
Limited Genetic Diversity Detected in Middle East Respiratory Syndrome-Related Coronavirus Variants Circulating in Dromedary Camels in Jordan

 , ,
, ,  , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
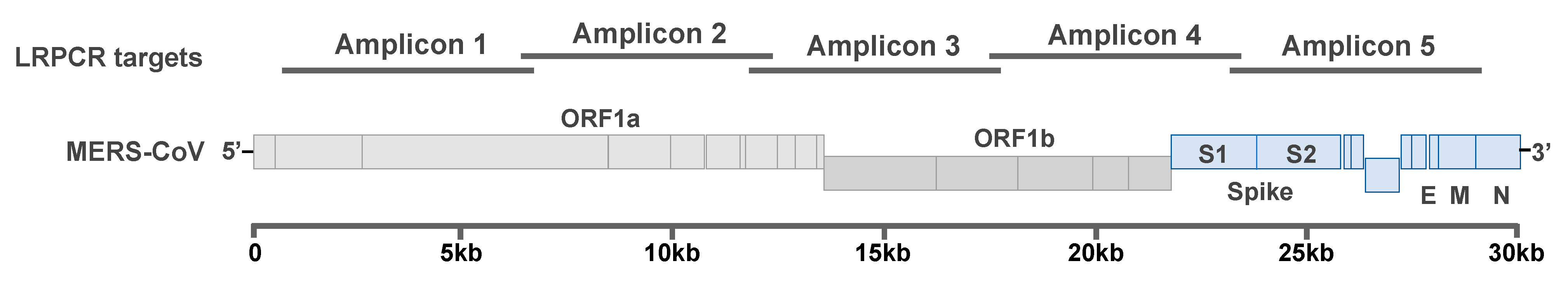
2.1. Amplification of MERS-CoV Genome
2.2. Library Preparation and Sequencing
2.3. Phylogenomic Analyses
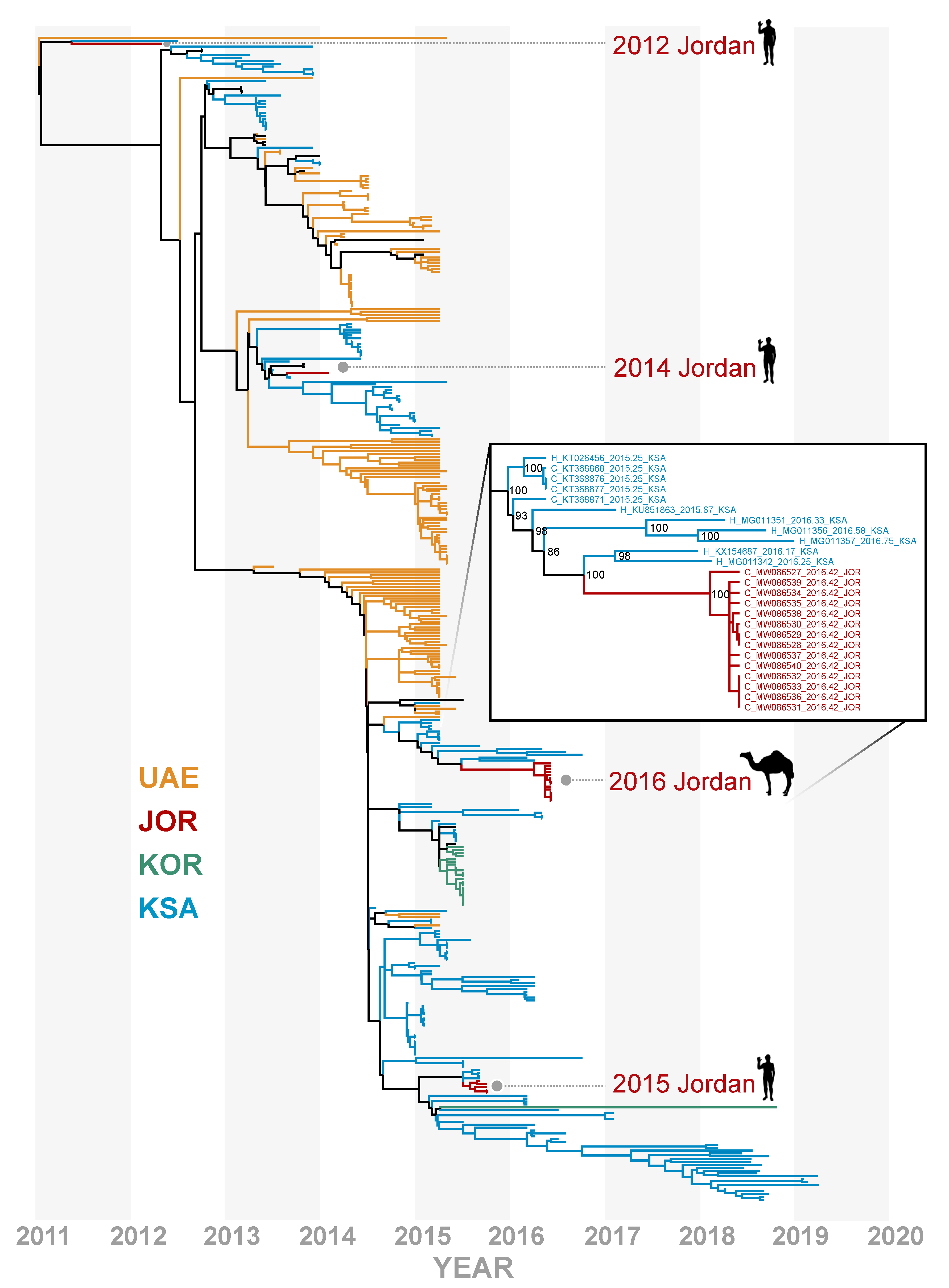
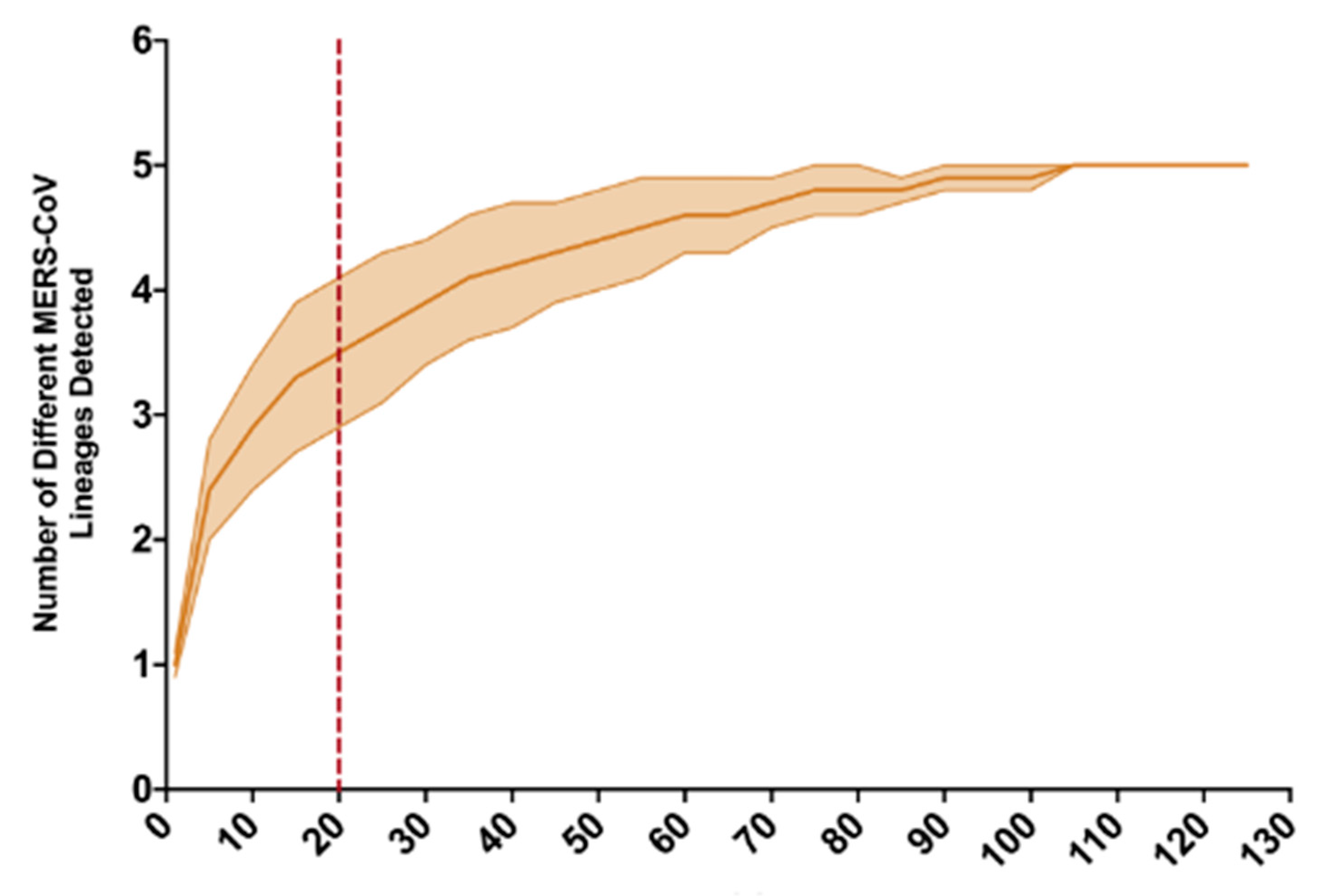
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. MERS Situation Update. In Eastern Mediterranean Regional Office; Egypt, C., Ed.; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Sabir, J.S.; Lam, T.T.; Ahmed, M.M.; Li, L.; Shen, Y.; Abo-Aba, S.E.; Qureshi, M.I.; Abu-Zeid, M.; Zhang, Y.; Khiyami, M.A.; et al. Co-circulation of three camel coronavirus species and recombination of MERS-CoVs in Saudi Arabia. Science 2016, 351, 81–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yusof, M.F.; Queen, K.; Eltahir, Y.M.; Paden, C.R.; Al Hammadi, Z.; Tao, Y.; Li, Y.; Khalafalla, A.I.; Shi, M.; Zhang, J.; et al. Diversity of Middle East respiratory syndrome coronaviruses in 109 dromedary camels based on full-genome sequencing, Abu Dhabi, United Arab Emirates. Emerg. Microbes Infect. 2017, 6, e101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudas, G.; Carvalho, L.M.; Rambaut, A.; Bedford, T. MERS-CoV spillover at the camel-human interface. Elife 2018, 7, e31257. [Google Scholar] [CrossRef] [PubMed]
