
The Role of Phylogenetics in Discerning HIV-1 Mixing among Vulnerable Populations and Geographic Regions in Sub-Saharan Africa: A Systematic Review

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Systematic Literature Review
2.1.1. Information Sources
2.1.2. Search Strategy
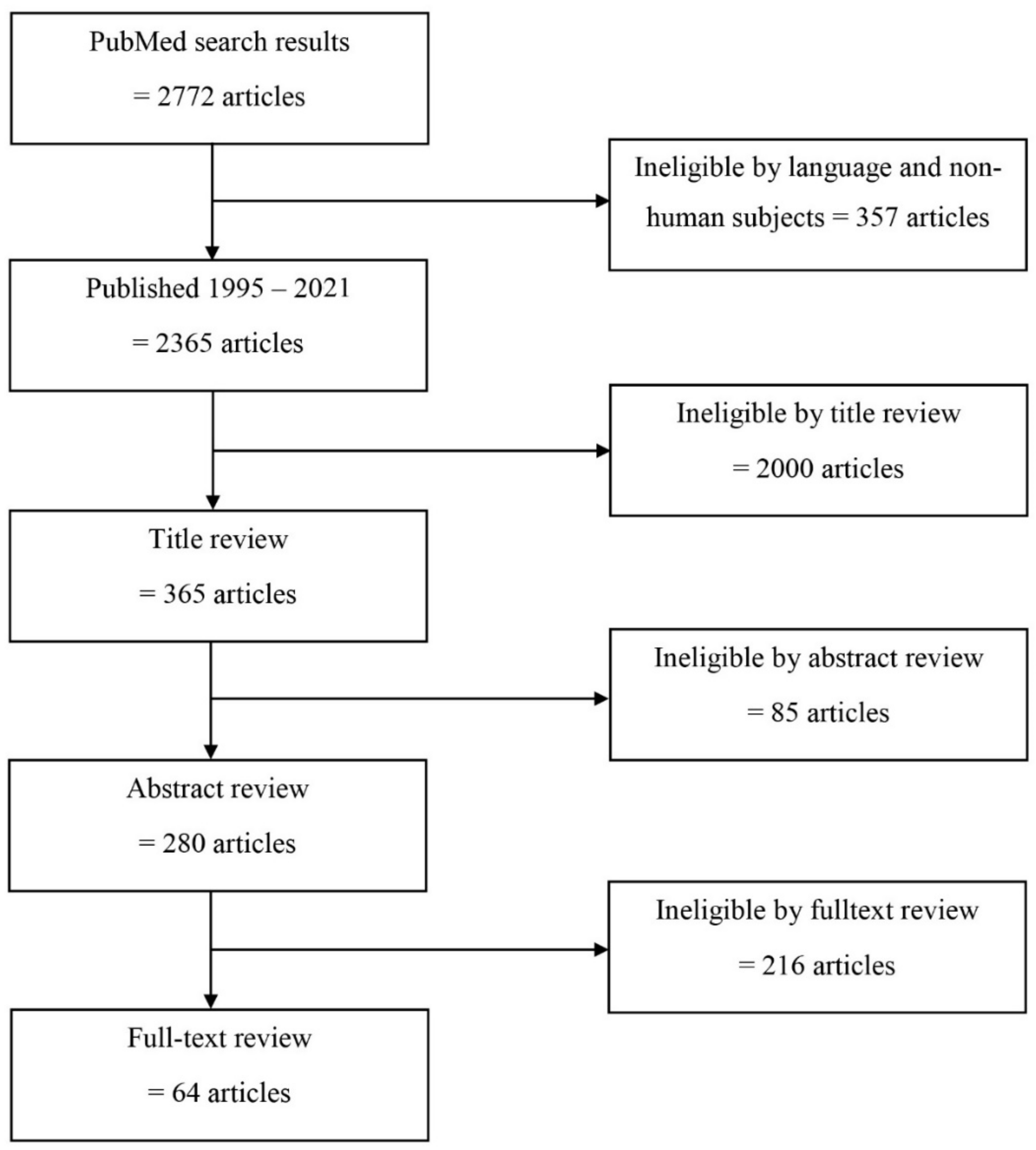
2.1.3. Selection Process
2.1.4. Data Extraction and Data Analysis
3. Results
3.1. HIV-1 Molecular Transmission Networks in sSA: What Has Been Done from a Geographic Perspective?
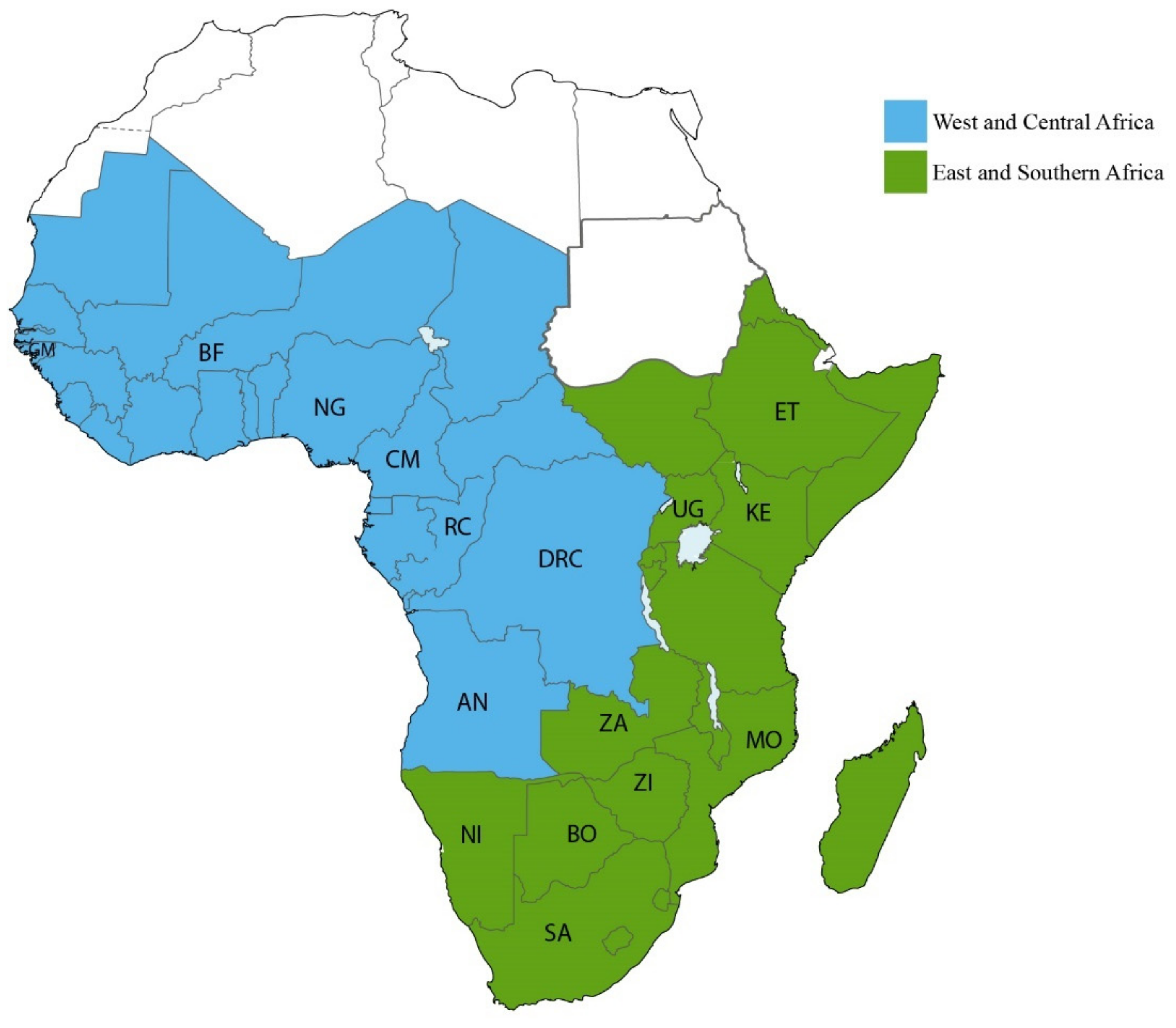
3.1.1. HIV-1 Transmission in West and Central African Countries
3.1.2. HIV-1 Transmission in East and Southern Africa Countries
3.1.3. HIV-1 Transmission beyond Borders
3.1.4. Conclusion Phylogeographic Linkages in sSA
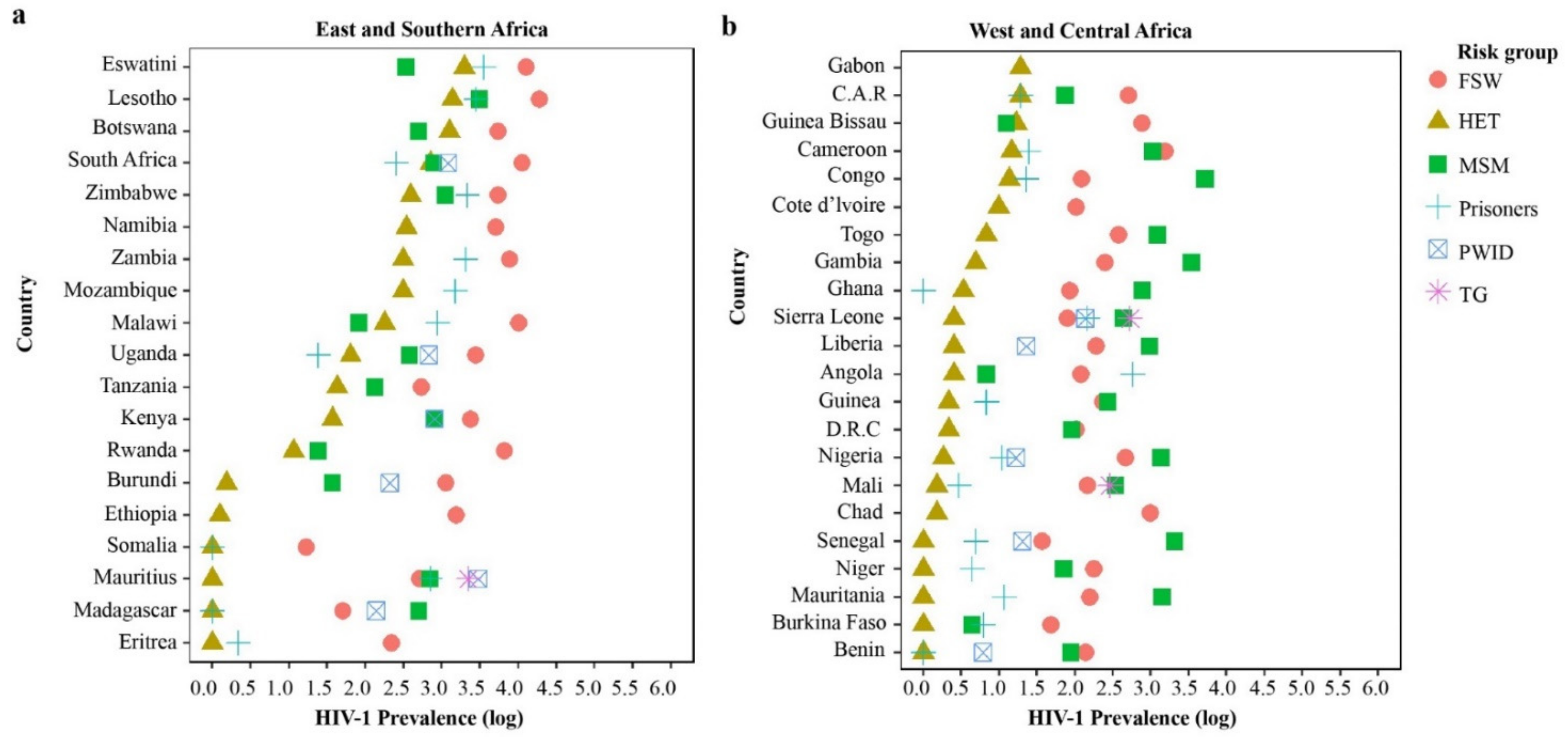
3.2. The Role of HIV-1 Key and Vulnerable Populations in Mixed HIV-1 Epidemics: A Risk Groups Perspective
3.2.1. HIV-1 Phylogenetic Linkages Involving Heterosexual Transmission
3.2.2. HIV-1 Phylogenetic Linkages among MSM
3.2.3. HIV-1 Phylogenetic Linkages among PWID
3.3. Phylogenetic Analysis to Examine HIV-1 Mixing between Risk Groups
3.4. Phylogenetic Analysis to Examine Sources and Direction of HIV-1 Transmission between HIV-1 Key and Vulnerable Populations and HET in sSA
4. Perspectives, Challenges, and Potential Solutions with Phylogenetic Inference in sSA
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Field, N.; Cohen, T.; Struelens, M.J.; Palm, D.; Cookson, B.; Glynn, J.R.; Gallo, V.; Ramsay, M.; Sonnenberg, P.; MacCannell, D. Strengthening the Reporting of Molecular Epidemiology for Infectious Diseases (STROME-ID): An extension of the STROBE statement. Lancet Infect. Dis. 2014, 14, 341–352. [Google Scholar] [CrossRef]
- Riley, L.W. Molecular Epidemiology of Infectious Diseases: Principles and Practices; American Society for Microbiology: Washington, DC, USA, 2004. [Google Scholar]
- Lemey, P.; Suchard, M.; Rambaut, A. Reconstructing the initial global spread of a human influenza pandemic: A Bayesian spatial-temporal model for the global spread of H1N1pdm. PLoS Curr. 2009, 1. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Worobey, M.; Pekar, J.; Larsen, B.B.; Nelson, M.I.; Hill, V.; Joy, J.B.; Rambaut, A.; Suchard, M.A.; Wertheim, J.O.; Lemey, P. The emergence of SARS-CoV-2 in Europe and North America. Science 2020, 370, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Lemey, P.; Hong, S.L.; Hill, V.; Baele, G.; Poletto, C.; Colizza, V.; O’Toole, Á.; McCrone, J.T.; Andersen, K.G.; Worobey, M. Accommodating individual travel history and unsampled diversity in Bayesian phylogeographic inference of SARS-CoV-2. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Bruhn, C.A.; Audelin, A.M.; Helleberg, M.; Bjorn-Mortensen, K.; Obel, N.; Gerstoft, J.; Nielsen, C.; Melbye, M.; Medstrand, P.; Gilbert, M.T.; et al. The origin and emergence of an HIV-1 epidemic: From introduction to endemicity. AIDS 2014, 28, 1031–1040. [Google Scholar] [CrossRef]
