
The Role of Phylogenetics in Unravelling Patterns of HIV Transmission towards Epidemic Control: The Quebec Experience (2002–2020)

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Study Design and Populations
HIV-TRACE and Microbe Trace Network Construction
2.2. Epidemiological Correlates of Transmission Clustering
3. Results
3.1. Molecular Network Analysis of the HIV-1 Epidemics in Quebec
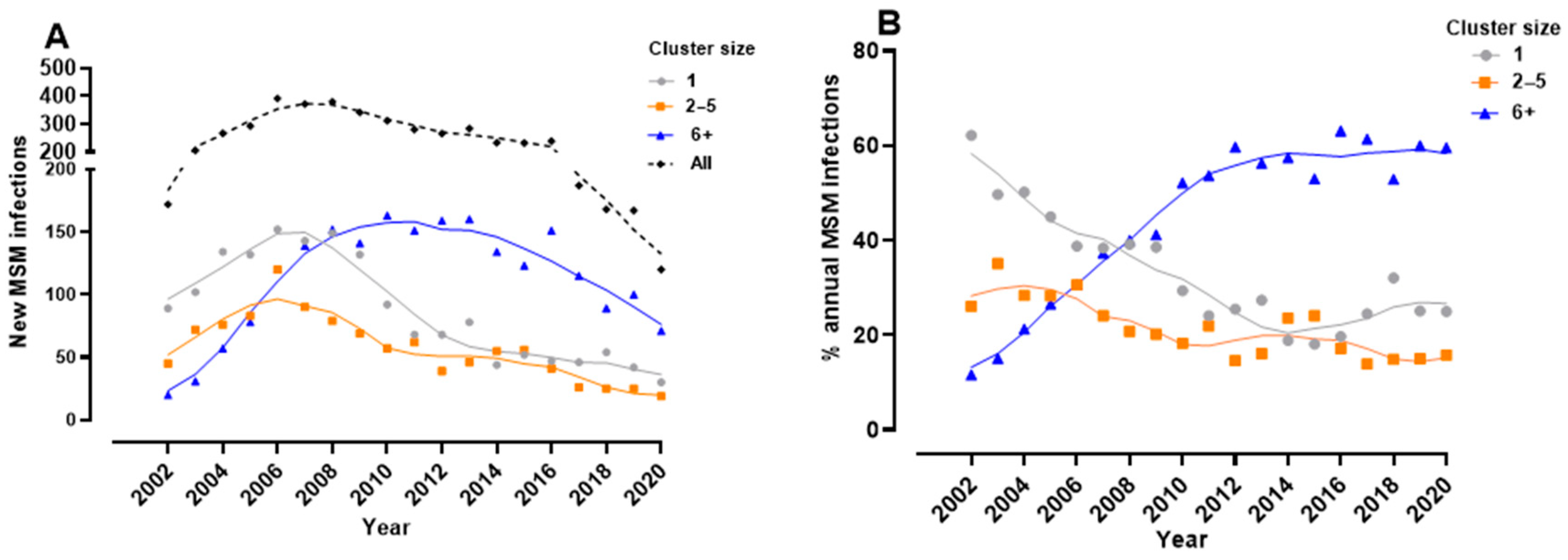
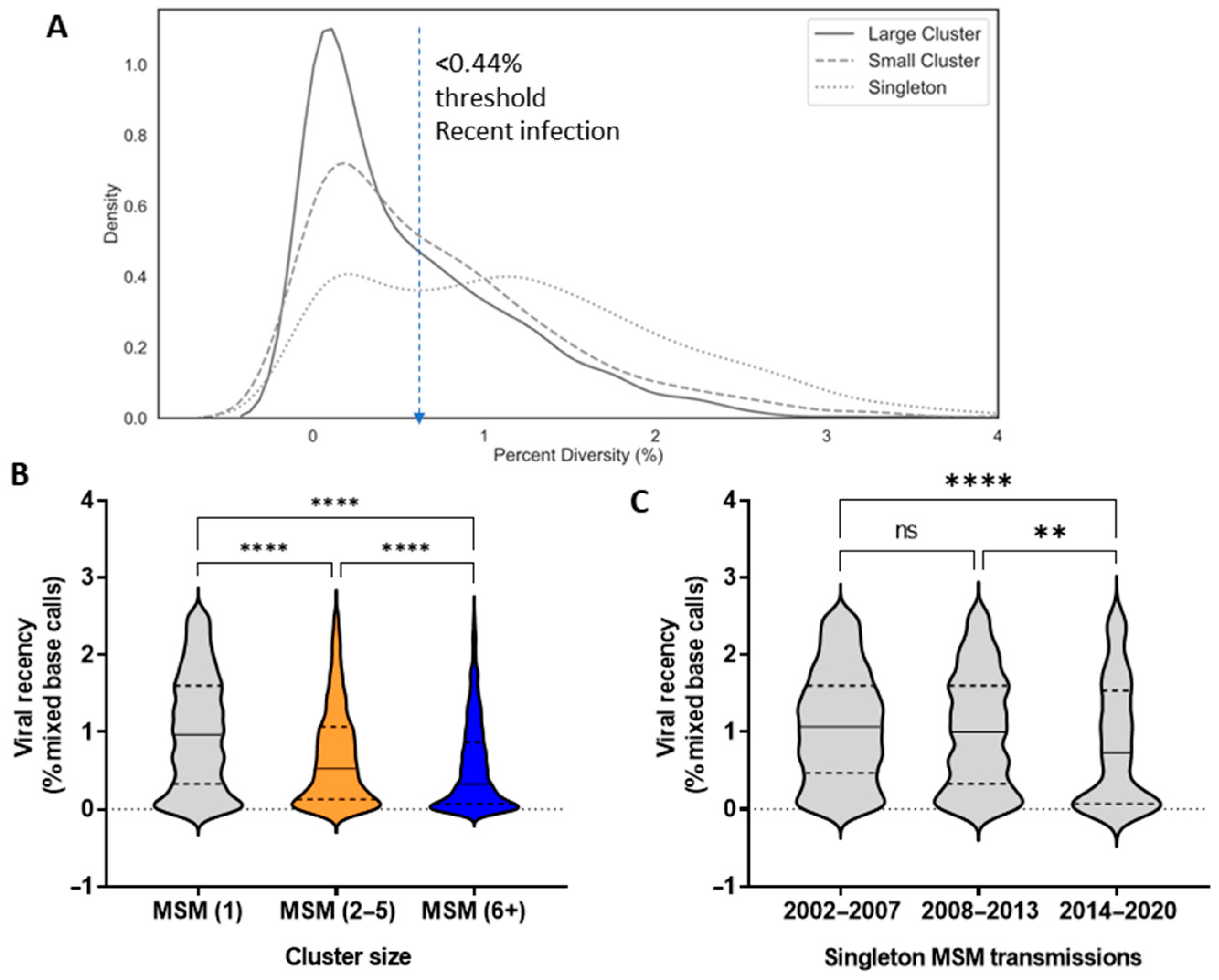
3.2. Phylodynamics Reveal Shifting Trends in the Spread of the Subtype B Epidemic among MSM