- van Doremalen, N.; Hijazeen, Z.S.; Holloway, P.; Al Omari, B.; McDowell, C.; Adney, D.; Talafha, H.A.; Guitian, J.; Steel, J.; Amarin, N.; et al. High Prevalence of Middle East Respiratory Coronavirus in Young Dromedary Camels in Jordan. Vector Borne Zoonotic Dis. 2017, 17, 155–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hijawi, B.; Abdallat, M.; Sayaydeh, A.; Alqasrawi, S.; Haddadin, A.; Jaarour, N.; Alsheikh, S.; Alsanouri, T. Novel coronavirus infections in Jordan, April 2012: Epidemiological findings from a retrospective investigation. East. Mediterr. Health J. 2013, 19 (Suppl. 1), S12–S18. [Google Scholar] [CrossRef] [PubMed]
- Lamers, M.M.; Raj, V.S.; Shafei, M.; Ali, S.S.; Abdallh, S.M.; Gazo, M.; Nofal, S.; Lu, X.; Erdman, D.D.; Koopmans, M.P.; et al. Deletion Variants of Middle East Respiratory Syndrome Coronavirus from Humans, Jordan, 2015. Emerg. Infect. Dis. 2016, 22, 716–719. [Google Scholar] [CrossRef] [PubMed]
- Seifert, S.N.; Schulz, J.E.; Matson, M.J.; Bushmaker, T.; Marzi, A.; Munster, V.J. Long-Range Polymerase Chain Reaction Method for Sequencing the Ebola Virus Genome from Ecological and Clinical Samples. J. Infect. Dis. 2018, 218 (Suppl. 5), S301–S304. [Google Scholar] [CrossRef] [PubMed]
- Yinda, C.K.; Seifert, S.N.; Macmenamin, P.; van Doremalen, N.; Kim, L.; Bushmaker, T.; de Wit, E.; Quinones, M.; Munster, V.J. A Novel Field-Deployable Method for Sequencing and Analyses of Henipavirus Genomes from Complex Samples on the MinION Platform. J. Infect. Dis. 2020, 221 (Suppl. 