- Faria, N.R.; Rambaut, A.; Suchard, M.A.; Baele, G.; Bedford, T.; Ward, M.J.; Tatem, A.J.; Sousa, J.D.; Arinaminpathy, N.; Pépin, J. The early spread and epidemic ignition of HIV-1 in human populations. Science 2014, 346, 56–61. [Google Scholar] [CrossRef] [Green Version]
- Faria, N.R.; Vidal, N.; Lourenco, J.; Raghwani, J.; Sigaloff, K.C.E.; Tatem, A.J.; van de Vijver, D.A.M.; Pineda-Peña, A.C.; Rose, R.; Wallis, C.L.; et al. Distinct rates and patterns of spread of the major HIV-1 subtypes in Central and East Africa. PLoS Pathog. 2019, 15, e1007976. [Google Scholar] [CrossRef] [Green Version]
- Hassan, A.S.; Esbjornsson, J.; Wahome, E.; Thiong’o, A.; Makau, G.N.; Price, M.A.; Sanders, E.J. HIV-1 subtype diversity, transmission networks and transmitted drug resistance amongst acute and early infected MSM populations from Coastal Kenya. PLoS ONE 2018, 13, e0206177. [Google Scholar] [CrossRef] [Green Version]
- Hassan, A.S.; Mwaringa, S.M.; Obonyo, C.A.; Nabwera, H.M.; Sanders, E.J.; Rinke de Wit, T.F.; Cane, P.A.; Berkley, J.A. Low prevalence of transmitted HIV type 1 drug resistance among antiretroviral-naive adults in a rural HIV clinic in Kenya. AIDS Res. Hum. Retrovir. 2013, 29, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Nazziwa, J.; Faria, N.R.; Chaplin, B.; Rawizza, H.; Kanki, P.; Dakum, P.; Abimiku, A.; Charurat, M.; Ndembi, N.; Esbjörnsson, J. Characterisation of HIV-1 Molecular Epidemiology in Nigeria: Origin, Diversity, Demography and Geographic Spread. Sci. Rep. 2020, 10, 3468. [Google Scholar] [CrossRef] [Green Version]
- Poon, A.F.; Gustafson, R.; Daly, P.; Zerr, L.; Demlow, S.E.; Wong, J.; Woods, C.K.; Hogg, R.S.; Krajden, M.; Moore, D. Near real-time monitoring of HIV transmission hotspots from routine HIV genotyping: An implementation case study. Lancet HIV 2016, 3, e231–e238. [Google Scholar] [CrossRef] [Green Version]
- Rodger, A.J.; Cambiano, V.; Bruun, T.; Vernazza, P.; Collins, S.; Degen, O.; Corbelli, G.M.; Estrada, V.; Geretti, A.M.; Beloukas, A. Risk of HIV transmission through condomless sex in serodifferent gay couples with the HIV-positive partner taking suppressive antiretroviral therapy (PARTNER): Final results of a multicentre, prospective, observational study. Lancet 2019, 393, 2428–2438. [Google Scholar] [CrossRef] [Green Version]
- Rodger, A.J.; Cambiano, V.; Bruun, T.; Vernazza, P.; Collins, S.; Van Lunzen, J.; Corbelli, G.M.; Estrada, V.; Geretti, A.M.; Beloukas, A. Sexual activity without condoms and risk of HIV transmission in serodifferent couples when the HIV-positive partner is using suppressive antiretroviral therapy. JAMA 2016, 316, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Vasylyeva, T.I.; Liulchuk, M.; Friedman, S.R.; Sazonova, I.; Faria, N.R.; Katzourakis, A.; Babii, N.; Scherbinska, A.; Thézé, J.; Pybus, O.G.; et al. Molecular epidemiology reveals the role of war in the spread of HIV in Ukraine. Proc. Natl. Acad. Sci. USA 2018, 115, 1051–1056. [Google Scholar] [CrossRef] [Green Version]
- Esbjörnsson, J.; Mild, M.; Audelin, A.; Fonager, J.; Skar, H.; Bruun Jørgensen, L.; Liitsola, K.; Björkman, P.; Bratt, G.; Gisslén, M. HIV-1 transmission between MSM and heterosexuals, and increasing proportions of circulating recombinant forms in the Nordic Countries. Virus Evol. 2016, 2, vew010. [Google Scholar] [CrossRef] [Green Version]