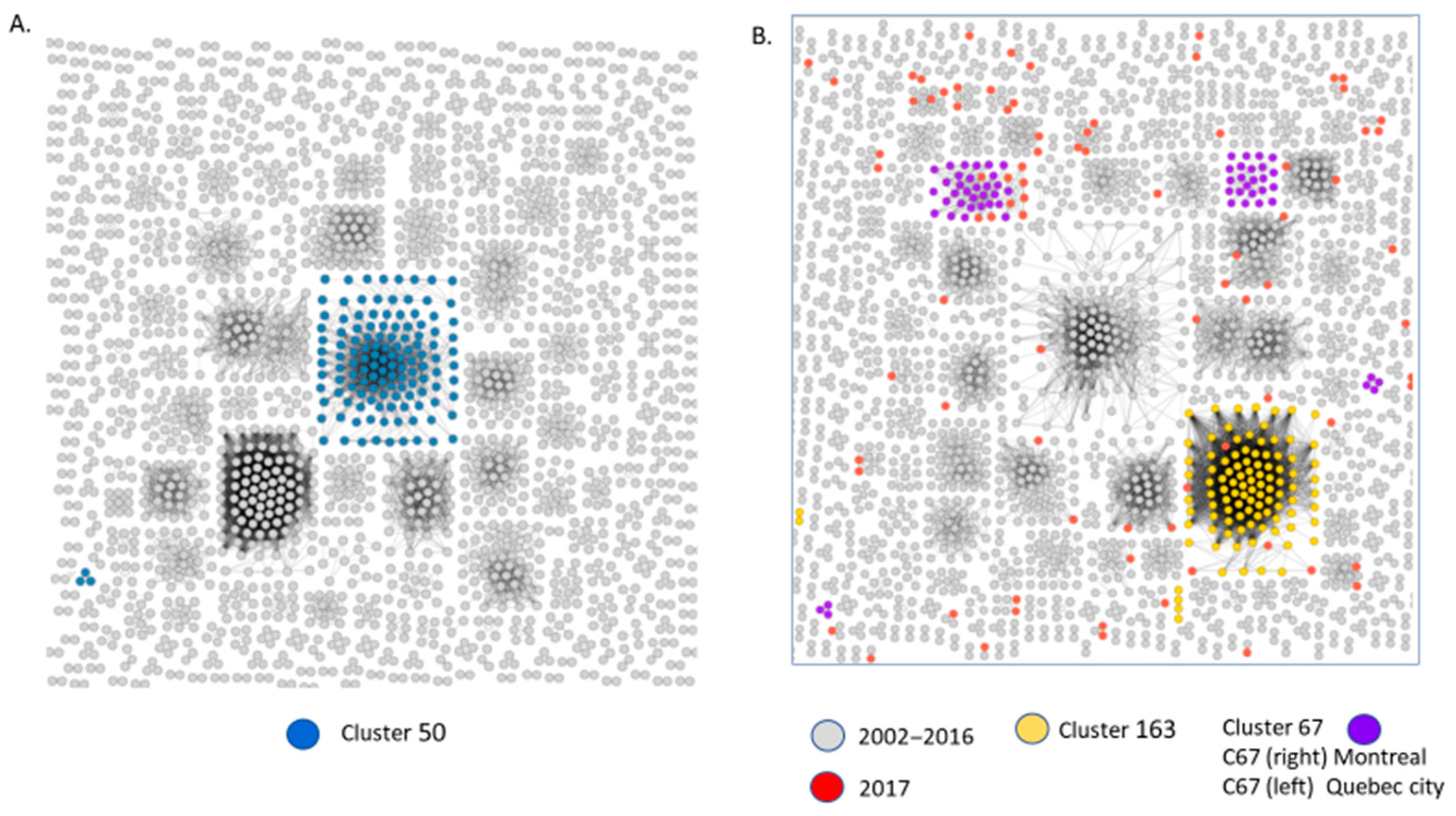
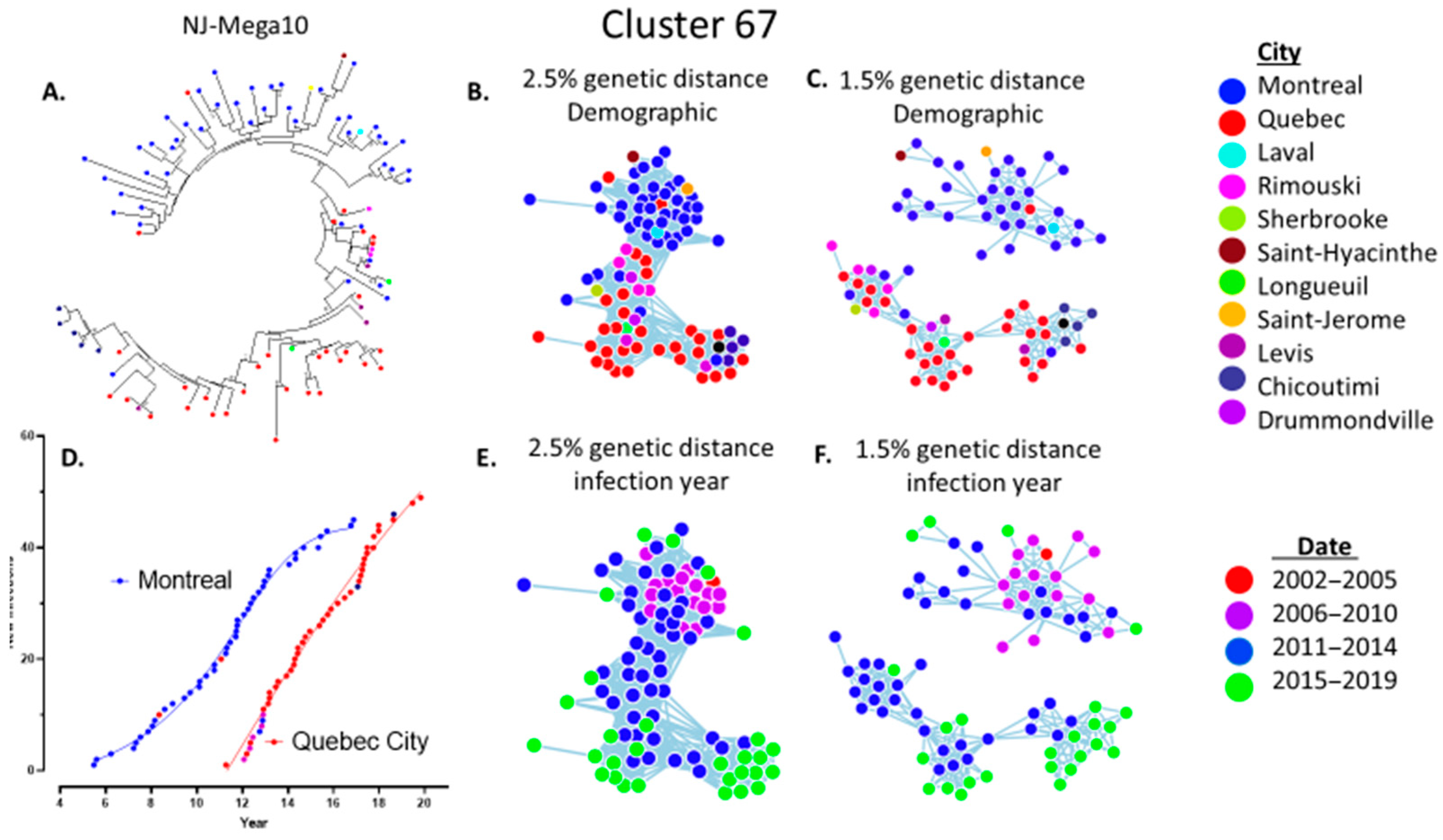
3.3. Molecular Network Analyses Using HIV-TRACE and Microbe Trace
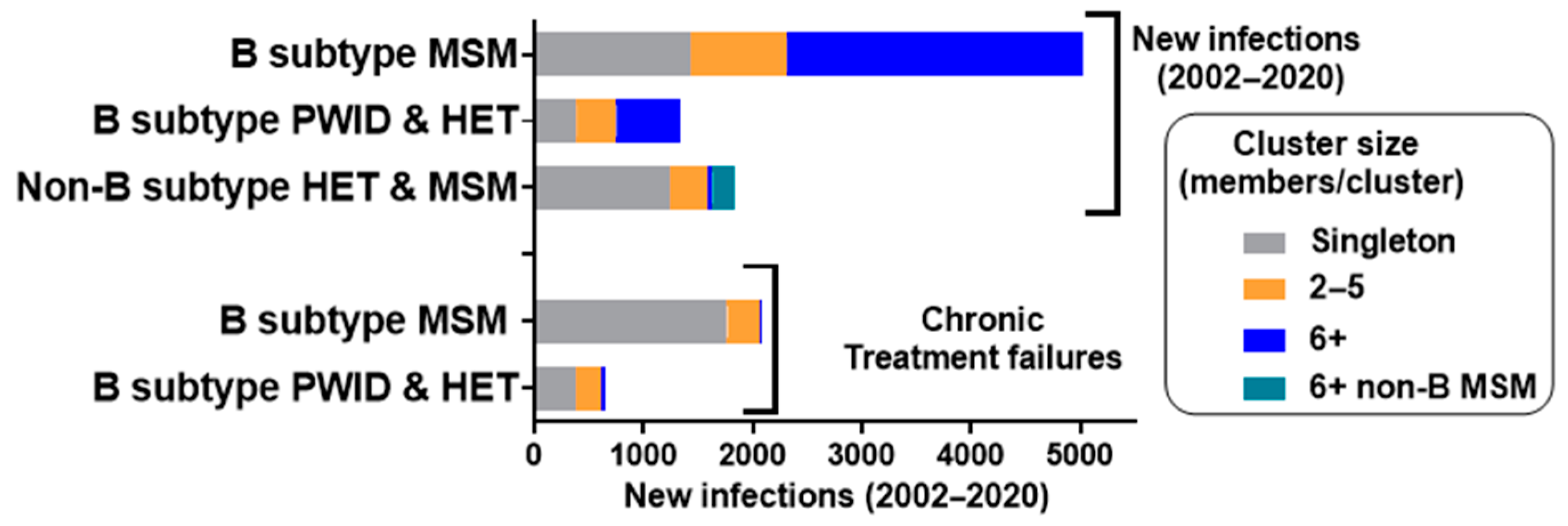
3.4. Epidemiological Features Implicated in the Growth Trajectories of