4), S383–S388. [Google Scholar] [CrossRef] [PubMed]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.K.W.; Hui, K.P.Y.; Perera, R.; Miguel, E.; Niemeyer, D.; Zhao, J.; Channappanavar, R.; Dudas, G.; Oladipo, J.O.; Traore, A.; et al. MERS coronaviruses from camels in Africa exhibit region-dependent genetic diversity. Proc. Natl. Acad. Sci. USA 2018, 115, 3144–3149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.; El-Shesheny, R.; Kandeil, A.; Shehata, M.; Elsokary, B.; Gomaa, M.; Hassan, N.; El Sayed, A.; El-Taweel, A.; Sobhy, H.; et al. Cross-sectional surveillance of Middle East respiratory syndrome coronavirus (MERS-CoV) in dromedary camels and other mammals in Egypt, August 2015 to January 2016. Eurosurveillance 2017, 22, 30487. [Google Scholar] [CrossRef] [PubMed]
- Ommeh, S.; Zhang, W.; Zohaib, A.; Chen, J.; Zhang, H.; Hu, B.; Ge, X.Y.; Yang, X.L.; Masika, M.; Obanda, V.; et al. Genetic Evidence of Middle East Respiratory Syndrome Coronavirus (MERS-Cov) and Widespread Seroprevalence among Camels in Kenya. Virol. Sin. 2018, 33, 484–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolah, A.M.; Al Masaudi, S.B.; El-Kafrawy, S.A.; Mirza, A.A.; Harakeh, S.M.; Hassan, A.M.; Alsaadi, M.A.; Alzahrani, A.A.; Alsaaidi, G.A.; Amor, N.M.S.; et al. Cross-sectional prevalence study of MERS-CoV in local and imported dromedary camels in Saudi Arabia, 2016–2018. PLoS ONE 2020, 15, e0232790. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Amplicon | Forward Primer Sequence (5′–3′) | Reverse Primer Sequence (5′–3′) | Round of LRPCR |
|---|---|---|---|
| 1A | AACGAACTTAAATAAAAGCCCTGTTGTTT | GGGCATCTTCAAACATAACATCACTT | Round 1 |
| 2A | CACTTTCACTGCTACCACTGCTGTA | CGCGAAGTTTATTTGAAGCACA | |
| 3A | CGCCTATGAGAAGGATAAGGCAGT | GATGCAGACGTTAATTCAAAGCCAT | |
| 4A | GCCAGTTGGTGTTGTAGACACTGA | TTGCTAGGGTAATAACCAACATGCAT | |