- Volz, E.M.; Ionides, E.; Romero-Severson, E.O.; Brandt, M.G.; Mokotoff, E.; Koopman, J.S. HIV-1 transmission during early infection in men who have sex with men: A phylodynamic analysis. PLoS Med. 2013, 10, e1001568. [Google Scholar] [CrossRef]
- Brenner, B.G.; Roger, M.; Routy, J.P.; Moisi, D.; Ntemgwa, M.; Matte, C.; Baril, J.G.; Thomas, R.; Rouleau, D.; Bruneau, J.; et al. High rates of forward transmission events after acute/early HIV-1 infection. J. Infect. Dis. 2007, 195, 951–959. [Google Scholar] [CrossRef]
- Ratmann, O.; Van Sighem, A.; Bezemer, D.; Gavryushkina, A.; Jurriaans, S.; Wensing, A.; De Wolf, F.; Reiss, P.; Fraser, C. Sources of HIV infection among men having sex with men and implications for prevention. Sci. Transl. Med. 2016, 8, 320ra2. [Google Scholar] [CrossRef] [Green Version]
- Sallam, M.; Esbjörnsson, J.; Baldvinsdóttir, G.; Indriðason, H.; Björnsdóttir, T.B.; Widell, A.; Gottfreðsson, M.; Löve, A.; Medstrand, P. Molecular epidemiology of HIV-1 in Iceland: Early introductions, transmission dynamics and recent outbreaks among injection drug users. Infect. Genet. Evol. 2017, 49, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Ragonnet-Cronin, M.; Lycett, S.J.; Hodcroft, E.B.; Hué, S.; Fearnhill, E.; Brown, A.E.; Delpech, V.; Dunn, D.; Leigh Brown, A.J. Transmission of Non-B HIV Subtypes in the United Kingdom Is Increasingly Driven by Large Non-Heterosexual Transmission Clusters. J. Infect. Dis. 2016, 213, 1410–1418. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Consolidated guidelines on HIV prevention, diagnosis, treatment and care for key populations: 2016 update. In Consolidated Guidelines on HIV Prevention, Diagnosis, Treatment and Care for Key Populations: 2016 Update; WHO: Geneva, Switzerland, 2016. [Google Scholar]
- Abeler-Dörner, L.; Grabowski, M.K.; Rambaut, A.; Pillay, D.; Fraser, C. PANGEA-HIV 2: Phylogenetics and networks for generalised epidemics in Africa. Curr. Opin. HIV AIDS 2019, 14, 173. [Google Scholar] [CrossRef] [Green Version]
- Dwyer-Lindgren, L.; Cork, M.A.; Sligar, A.; Steuben, K.M.; Wilson, K.F.; Provost, N.R.; Mayala, B.K.; VanderHeide, J.D.; Collison, M.L.; Hall, J.B.; et al. Mapping HIV prevalence in sub-Saharan Africa between 2000 and 2017. Nature 2019, 570, 189–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanser, F.; de Oliveira, T.; Maheu-Giroux, M.; Bärnighausen, T. Concentrated HIV sub-epidemics in generalized epidemic settings. Curr. Opin. HIV AIDS 2014, 9, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makofane, K.; van der Elst, E.M.; Walimbwa, J.; Nemande, S.; Baral, S.D. From general to specific: Moving past the general population in the HIV response across sub-Saharan Africa. J. Int. AIDS Soc. 2020, 23, e25605. [Google Scholar] [CrossRef] [PubMed]
- Joint United Nations Programme on HIV/AIDS (UNAIDS). UNAIDS. Global AIDS Report. 2020:384. Seizing the Moment 7/6/2020. Available online: https://www.unaids.org/sites/default/files/media_asset/2020_global-aids-report_en.pdf (accessed on 12 May 2020).