Large Cluster Networks Fuelling MSM Epidemics
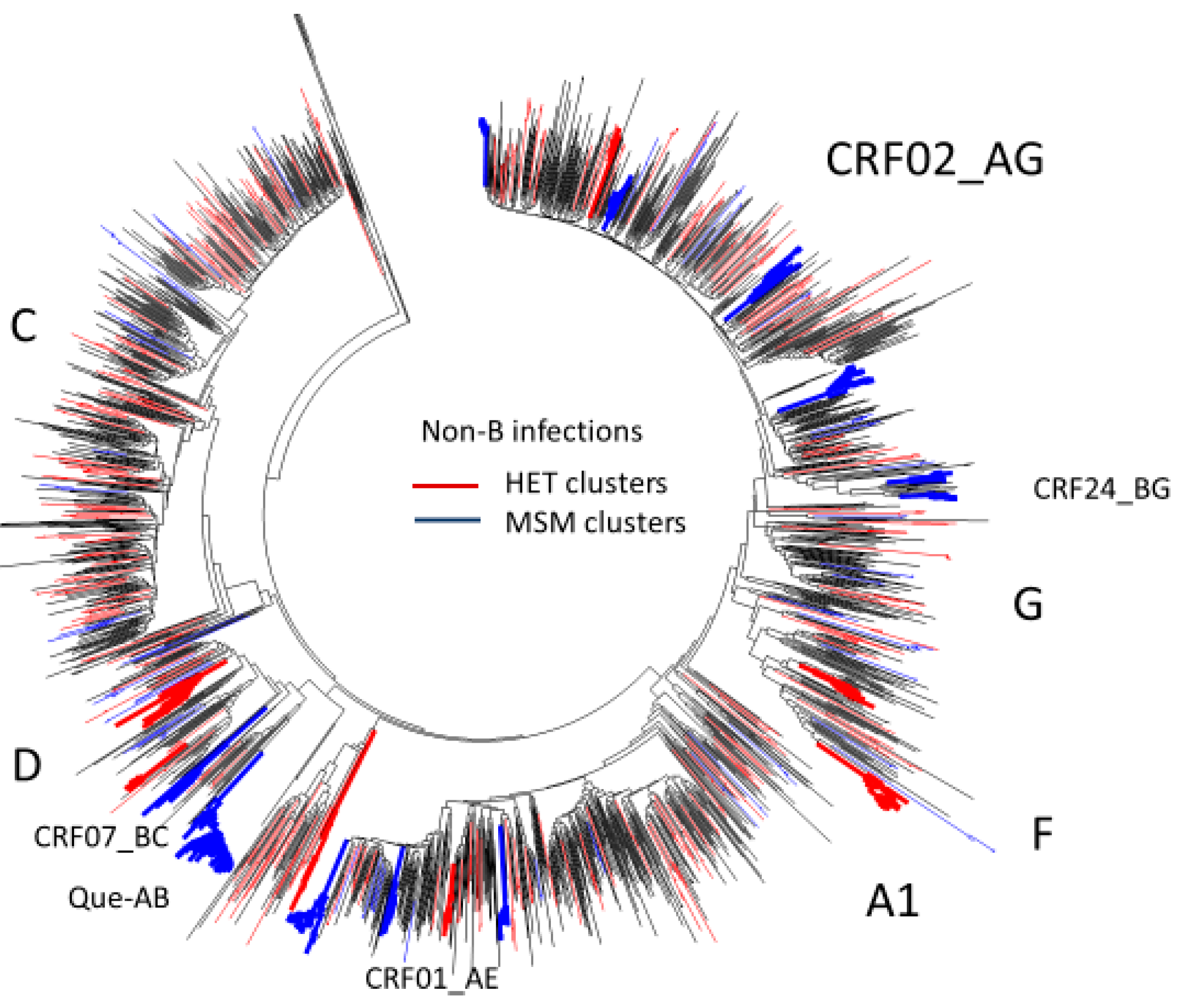
3.5. Phylogenetic Inferences on the Spread of Non-Subtype B Epidemics
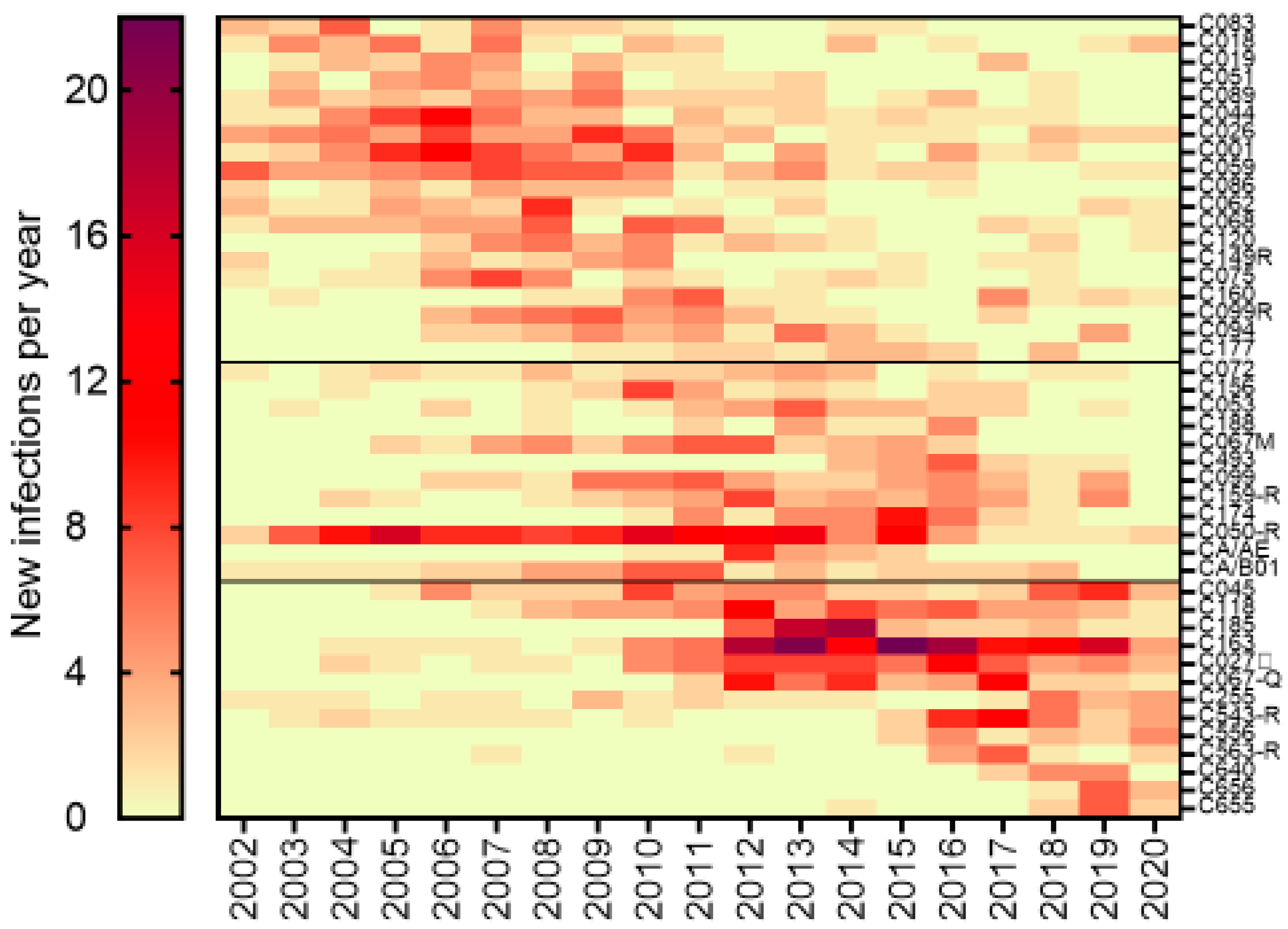
3.6. Phylogenetic Inferences of Cluster Dynamics Using Integrase Sequences
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- 2020 Global AIDS Update—Seizing the Moment—Tackling Entrenched Inequalities to End Epidemics. Available online: https://www.unaids.org/sites/default/files/media_asset/2020_global-aids-report_en.pdf (accessed on 11 November 2020).
