for Antiviral Therapy through Small Molecule Agonists of the RLR Pathway. J. Virol. 2015, 90, 2372–2387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, R.R.; Wilkins, C.; Pattabhi, S.; Dong, R.; Loo, Y.; Gale, M., Jr. Transcriptional analysis of antiviral small molecule therapeutics as agonists of the RLR pathway. Genom. Data 2016, 7, 290–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Goyal, A.; Perelson, A.S.; Ishida, Y.; Saito, T.; Gale, M., Jr. Suppression of hepatitis B virus through therapeutic activation of RIG-I and IRF3 signaling in hepatocytes. iScience 2021, 24, 101969. [Google Scholar] [CrossRef]
- Probst, P.; Grigg, J.B.; Wang, M.; Munoz, E.; Loo, Y.M.; Ireton, R.C.; Gale, M., Jr.; Iadonato, S.P.; Bedard, K.M. A small-molecule IRF3 agonist functions as an influenza vaccine adjuvant by modulating the antiviral immune response. Vaccine 2017, 35, 1964–1971. [Google Scholar] [CrossRef] [PubMed]
- Bonam, S.R.; Muller, S.; Bayry, J.; Klionsky, D.J. Autophagy as an emerging target for COVID-19: Lessons from an old friend, chloroquine. Autophagy 2020, 16, 2260–2266. [Google Scholar] [CrossRef] [PubMed]
- Hardy, S.; Jackson, B.; Goodbourn, S.; Seago, J. Classical swine fever virus N(pro) antagonises IRF3 to prevent IFN-independent TLR3 and RIG-I-mediated apoptosis. J. Virol. 2020. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glanz, A.; Chakravarty, S.; Varghese, M.; Kottapalli, A.; Fan, S.; Chakravarti, R.; Chattopadhyay, S. Transcriptional and Non-Transcriptional Activation, Posttranslational Modifications, and Antiviral Functions of Interferon Regulatory Factor 3 and Viral Antagonism by the SARS-Coronavirus. Viruses 2021, 13, 575. https://0-doi-org.brum.beds.ac.uk/10.3390/v13040575
Glanz A, Chakravarty S, Varghese M, Kottapalli A, Fan S, Chakravarti R, Chattopadhyay S. Transcriptional and Non-Transcriptional Activation, Posttranslational Modifications, and Antiviral Functions of Interferon Regulatory Factor 3 and Viral Antagonism by the SARS-Coronavirus. Viruses. 2021; 13(4):575. https://0-doi-org.brum.beds.ac.uk/10.3390/v13040575
Chicago/Turabian StyleGlanz, Anna, Sukanya Chakravarty, Merina Varghese, Anita Kottapalli, Shumin Fan, Ritu Chakravarti, and Saurabh Chattopadhyay. 2021. "Transcriptional and Non-Transcriptional Activation, Posttranslational Modifications, and Antiviral Functions of Interferon Regulatory Factor 3 and Viral Antagonism by the SARS-Coronavirus" Viruses 13, no. 4: 575. https://0-doi-org.brum.beds.ac.uk/10.3390/v13040575