- Anderson, S.-J.; Cherutich, P.; Kilonzo, N.; Cremin, I.; Fecht, D.; Kimanga, D.; Harper, M.; Masha, R.L.; Ngongo, P.B.; Maina, W. Maximising the effect of combination HIV prevention through prioritisation of the people and places in greatest need: A modelling study. Lancet 2014, 384, 249–256. [Google Scholar] [CrossRef] [Green Version]
- Kelly, S.L.; Martin-Hughes, R.; Stuart, R.M.; Yap, X.F.; Kedziora, D.J.; Grantham, K.L.; Hussain, S.A.; Reporter, I.; Shattock, A.J.; Grobicki, L. The global Optima HIV allocative efficiency model: Targeting resources in efforts to end AIDS. Lancet HIV 2018, 5, e190–e198. [Google Scholar] [CrossRef]
- Wilson, D.; Halperin, D.T. “Know your epidemic, know your response”: A useful approach, if we get it right. Lancet 2008, 372, 423–426. [Google Scholar] [CrossRef]
- Dennis, A.M.; Herbeck, J.T.; Brown, A.L.; Kellam, P.; de Oliveira, T.; Pillay, D.; Fraser, C.; Cohen, M.S. Phylogenetic studies of transmission dynamics in generalized HIV epidemics: An essential tool where the burden is greatest? J. Acquir. Immune Defic. Syndr. 2014, 67, 181–195. [Google Scholar] [CrossRef] [Green Version]
- Lihana, R.W.; Ssemwanga, D.; Abimiku, A.; Ndembi, N. Update on HIV-1 diversity in Africa: A decade in review. AIDS Rev. 2012, 14, 83–100. [Google Scholar]
- Bbosa, N.; Kaleebu, P.; Ssemwanga, D. HIV subtype diversity worldwide. Curr. Opin. HIV AIDS 2019, 14, 153–160. [Google Scholar] [CrossRef]
- Hemelaar, J.; Elangovan, R.; Yun, J.; Dickson-Tetteh, L.; Fleminger, I.; Kirtley, S.; Williams, B.; Gouws-Williams, E.; Ghys, P.D.; Abimiku, A.L.G.; et al. Global and regional molecular epidemiology of HIV-1, 1990–2015: A systematic review, global survey, and trend analysis. Lancet Infect. Dis. 2019, 19, 143–155. [Google Scholar] [CrossRef]
- Esbjörnsson, J.; Mild, M.; Månsson, F.; Norrgren, H.; Medstrand, P. HIV-1 molecular epidemiology in Guinea-Bissau, West Africa: Origin, demography and migrations. PLoS ONE 2011, 6, e17025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemelaar, J.; Loganathan, S.; Elangovan, R.; Yun, J.; Dickson-Tetteh, L.; Kirtley, S. Country Level Diversity of the HIV-1 Pandemic between 1990 and 2015. J. Virol. 2020, 95, e01580–e01620. [Google Scholar] [CrossRef] [PubMed]
- Véras, N.M.; Santoro, M.M.; Gray, R.R.; Tatem, A.J.; Lo Presti, A.; Olearo, F.; Cappelli, G.; Colizzi, V.; Takou, D.; Torimiro, J.; et al. Molecular epidemiology of HIV type 1 CRF02_AG in Cameroon and African patients living in Italy. Aids Res. Hum. Retrovir. 2011, 27, 1173–1182. [Google Scholar] [CrossRef] [Green Version]
- Faria, N.R.; Suchard, M.A.; Abecasis, A.; Sousa, J.D.; Ndembi, N.; Bonfim, I.; Camacho, R.J.; Vandamme, A.M.; Lemey, P. Phylodynamics of the HIV-1 CRF02_AG clade in Cameroon. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2012, 12, 453–460. [Google Scholar] [CrossRef] [Green Version]
- Gill, M.S.; Tung Ho, L.S.; Baele, G.; Lemey, P.; Suchard, M.A. A Relaxed Directional Random Walk Model for Phylogenetic Trait Evolution. Syst. Biol. 2016, syw093. [Google Scholar] [CrossRef] [Green Version]
- Bártolo, I.; Calado, R.; Borrego, P.; Leitner, T.; Taveira, N. Rare HIV-1 Subtype J Genomes and a New H/U/CRF02_AG Recombinant Genome Suggests an Ancient Origin of HIV-1 in Angola. AIDS Res. Hum. Retrovir. 2016, 32, 822–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bártolo, I.; Zakovic, S.; Martin, F.; Palladino, C.; Carvalho, P.; Camacho, R.; Thamm, S.; Clemente, S.; Taveira, N. HIV-1 diversity, transmission dynamics and primary drug resistance in Angola. PLoS ONE 2014, 9, e113626. [Google Scholar] [CrossRef] [Green Version]
- Hué, S.; Hassan, A.S.; Nabwera, H.; Sanders, E.J.; Pillay, D.; Berkley, J.A.; Cane, P.A. HIV type 1 in a rural coastal town in Kenya shows multiple introductions with many subtypes and much recombination. AIDS Res. Hum. Retrovir. 2012, 28, 220–224. [Google Scholar] [CrossRef]
- Nduva, G.M.; Hassan, A.S.; Nazziwa, J.; Graham, S.M.; Esbjörnsson, J.; Sanders, E.J. HIV-1 Transmission Patterns Within and Between Risk Groups in Coastal Kenya. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yebra, G.; Ragonnet-Cronin, M.; Ssemwanga, D.; Parry, C.M.; Logue, C.H.; Cane, P.A.; Kaleebu, P.; Brown, A.J. Analysis of the history and spread of HIV-1 in Uganda using phylodynamics. J. Gen. Virol. 2015, 96, 1890–1898. [Google Scholar] [CrossRef] [PubMed]
- Ssemwanga, D.; Bbosa, N.; Nsubuga, R.N.; Ssekagiri, A.; Kapaata, A.; Nannyonjo, M.; Nassolo, F.; Karabarinde, A.; Mugisha, J.; Seeley, J.; et al. The Molecular Epidemiology and Transmission Dynamics of HIV Type 1 in a General Population Cohort in Uganda. Viruses 2020, 12, 1283. [Google Scholar] [CrossRef] [PubMed]
- Arimide, D.A.; Abebe, A.; Kebede, Y.; Adugna, F.; Tilahun, T.; Kassa, D.; Assefa, Y.; Balcha, T.T.; Björkman, P.; Medstrand, P. HIV-genetic diversity and drug resistance transmission clusters in Gondar, Northern Ethiopia, 2003-2013. PLoS ONE 2018, 13, e0205446. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, E.; Engelbrecht, S.; de Oliveira, T. History and origin of the HIV-1 subtype C epidemic in South Africa and the greater southern African region. Sci. Rep. 2015, 5, 16897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, E.; Rasmussen, D.; Ratmann, O.; Stadler, T.; Engelbrecht, S.; de Oliveira, T. Origin, imports and exports of HIV-1 subtype C in South Africa: A historical perspective. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2016, 46, 200–208. [Google Scholar] [CrossRef]
- Wilkinson, E.; Junqueira, D.M.; Lessells, R.; Engelbrecht, S.; van Zyl, G.; de Oliveira, T.; Salemi, M. The effect of interventions on the transmission and spread of HIV in South Africa: A phylodynamic analysis. Sci. Rep. 2019, 9, 2640. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, D.A.; Wilkinson, E.; Vandormael, A.; Tanser, F.; Pillay, D.; Stadler, T.; De Oliveira, T. Tracking external introductions of HIV using phylodynamics reveals a major source of infections in rural KwaZulu-Natal, South Africa. Virus Evol. 2018, 4. [Google Scholar] [CrossRef]