- Beyrer, C.; Sullivan, P.; Sanchez, J.; Baral, S.D.; Collins, C.; Wirtz, A.L.; Altman, D.; Trapence, G.; Mayer, K. The increase in global HIV epidemics in MSM. AIDS 2013, 27, 2665–2678. [Google Scholar] [CrossRef]
- Paraskevis, D.; Pybus, O.; Magiorkinis, G.; Hatzakis, A.; Wensing, A.M.; van de Vijver, D.A.; Albert, J.; Angarano, G.; Åsjö, B.; Balotta, C.; et al. Tracing the HIV-1 subtype B mobility in Europe: A phylogeographic approach. Retrovirology 2009, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Brenner, B.G.; Ibanescu, R.I.; Hardy, I.; Roger, M. Genotypic and Phylogenetic Insights on Prevention of the Spread of HIV-1 and Drug Resistance in “Real-World” Settings. Viruses 2017, 10, 10. [Google Scholar] [CrossRef] [Green Version]
- Nduva, G.M.; Nazziwa, J.; Hassan, A.S.; Sanders, E.J.; Esbjornsson, J. The Role of Phylogenetics in Discerning HIV-1 Mixing among Vulnerable Populations and Geographic Regions in Sub-Saharan Africa: A Systematic Review. Viruses 2021, 13, 1174. [Google Scholar] [CrossRef]
- Dauwe, K.; Mortier, V.; Schauvliege, M.; Van Den Heuvel, A.; Fransen, K.; Servais, J.Y.; Bercoff, D.P.; Seguin-Devaux, C.; Verhofstede, C. Characteristics and spread to the native population of HIV-1 non-B subtypes in two European countries with high migration rate. BMC Infect. Dis. 2015, 15, 524. [Google Scholar] [CrossRef] [Green Version]
- von Wyl, V.; Kouyos, R.D.; Yerly, S.; Boni, J.; Shah, C.; Burgisser, P.; Klimkait, T.; Weber, R.; Hirschel, B.; Cavassini, M.; et al. The role of migration and domestic transmission in the spread of HIV-1 non-B subtypes in Switzerland. J. Infect. Dis. 2011, 204, 1095–1103. [Google Scholar] [CrossRef]
- Marsh, K.; Eaton, J.W.; Mahy, M.; Sabin, K.; Autenrieth, C.S.; Wanyeki, I.; Daher, J.; Ghys, P.D. Global, regional and country-level 90-90-90 estimates for 2018: Assessing progress towards the 2020 target. AIDS 2019, 33 (Suppl. 3), S213–S226. [Google Scholar] [CrossRef]
- Saag, M.S.; Gandhi, R.T.; Hoy, J.F.; Landovitz, R.J.; Thompson, M.A.; Sax, P.E.; Smith, D.M.; Benson, C.A.; Buchbinder, S.P.; Del Rio, C.; et al. Antiretroviral Drugs for Treatment and Prevention of HIV Infection in Adults: 2020 Recommendations of the International Antiviral Society-USA Panel. JAMA 2020, 324, 1651–1669. [Google Scholar] [CrossRef]
- Porter, K.; Gourlay, A.; Attawell, K.; Hales, D.; Supervie, V.; Touloumi, G.; Rosinska, M.; Vourli, G.; van Sighem, A.; Pharris, A.; et al. Substantial Heterogeneity in Progress toward Reaching the 90-90-90 HIV Target in the WHO European Region. J. Acquir. Immune Defic. Syndr. 2018, 79, 28–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haddad, N.; Li, J.S.; Totten, S.; McGuire, M. HIV in Canada-Surveillance Report, 2017. Can. Commun. Dis. Rep. 2018, 44, 348–356. [Google Scholar] [CrossRef]
- Haddad, N.; Robert, A.; Weeks, A.; Popovic, N.; Siu, W.; Archibald, C. HIV in Canada-Surveillance Report, 2018. Can. Commun. Dis. Rep. 2019, 45, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.; Wainberg, M.A.; Roger, M. Phylogenetic inferences on HIV-1 transmission: Implications for the design of prevention and treatment interventions. AIDS 2013, 27, 1045–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, B.G.; Wainberg, M.A. Future of phylogeny in HIV prevention. J. Acquir. Immune Defic. Syndr. 2013, 63 (Suppl. 2), S248–S254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, B.G.; Ibanescu, R.I.; Hardy, I.; Stephens, D.; Otis, J.; Moodie, E.; Grossman, Z.; Vandamme, A.M.; Roger, M.; Wainberg, M.A.; et al. Large cluster outbreaks sustain the HIV epidemic among MSM in Quebec. AIDS 2017, 31, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Wertheim, J.O.; Murrell, B.; Mehta, S.R.; Forgione, L.A.; Kosakovsky Pond, S.L.; Smith, D.M.; Torian, L.V. Growth of HIV-1 Molecular Transmission Clusters in New York City. J. Infect. Dis. 2018, 218, 1943–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezemer, D.; Cori, A.; Ratmann, O.; van Sighem, A.; Hermanides, H.S.; Dutilh, B.E.; Gras, L.; Rodrigues Faria, N.; van den Hengel, R.; Duits, A.J.; et al. Dispersion of the HIV-1 Epidemic in Men Who Have Sex with Men in the Netherlands: A Combined Mathematical Model and Phylogenetic Analysis. PLoS Med. 2015, 12, e1001898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, B.G.; Roger, M.; Routy, J.P.; Moisi, D.; Ntemgwa, M.; Matte, C.; Baril, J.G.; Thomas, R.; Rouleau, D.; Bruneau, J.; et al. High rates of forward transmission events after acute/early HIV-1 infection. J. Infect. Dis. 2007, 195, 951–959. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Weaver, S.; Leigh Brown, A.J.; Wertheim, J.O. HIV-TRACE (TRAnsmission Cluster Engine): A Tool for Large Scale Molecular Epidemiology of HIV-1 and Other Rapidly Evolving Pathogens. Mol. Biol. Evol. 2018, 35, 1812–1819. [Google Scholar] [CrossRef] [Green Version]
- Campbell, E.M.; Patala, A.; Shankar, A.; Li, J.F.; Johnson, J.A.; Westheimer, E.; Gay, C.L.; Cohen, S.E.; Switzer, W.M.; Peters, P.J. Phylodynamic Analysis Complements Partner Services by Identifying Acute and Unreported HIV Transmission. Viruses 2020, 12, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, B.; Moodie, E.E. HIV Sexual Networks: The Montreal experience. Stat. Commun. Infect. Dis. 2012, 4, 1–22. [Google Scholar] [CrossRef]
- Brenner, B.G.; Roger, M.; Stephens, D.; Moisi, D.; Hardy, I.; Weinberg, J.; Turgel, R.; Charest, H.; Koopman, J.; Wainberg, M.A. Transmission clustering drives the onward spread of the HIV epidemic among men who have sex with men in Quebec. J. Infect. Dis. 2011, 204, 1115–1119. [Google Scholar] [CrossRef]
- Brenner, B.G.; Roger, M.; Moisi, D.D.; Oliveira, M.; Hardy, I.; Turgel, R.; Charest, H.; Routy, J.P.; Wainberg, M.A. Transmission networks of drug resistance acquired in primary/early stage HIV infection. AIDS 2008, 22, 2509–2515. [Google Scholar] [CrossRef]
- Programme de Surveillance de L’infection par le Virus de L’immunodéfience Humaine (VIH) au Québec. Rapport 2019. Available online: https://www.inspq.qc.ca/sites/default/files/publications/2706_programme_surveillance_infection_2019_0.pdf (accessed on 16 August 2021).