| 5A | GCTCGTGATCTTATTTGTGCTCAA | GCAGAGGTGACAGTCTTTAACATTCTCT | |
| 1B | AACGAACTTAAATAAAAGCCCTGTTGTTT | ATACTTAAATCAACAGCAGCAGTGCAA | Round 2 |
| 2B | CACTTTCACTGCTACCACTGCTGTA | CTAAGAGGTATACAACCATTCCTAGCGTT | |
| 3B | CGCCTATGAGAAGGATAAGGCAGT | ACGAGGTGCTTAAACTGTTCACCT | |
| 4B | GCCAGTTGGTGTTGTAGACACTGA | CTGCTATGCTGCCAAGCAAA | |
| 5B | GCTCGTGATCTTATTTGTGCTCAA | TGTGCAAGAGTGGACAAGCGAT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seifert, S.N.; Schulz, J.E.; Ricklefs, S.; Letko, M.; Yabba, E.; Hijazeen, Z.S.; Holloway, P.; Al-Omari, B.; Talafha, H.A.; Tibbo, M.; et al. Limited Genetic Diversity Detected in Middle East Respiratory Syndrome-Related Coronavirus Variants Circulating in Dromedary Camels in Jordan. Viruses 2021, 13, 592. https://0-doi-org.brum.beds.ac.uk/10.3390/v13040592
Seifert SN, Schulz JE, Ricklefs S, Letko M, Yabba E, Hijazeen ZS, Holloway P, Al-Omari B, Talafha HA, Tibbo M, et al. Limited Genetic Diversity Detected in Middle East Respiratory Syndrome-Related Coronavirus Variants Circulating in Dromedary Camels in Jordan. Viruses. 2021; 13(4):592. https://0-doi-org.brum.beds.ac.uk/10.3390/v13040592
Chicago/Turabian StyleSeifert, Stephanie N., Jonathan E. Schulz, Stacy Ricklefs, Michael Letko, Elangeni Yabba, Zaidoun S. Hijazeen, Peter Holloway, Bilal Al-Omari, Hani A. Talafha, Markos Tibbo, and et al. 2021. "Limited Genetic Diversity Detected in Middle East Respiratory Syndrome-Related Coronavirus Variants Circulating in Dromedary Camels in Jordan" Viruses 13, no. 4: 592. https://0-doi-org.brum.beds.ac.uk/10.3390/v13040592