- Novitsky, V.; Bussmann, H.; Logan, A.; Moyo, S.; van Widenfelt, E.; Okui, L.; Mmalane, M.; Baca, J.; Buck, L.; Phillips, E.; et al. Phylogenetic relatedness of circulating HIV-1C variants in Mochudi, Botswana. PLoS ONE 2013, 8, e80589. [Google Scholar] [CrossRef]
- Yebra, G.; Kalish, M.L.; Leigh Brown, A.J. Reconstructing the HIV-1 CRF02_AG and CRF06_cpx epidemics in Burkina Faso and West Africa using early samples. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2016, 46, 209–218. [Google Scholar] [CrossRef] [Green Version]
- Mir, D.; Jung, M.; Delatorre, E.; Vidal, N.; Peeters, M.; Bello, G. Phylodynamics of the major HIV-1 CRF02_AG African lineages and its global dissemination. Infect. Genet. Evol. 2016, 46, 190–199. [Google Scholar] [CrossRef] [Green Version]
- Delatorre, E.; Bello, G. Spatiotemporal dynamics of the HIV-1 CRF06_cpx epidemic in western Africa. AIDS 2013, 27, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Delatorre, E.; Bello, G. Time-scale of minor HIV-1 complex circulating recombinant forms from Central and West Africa. BMC Evol. Biol. 2016, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delatorre, E.; Mir, D.; Bello, G. Spatiotemporal Dynamics of the HIV-1 Subtype G Epidemic in West and Central Africa. PLoS ONE 2014, 9, e98908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delatorre, E.O.; Bello, G. Phylodynamics of HIV-1 subtype C epidemic in east Africa. PLoS ONE 2012, 7, e41904. [Google Scholar]
- Mir, D.; Gräf, T.; Esteves de Matos Almeida, S.; Pinto, A.R.; Delatorre, E.; Bello, G. Inferring population dynamics of HIV-1 subtype C epidemics in Eastern Africa and Southern Brazil applying different Bayesian phylodynamics approaches. Sci. Rep. 2018, 8, 8778. [Google Scholar] [CrossRef] [Green Version]
- Gray, R.R.; Tatem, A.J.; Lamers, S.; Hou, W.; Laeyendecker, O.; Serwadda, D.; Sewankambo, N.; Gray, R.H.; Wawer, M.; Quinn, T.C.; et al. Spatial phylodynamics of HIV-1 epidemic emergence in east Africa. AIDS 2009, 23, F9–F17. [Google Scholar] [CrossRef]
- Dalai, S.C.; de Oliveira, T.; Harkins, G.W.; Kassaye, S.G.; Lint, J.; Manasa, J.; Johnston, E.; Katzenstein, D. Evolution and molecular epidemiology of subtype C HIV-1 in Zimbabwe. AIDS 2009, 23, 2523–2532. [Google Scholar] [CrossRef] [Green Version]
- Afonso, J.M.; Morgado, M.G.; Bello, G. Evidence of multiple introductions of HIV-1 subtype C in Angola. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2012, 12, 1458–1465. [Google Scholar] [CrossRef]
- Grabowski, M.K.; Lessler, J.; Bazaale, J.; Nabukalu, D.; Nankinga, J.; Nantume, B.; Ssekasanvu, J.; Reynolds, S.J.; Ssekubugu, R.; Nalugoda, F. Migration, hotspots, and dispersal of HIV infection in Rakai, Uganda. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kreiss, J.K.; Koech, D.; Plummer, F.A.; Holmes, K.K.; Lightfoote, M.; Piot, P.; Ronald, A.R.; Ndinya-Achola, J.O.; D’Costa, L.J.; Roberts, P.; et al. AIDS virus infection in Nairobi prostitutes. Spread of the epidemic to East Africa. N. Engl. J. Med. 1986, 314, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D.; Tapsoba, P.; Peshu, N.; Sanders, E.J.; Jaffe, H.W. Men who have sex with men and HIV/AIDS in sub-Saharan Africa. Lancet 2009, 374, 416–422. [Google Scholar] [CrossRef]
- Rakwar, J.; Lavreys, L.; Thompson, M.L.; Jackson, D.; Bwayo, J.; Hassanali, S.; Mandaliya, K.; Ndinya-Achola, J.; Kreiss, J. Cofactors for the acquisition of HIV-1 among heterosexual men: Prospective cohort study of trucking company workers in Kenya. AIDS 1999, 13, 607–614. [Google Scholar] [CrossRef] [Green Version]
- Biggar, R. The AIDS problem in Africa. Lancet 1986, 327, 79–83. [Google Scholar] [CrossRef]
- Smith, A.D.; Muhaari, A.D.; Agwanda, C.; Kowuor, D.; van der Elst, E.; Davies, A.; Graham, S.M.; Jaffe, H.W.; Sanders, E.J. Heterosexual behaviours among men who sell sex to men in coastal Kenya. AIDS 2015, 29 (Suppl. 3), S201–S210. [Google Scholar] [CrossRef]
- Parker, R.; Khan, S.; Aggleton, P. Conspicuous by their absence? Men who have sex with men (msm) in developing countries: Implications for HIV prevention. Crit. Public Health 1998, 8, 329–346. [Google Scholar] [CrossRef]
- Carroll, A.; Mendos, L.R. State Sponsored Homophobia 2017: A World Survey of Sexual Orientation Laws: Criminalisation, Protection and Recognition; International Lesbian, Gay, Bisexual, Trans and Intersex Association (ILGA): Geneva, Switzerland, 2017; pp. 26–191. [Google Scholar]
- Novitsky, V.; Kühnert, D.; Moyo, S.; Widenfelt, E.; Okui, L.; Essex, M. Phylodynamic analysis of HIV sub-epidemics in Mochudi, Botswana. Epidemics 2015, 13, 44–55. [Google Scholar] [CrossRef] [Green Version]
- Sivay, M.V.; Hudelson, S.E.; Wang, J.; Agyei, Y.; Hamilton, E.L.; Selin, A.; Dennis, A.; Kahn, K.; Gomez-Olive, F.X.; MacPhail, C.; et al. HIV-1 diversity among young women in rural South Africa: HPTN 068. PLoS ONE 2018, 13, e0198999. [Google Scholar] [CrossRef]
- Trask, S.A.; Derdeyn, C.A.; Fideli, U.; Chen, Y.; Meleth, S.; Kasolo, F.; Musonda, R.; Hunter, E.; Gao, F.; Allen, S.; et al. Molecular epidemiology of human immunodeficiency virus type 1 transmission in a heterosexual cohort of discordant couples in Zambia. J. Virol. 2002, 76, 397–405. [Google Scholar] [CrossRef] [Green Version]
- Rusine, J.; Jurriaans, S.; van de Wijgert, J.; Cornelissen, M.; Kateera, B.; Boer, K.; Karita, E.; Mukabayire, O.; de Jong, M.; Ondoa, P. Molecular and phylogeographic analysis of human immuno-deficiency virus type 1 strains infecting treatment-naive patients from Kigali, Rwanda. PLoS ONE 2012, 7, e42557. [Google Scholar] [CrossRef] [Green Version]