- Hassan, A.S.; Pybus, O.G.; Sanders, E.J.; Albert, J.; Esbjornsson, J. Defining HIV-1 transmission clusters based on sequence data. AIDS 2017, 31, 1211–1222. [Google Scholar] [CrossRef]
- Novitsky, V.; Steingrimsson, J.A.; Howison, M.; Gillani, F.S.; Li, Y.; Manne, A.; Fulton, J.; Spence, M.; Parillo, Z.; Marak, T.; et al. Empirical comparison of analytical approaches for identifying molecular HIV-1 clusters. Sci. Rep. 2020, 10, 18547. [Google Scholar] [CrossRef]
- Erly, S.J.; Herbeck, J.T.; Kerani, R.P.; Reuer, J.R. Characterization of Molecular Cluster Detection and Evaluation of Cluster Investigation Criteria Using Machine Learning Methods and Statewide Surveillance Data in Washington State. Viruses 2020, 12, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouyos, R.D.; von Wyl, V.; Yerly, S.; Boni, J.; Rieder, P.; Joos, B.; Taffe, P.; Shah, C.; Burgisser, P.; Klimkait, T.; et al. Ambiguous nucleotide calls from population-based sequencing of HIV-1 are a marker for viral diversity and the age of infection. Clin. Infect. Dis. 2011, 52, 532–539. [Google Scholar] [CrossRef] [Green Version]
- Andersson, E.; Shao, W.; Bontell, I.; Cham, F.; do Cuong, D.; Wondwossen, A.; Morris, L.; Hunt, G.; Sonnerborg, A.; Bertagnolio, S.; et al. Evaluation of sequence ambiguities of the HIV-1 pol gene as a method to identify recent HIV-1 infection in transmitted drug resistance surveys. Infect. Genet. Evol. 2013, 18, 125–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragonnet-Cronin, M.; Aris-Brosou, S.; Joanisse, I.; Merks, H.; Vallee, D.; Caminiti, K.; Rekart, M.; Krajden, M.; Cook, D.; Kim, J.; et al. Genetic diversity as a marker for timing infection in HIV-infected patients: Evaluation of a 6-month window and comparison with BED. J. Infect. Dis. 2012, 206, 756–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragonnet-Cronin, M.; Golubchik, T.; Moyo, S.; Fraser, C.; Essex, M.; Novitsky, V.; Volz, E.; PANGEA Consortium. HIV genetic diversity informs stage of HIV-1 infection among patients receiving antiretroviral therapy in Botswana. J. Infect. Dis. 2021, jiab293. [Google Scholar] [CrossRef]
- Park, H.; Brenner, B.; Ibanescu, R.-I.; Cox, J.; Weiss, K.; Klein, M.B.; Hardy, I.; Narasiah, L.; Roger, M.; Kronfli, N. Phylogenetic clustering among asylum seekers with new HIV diagnoses in Montreal, Quebec, Canada. Viruses 2021, 13, 601. [Google Scholar] [CrossRef]
- Novitsky, V.; Steingrimsson, J.; Howison, M.; Dunn, C.; Gillani, F.S.; Manne, A.; Li, Y.; Spence, M.; Parillo, Z.; Fulton, J.; et al. Longitudinal typing of molecular HIV clusters in a statewide epidemic. AIDS 2021, 35, 1711–1722. [Google Scholar] [CrossRef]
- Villandre, L.; Labbe, A.; Brenner, B.; Ibanescu, R.I.; Roger, M.; Stephens, D.A. Assessing the role of transmission chains in the spread of HIV-1 among men who have sex with men in Quebec, Canada. PLoS ONE 2019, 14, e0213366. [Google Scholar] [CrossRef]
- Villandre, L.; Stephens, D.A.; Labbe, A.; Gunthard, H.F.; Kouyos, R.; Stadler, T.; Swiss HIV Cohort Study. Assessment of Overlap of Phylogenetic Transmission Clusters and Communities in Simple Sexual Contact Networks: Applications to HIV-1. PLoS ONE 2016, 11, e0148459. [Google Scholar] [CrossRef] [Green Version]
- Villandre, L.; Labbe, A.; Brenner, B.; Roger, M.; Stephens, D.A. DM-PhyClus: A Bayesian phylogenetic algorithm for infectious disease transmission cluster inference. BMC Bioinform. 2018, 19, 324. [Google Scholar] [CrossRef]
- Charest, H.; Doualla-Bell, F.; Cantin, R.; Murphy, D.G.; Lemieux, L.; Brenner, B.; Hardy, I.; Moisi, D.; Lo, E.; Baril, J.G.; et al. A Significant Reduction in the Frequency of HIV-1 Drug Resistance in Quebec from 2001 to 2011 Is Associated with a Decrease in the Monitored Viral Load. PLoS ONE 2014, 9, e109420. [Google Scholar] [CrossRef] [Green Version]
- Parveen, N.; Moodie, E.E.; Cox, J.; Lambert, G.; Otis, J.; Roger, M.; Brenner, B. New challenges in HIV research: Combining phylogenetic cluster size and epidemiological data. Epidemiol. Methods 2018, 7. [Google Scholar] [CrossRef]
- Otis, J.; Lesage, D.; Godin, G.; Brown, B.; Farley, C.; Lambert, J. Predicting and reinforcing children’s intentions to wear protective helmets while bicycling. Public Health Rep. 1992, 107, 283–289. [Google Scholar] [PubMed]
- Marzel, A.; Shilaih, M.; Yang, W.L.; Boni, J.; Yerly, S.; Klimkait, T.; Aubert, V.; Braun, D.L.; Calmy, A.; Furrer, H.; et al. HIV-1 Transmission during Recent Infection and during Treatment Interruptions as Major Drivers of New Infections in the Swiss HIV Cohort Study. Clin. Infect. Dis. 2016, 62, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Wertheim, J.O.; Oster, A.M.; Switzer, W.M.; Zhang, C.; Panneer, N.; Campbell, E.; Saduvala, N.; Johnson, J.A.; Heneine, W. Natural selection favoring more transmissible HIV detected in United States molecular transmission network. Nat. Commun. 2019, 10, 5788. [Google Scholar] [CrossRef]
- Brenner, B.G.; Ibanescu, R.I.; Oliveira, M.; Roger, M.; Hardy, I.; Routy, J.P.; Kyeyune, F.; Quinones-Mateu, M.E.; Wainberg, M.A.; Montreal PHI Cohort Study Group. HIV-1 strains belonging to large phylogenetic clusters show accelerated escape from integrase inhibitors in cell culture compared with viral isolates from singleton/small clusters. J. Antimicrob. Chemother. 2017, 72, 2171–2183. [Google Scholar] [CrossRef]
- Novitsky, V.; Ndung’u, T.; Wang, R.; Bussmann, H.; Chonco, F.; Makhema, J.; De Gruttola, V.; Walker, B.D.; Essex, M. Extended high viremics: A substantial fraction of individuals maintain high plasma viral RNA levels after acute HIV-1 subtype C infection. AIDS 2011, 25, 1515–1522. [Google Scholar] [CrossRef] [PubMed]
- Oster, A.M.; France, A.M.; Panneer, N.; Banez Ocfemia, M.C.; Campbell, E.; Dasgupta, S.; Switzer, W.M.; Wertheim, J.O.; Hernandez, A.L. Identifying Clusters of Recent and Rapid HIV Transmission through Analysis of Molecular Surveillance Data. J. Acquir. Immune Defic. Syndr. 2018, 79, 543–550. [Google Scholar] [CrossRef]
- Adrien, A.; Cox, J.; Leclerc, P.; Boivin, J.F.; Archibald, C.; Boulos, D.; Jean-Gilles, J.; Joseph, G.; Tremblay, C. Behavioural risks for HIV infection among Quebec residents of Haitian origin. J. Immigr. Minor. Health 2010, 12, 894–899. [Google Scholar] [CrossRef]
- Greenwald, Z.R.; Maheu-Giroux, M.; Szabo, J.; Robin, J.A.B.; Boissonnault, M.; Nguyen, V.K.; Thomas, R. Cohort profile: L’Actuel Pre-Exposure Prophylaxis (PrEP) Cohort study in Montreal, Canada. BMJ Open 2019, 9, e028768. [Google Scholar] [CrossRef] [Green Version]
- Bbosa, N.; Ssemwanga, D.; Nsubuga, R.N.; Kiwanuka, N.; Bagaya, B.S.; Kitayimbwa, J.M.; Ssekagiri, A.; Yebra, G.; Kaleebu, P.; Leigh-Brown, A. Phylogenetic Networks and Parameters Inferred from HIV Nucleotide Sequences of High-Risk and General Population Groups in Uganda: Implications for Epidemic Control. Viruses 2021, 13, 970. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brenner, B.G.; Ibanescu, R.-I.; Osman, N.; Cuadra-Foy, E.; Oliveira, M.; Chaillon, A.; Stephens, D.; Hardy, I.; Routy, J.-P.; Thomas, R.; et al. The Role of Phylogenetics in Unravelling Patterns of HIV Transmission towards Epidemic Control: The Quebec Experience (2002–2020). Viruses 2021, 13, 1643. https://0-doi-org.brum.beds.ac.uk/10.3390/v13081643
Brenner BG, Ibanescu R-I, Osman N, Cuadra-Foy E, Oliveira M, Chaillon A, Stephens D, Hardy I, Routy J-P, Thomas R, et al. The Role of Phylogenetics in Unravelling Patterns of HIV Transmission towards Epidemic Control: The Quebec Experience (2002–2020). Viruses. 2021; 13(8):1643. https://0-doi-org.brum.beds.ac.uk/10.3390/v13081643
Chicago/Turabian StyleBrenner, Bluma G., Ruxandra-Ilinca Ibanescu, Nathan Osman, Ernesto Cuadra-Foy, Maureen Oliveira, Antoine Chaillon, David Stephens, Isabelle Hardy, Jean-Pierre Routy, Réjean Thomas, and et al. 2021. "The Role of Phylogenetics in Unravelling Patterns of HIV Transmission towards Epidemic Control: The Quebec Experience (2002–2020)" Viruses 13, no. 8: 1643. https://0-doi-org.brum.beds.ac.uk/10.3390/v13081643