- Rubio-Garrido, M.; González-Alba, J.M.; Reina, G.; Ndarabu, A.; Barquín, D.; Carlos, S.; Galán, J.C.; Holguín, Á. Current and historic HIV-1 molecular epidemiology in paediatric and adult population from Kinshasa in the Democratic Republic of Congo. Sci. Rep. 2020, 10, 18461. [Google Scholar] [CrossRef]
- Novitsky, V.; Moyo, S.; Lei, Q.; DeGruttola, V.; Essex, M. Impact of sampling density on the extent of HIV clustering. AIDS Res. Hum. Retrovir. 2014, 30, 1226–1235. [Google Scholar] [CrossRef] [Green Version]
- Bezemer, D.; Faria, N.R.; Hassan, A.; Hamers, R.L.; Mutua, G.; Anzala, O.; Mandaliya, K.; Cane, P.; Berkley, J.A.; Rinke de Wit, T.F. HIV Type 1 transmission networks among men having sex with men and heterosexuals in Kenya. AIDS Res. Hum. Retrovir. 2014, 30, 118–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osman, S.; Lihana, R.W.; Kibaya, R.M.; Ishizaki, A.; Bi, X.; Okoth, F.A.; Ichimura, H.; Lwembe, R.M. Diversity of HIV type 1 and drug resistance mutations among injecting drug users in Kenya. AIDS Res. Hum. Retrovir. 2013, 29, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Billings, E.; Kijak, G.H.; Sanders-Buell, E.; Ndembi, N.; OʼSullivan, A.M.; Adebajo, S.; Kokogho, A.; Milazzo, M.; Lombardi, K.; Baral, S.; et al. New Subtype B Containing HIV-1 Circulating Recombinant of sub-Saharan Africa Origin in Nigerian Men Who Have Sex with Men. J. Acquir. Immune Defic. Syndr. 2019, 81, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, H.; Ramadhani, H.O.; Ndembi, N.; Crowell, T.A.; Kijak, G.; Robb, M.L.; Ake, J.A.; Kokogho, A.; Nowak, R.G.; et al. Genetic clustering analysis for HIV infection among MSM in Nigeria: Implications for intervention. AIDS 2020, 34, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Ndiaye, H.D.; Toure-Kane, C.; Vidal, N.; Niama, F.R.; Niang-Diallo, P.A.; Dièye, T.; Gaye-Diallo, A.; Wade, A.S.; Peeters, M.; Mboup, S. Surprisingly High Prevalence of Subtype C and Specific HIV-1 Subtype/CRF Distribution in Men Having Sex With Men in Senegal. J. Acquir. Immune Defic. Syndr. 2009, 52, 249–252. [Google Scholar] [CrossRef]
- Ndiaye, H.D.; Tchiakpe, E.; Vidal, N.; Ndiaye, O.; Diop, A.K.; Peeters, M.; Mboup, S.; Toure-Kane, C. HIV type 1 subtype C remains the predominant subtype in men having sex with men in Senegal. AIDS Res. Hum. Retrovir. 2013, 29, 1265–1272. [Google Scholar] [CrossRef]
- Skar, H.; Axelsson, M.; Berggren, I.; Thalme, A.; Gyllensten, K.; Liitsola, K.; Brummer-Korvenkontio, H.; Kivela, P.; Spangberg, E.; Leitner, T.; et al. Dynamics of Two Separate but Linked HIV-1 CRF01_AE Outbreaks among Injection Drug Users in Stockholm, Sweden, and Helsinki, Finland. J. Virol. 2011, 85, 510–518. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, E.; Engelbrecht, S.; de Oliveira, T. Detection of transmission clusters of HIV-1 subtype C over a 21-year period in Cape Town, South Africa. PLoS ONE 2014, 9, e109296. [Google Scholar] [CrossRef]
- Middelkoop, K.; Rademeyer, C.; Brown, B.B.; Cashmore, T.J.; Marais, J.C.; Scheibe, A.P.; Bandawe, G.P.; Myer, L.; Fuchs, J.D.; Williamson, C.; et al. Epidemiology of HIV-1 Subtypes Among Men Who Have Sex With Men in Cape Town, South Africa. J. Acquir. Immune Defic. Syndr. 2014, 65, 473–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leye, N.; Vidal, N.; Ndiaye, O.; Diop-Ndiaye, H.; Wade, A.S.; Mboup, S.; Delaporte, E.; Toure-Kane, C.; Peeters, M. High frequency of HIV-1 infections with multiple HIV-1 strains in men having sex with men (MSM) in Senegal. Infect. Genet. Evol. 2013, 20, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Konou, A.A.; Vidal, N.; Salou, M.; Anato, S.; Singo-Tokofaï, A.; Ekouevi, D.K.; Pitché, P.; Prince-David, M.; Delaporte, E.; Peeters, M.; et al. Genetic diversity and transmission networks of HIV-1 strains among men having sex with men (MSM) in Lomé, Togo. Infect. Genet. Evol. 2016, 46, 279–285. [Google Scholar] [CrossRef]
- Nazziwa, J.; Njai, H.F.; Ndembi, N.; Birungi, J.; Lyagoba, F.; Gershim, A.; Nakiyingi-Miiro, J.; Nielsen, L.; Mpendo, J.; Nanvubya, A.; et al. Short communication: HIV type 1 transmitted drug resistance and evidence of transmission clusters among recently infected antiretroviral-naive individuals from Ugandan fishing communities of Lake Victoria. AIDS Res. Hum. Retrovir. 2013, 29, 788–795. [Google Scholar] [CrossRef] [PubMed]
- Kiwuwa-Muyingo, S.; Nazziwa, J.; Ssemwanga, D.; Ilmonen, P.; Njai, H.; Ndembi, N.; Parry, C.; Kitandwe, P.K.; Gershim, A.; Mpendo, J.; et al. HIV-1 transmission networks in high risk fishing communities on the shores of Lake Victoria in Uganda: A phylogenetic and epidemiological approach. PLoS ONE 2017, 12, e0185818. [Google Scholar] [CrossRef] [Green Version]
- Bbosa, N.; Ssemwanga, D.; Nsubuga, R.N.; Salazar-Gonzalez, J.F.; Salazar, M.G.; Nanyonjo, M.; Kuteesa, M.; Seeley, J.; Kiwanuka, N.; Bagaya, B.S. Phylogeography of HIV-1 suggests that Ugandan fishing communities are a sink for, not a source of, virus from general populations. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Grabowski, M.K.; Lessler, J.; Redd, A.D.; Kagaayi, J.; Laeyendecker, O.; Ndyanabo, A.; Nelson, M.I.; Cummings, D.A.; Bwanika, J.B.; Mueller, A.C. The role of viral introductions in sustaining community-based HIV epidemics in rural Uganda: Evidence from spatial clustering, phylogenetics, and egocentric transmission models. PLoS Med. 2014, 11, e1001610. [Google Scholar] [CrossRef] [Green Version]
- Ssemwanga, D.; Ndembi, N.; Lyagoba, F.; Bukenya, J.; Seeley, J.; Vandepitte, J.; Grosskurth, H.; Kaleebu, P. HIV type 1 subtype distribution, multiple infections, sexual networks, and partnership histories in female sex workers in Kampala, Uganda. Aids Res. Hum. Retrovir. 2012, 28, 357–365. [Google Scholar] [CrossRef]
- Ratmann, O.; Kagaayi, J.; Hall, M.; Golubchick, T.; Kigozi, G.; Xi, X.; Wymant, C.; Nakigozi, G.; Abeler-Dörner, L.; Bonsall, D. Quantifying HIV transmission flow between high-prevalence hotspots and surrounding communities: A population-based study in Rakai, Uganda. Lancet HIV 2020, 7, e173–e183. [Google Scholar] [CrossRef] [Green Version]
- Nascimento, F.F.; Baral, S.; Geidelberg, L.; Mukandavire, C.; Schwartz, S.R.; Turpin, G.; Turpin, N.; Diouf, D.; Diouf, N.L.; Coly, K.; et al. Phylodynamic analysis of HIV-1 subtypes B, C and CRF 02_AG in Senegal. Epidemics 2020, 30, 100376. [Google Scholar] [CrossRef]
- Volz, E.M.; Ndembi, N.; Nowak, R.; Kijak, G.H.; Idoko, J.; Dakum, P.; Royal, W.; Baral, S.; Dybul, M.; Blattner, W.A.; et al. Phylodynamic analysis to inform prevention efforts in mixed HIV epidemics. Virus Evol. 2017, 3, vex014. [Google Scholar] [CrossRef] [Green Version]
- Dennis, A.M.; Cohen, M.S.; Rucinski, K.B.; Rutstein, S.E.; Powers, K.A.; Pasquale, D.K.; Phiri, S.; Hosseinipour, M.C.; Kamanga, G.; Nsona, D.; et al. Human Immunodeficiency Virus (HIV)-1 Transmission Among Persons With Acute HIV-1 Infection in Malawi: Demographic, Behavioral, and Phylogenetic Relationships. Clin. Infect. Dis. 2019, 69, 853–860. [Google Scholar] [CrossRef]
- Jennes, W.; Kyongo, J.K.; Vanhommerig, E.; Camara, M.; Coppens, S.; Seydi, M.; Mboup, S.; Heyndrickx, L.; Kestens, L. Molecular epidemiology of HIV-1 transmission in a cohort of HIV-1 concordant heterosexual couples from Dakar, Senegal. PLoS ONE 2012, 7, e37402. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira, T.; Kharsany, A.B.; Gräf, T.; Cawood, C.; Khanyile, D.; Grobler, A.; Puren, A.; Madurai, S.; Baxter, C.; Karim, Q.A. Transmission networks and risk of HIV infection in KwaZulu-Natal, South Africa: A community-wide phylogenetic study. Lancet HIV 2017, 4, e41–e50. [Google Scholar] [CrossRef] [Green Version]
- Novitsky, V.; Zahralban-Steele, M.; Moyo, S.; Nkhisang, T.; Maruapula, D.; McLane, M.F.; Leidner, J.; Bennett, K.; Wirth, K.E. Mapping of HIV-1C Transmission Networks Reveals Extensive Spread of Viral Lineages Across Villages in Botswana Treatment-as-Prevention Trial. J. Infect. Dis. 2020, 222, 1670–1680. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, M.K.; Lessler, J. Phylogenetic insights into age-disparate partnerships and HIV. Lancet HIV 2017, 4, e8–e9. [Google Scholar] [CrossRef]
- Ihantamalala, F.A.; Herbreteau, V.; Rakotoarimanana, F.M.J.; Rakotondramanga, J.M.; Cauchemez, S.; Rahoilijaona, B.; Pennober, G.; Buckee, C.O.; Rogier, C.; Metcalf, C.J.E.; et al. Estimating sources and sinks of malaria parasites in Madagascar. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Wesolowski, A.; Eagle, N.; Tatem, A.J.; Smith, D.L.; Noor, A.M.; Snow, R.W.; Buckee, C.O. Quantifying the Impact of Human Mobility on Malaria. Science 2012, 338, 267–270. [Google Scholar] [CrossRef] [Green Version]
- Okano, J.T.; Sharp, K.; Valdano, E.; Palk, L.; Blower, S. HIV transmission and source-sink dynamics in sub-Saharan Africa. Lancet. HIV 2020, 7, e209–e214. [Google Scholar] [CrossRef]
- Ragonnet-Cronin, M.; Hué, S.; Hodcroft, E.B.; Tostevin, A.; Dunn, D.; Fawcett, T.; Pozniak, A.; Brown, A.E.; Delpech, V.; Brown, A.J.L. Non-disclosed men who have sex with men in UK HIV transmission networks: Phylogenetic analysis of surveillance data. Lancet HIV 2018, 5, e309–e316. [Google Scholar] [CrossRef] [Green Version]
- Fisher, M.; Pao, D.; Brown, A.E.; Sudarshi, D.; Gill, O.N.; Cane, P.; Buckton, A.J.; Parry, J.V.; Johnson, A.M.; Sabin, C. Determinants of HIV-1 transmission in men who have sex with men: A combined clinical, epidemiological and phylogenetic approach. AIDS 2010, 24, 1739–1747. [Google Scholar] [CrossRef] [PubMed]
- Kouyos, R.D.; Von Wyl, V.; Yerly, S.; Böni, J.; Taffé, P.; Shah, C.; Börgisser, P.; Klimkait, T.; Weber, R.; Hirschel, B. Molecular epidemiology reveals long-term changes in HIV type 1 subtype B transmission in Switzerland. J. Infect. Dis. 2010, 201, 1488–1497. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Country | HIV-1 Subtype | Estimated Date of Introduction | Summary of the Main Findings | PMID 1 |
|---|---|---|---|---|
| Central and West African countries | ||||
| F1 | 1958 (1934–1973) | Spread from DRC, derived from a single founder event. “Pure” F1 variants are most common in Angola. | 19386115 | |
| Angola | F1 | 1983 (1978–1989) | The Angolan civil war was associated with a wave of emigration and a phase of negative migratory outflow during 1960–1980. | 22484759 |
| C | 1978 (1973–1985) 1979 (1973–1985) 1983 (1977–1990) 1990 (1982–1997) 1994 (1989–1998) 2005 (2002–2008) | HIV-1 subtype C epidemic in Angola originated from multiple independent introductions from Burundi, Zambia, Zimbabwe, and South Africa. The civil war (1974–2002) may have contributed to the emergence of the HIV-1 epidemic in Angola. | 22634597 | |
| J, H | Not available | HIV-1 subtypes J and H seem to have been present in Angola since at least 1993. | 27098898 | |
| Group M | 1978 (1975–1985) | The majority of sequences sampled in 2008–2010 in Luanda clustered together which is consistent with a locally fuelled epidemic. | 25479241 | |
| Cameroon | CRF02_AG | 1973 (1972–1975) | Two distinct lineages of CRF02_AG seem to have ignited in the urban centre of Cameroon. Ethnographic data suggests that well-supported HIV-1 migration was related to chance exportation events rather than by sustained human migratory flows. | 21565285 |
| CRF02_AG | 1976 (1966–1984) 1976 (1968–1986) 1979 (1953–1989) | Three monophyletic variants were identified and emerged in the mid-1970’s and spread slowly over 30 years. Continuous exchange of HIV-1 strains between Cameroon and other African countries. | 21453131 | |
| DRC 2 | A1, C, D | The 1960s | HIV-1 subtype C origin was estimated to originate in Mbuji-Mayi in the 1950s and subtypes A1, D originated in Kinshasa. The earliest dispersal events of subtype C occurred in a mining region close to Mbuji-Mayi and Lubumbashi. Subtype C spread at least three-fold faster than other subtypes circulating in Central and East Africa. | 31809523 |
| DRC 2, RC 3 | Group M | 1920 (1909–1930) | Kinshasa estimated to be the origin of the HIV-1 group M pandemic. HIV-1 spread to Brazzaville in the Republic of the Congo, and Lubumbashi and Mbuji-Mayi in the 1930s, which were better connected to Kinshasa, indicating a critical role of mobility networks in the early spread and establishment of the HIV-1 epidemic from the epicentre. | 25278604 |
| DRC 2, RC 3 | General Eastward and Southward trends in the spread of HIV-1 from the Kinshasa–Brazzaville and the Pointe-Noire areas to other population centres. | 27798403 | ||
| Guinea-Bissau | CRF02_AG A3 | 1981 (1974–1986) 1976 (1968–1982) 1980 (1974–1984) 1979 (1972–1984) 1981 (1975–1985) 1979 (1960–1988) | Multiple introductions of CRF02_AG 1976–1981, and a single introduction of sub-subtype A3 in 1979 (median estimates). HIV-1 was introduced into the urban centre (the Capital Bissau) from where it spread to rural areas. | 21365013 |
| Nigeria | G CRF02_AG CRF43_02G | 1975 (1969–1982) 1963 (1948–1974) 1970 (1960–1980) 1960 (1947–1974) 1971 (1952–1983) | Urban areas (Abuja and Lagos) were the major hubs of HIV-1 transmission in Nigeria. HIV-1 first emerged and expanded within large urban centres before migrating to smaller rural areas. | 32103028 |
| East and Southern African countries | ||||
| Botswana | C | 1996–2002 | Presence of multiple phylogenetically distinct HIV-1 subtype C variants (subepidemics) circulating in Mochudi with limited lifespans and temporal dominance. None of the sequences from a rural community of Mochudi clustered with non-Botswana sequences. | 26616041 24349005 |
| Ethiopia | C | 1965 (1959–1973) | Reconstruction of the epidemic history in Ethiopia revealed that subtype C likely originated from a single lineage in the late 1960s. | 20539092 |
| C | 1980 | Evidence of clustering between Gondar sequences and sequences from East Africa. | 30304061 | |
| Kenya | A1 | 1985–2012 | Kilifi sequences clustered closely with sequences from Kenya and other parts of Africa, including West Africa. HIV-1 has been introduced in coastal Kenya multiple times. | 32317722 |
| South Africa | C | 1960 (1956–1964) | Johannesburg was identified as the hub of HIV-1 dissemination in South Africa. The central region of KwaZulu-Natal was identified as the most likely ancestral location for HIV-1transmission in South Africa for 2 of 14 variants. | 26574165 |
| C | 1979–1992 | The HIV-1 epidemic in South Africa is suggested to have multiple, parallel subepidemics spreading in the country at the same time. | 30804361 | |
| C | 1990–2000 | Early HIV-1 epidemic dynamics in KwaZulu-Natal were largely driven by external introductions. | 30555720 | |
| Uganda | A1 | 1960 (1950–1968) | Ugandan epidemics originated in rural Southwestern Uganda with subsequent spread to other locations without any substantial HIV-1 introductions into this location suggesting that emerging infections from this low-incidence location are mostly from within the region. | 25724670 |
| D | 1973 (1970–1977) | 33182587 | ||
| Beyond borders | ||||
| West and Central Africa | CRF02_AG | 1980 (1978–1981) | CRF02_AG originated from Cameroon from where it spread to other Central and West African countries. | 27063411 |
| West and Central Africa | CRF02_AG | 1967 (1961–1974) West African | Five different CRF02_AG variants, four of which were restricted to Cameroon and one that grew out into West Africa. | 27180893 |
| West and Central Africa | CRF11_cpx | 1957 (1950–1966) | Cameroon as the epicentre of dissemination of CRF11_cpx to Central African Republic, Chad, Gabon, and Equatorial Guinea. | 27852214 |
| West Africa | CRF06_cpx | 1979 (1970–1985) | Burkina Faso was the hub of dissemination of CRF06_cpx to Mali, Nigeria, and the rest of western Central Africa. | 23343915 |
| West and Central Africa | G | 1974 (1966–1981) 1979 (1973–1984) | Subtype G epidemic clustered into two clusters according to sequence location, i.e., either West or Central Africa. Sequences from West Africa were further subdivided into two large monophyletic clusters that were nested within the Central African variant. | 24918930 |
| East Africa | C | 1962 (1942–1975) | Subtype C sequences from East Africa (Burundi, Ethiopia, Kenya, Tanzania, and Uganda) formed one large monophyletic cluster separate from sequences from Southern Africa. | 22848653 29884822 |
| East Africa | A1 D | 1948 (1958–1967) | Both subtype A1 and subtype D were suggested to have spread exponentially during the 1970s. | 19644346 |
| East and Southern Africa | C | Not available | The largest number of HIV-1 introductions into South Africa came from Zambia, followed by Botswana, Malawi, and Zimbabwe between 1985 and 2000, a period of mass inward immigration from neighbouring countries into South Africa. | 27421210 |
| Zimbabwe | C | 1972 (1979–1981) | Multiple cross-border independent introductions of subtype C HIV-1 into Zimbabwe between 1979 and 1981. | 19770693 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nduva, G.M.; Nazziwa, J.; Hassan, A.S.; Sanders, E.J.; Esbjörnsson, J. The Role of Phylogenetics in Discerning HIV-1 Mixing among Vulnerable Populations and Geographic Regions in Sub-Saharan Africa: A Systematic Review. Viruses 2021, 13, 1174. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061174
Nduva GM, Nazziwa J, Hassan AS, Sanders EJ, Esbjörnsson J. The Role of Phylogenetics in Discerning HIV-1 Mixing among Vulnerable Populations and Geographic Regions in Sub-Saharan Africa: A Systematic Review. Viruses. 2021; 13(6):1174. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061174
Chicago/Turabian StyleNduva, George M., Jamirah Nazziwa, Amin S. Hassan, Eduard J. Sanders, and Joakim Esbjörnsson. 2021. "The Role of Phylogenetics in Discerning HIV-1 Mixing among Vulnerable Populations and Geographic Regions in Sub-Saharan Africa: A Systematic Review" Viruses 13, no. 6: 1174. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061174