
Potential Utility of Natural Killer Cells for Eliminating Cells Harboring Reactivated Latent HIV-1 Following the Removal of CD8+ T Cell-Mediated Pro-Latency Effect(s)

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The LRA Landscape
3. Clinical Trials of the “Shock and Kill” Approach
4. Evidence That CD8+ T Cells Promote HIV-1, SIV, and SHIV Latency during ART and in the Presence of LRAs
4.1. CD8+ T Cells in HIV-1, SIV, and SHIV Infections
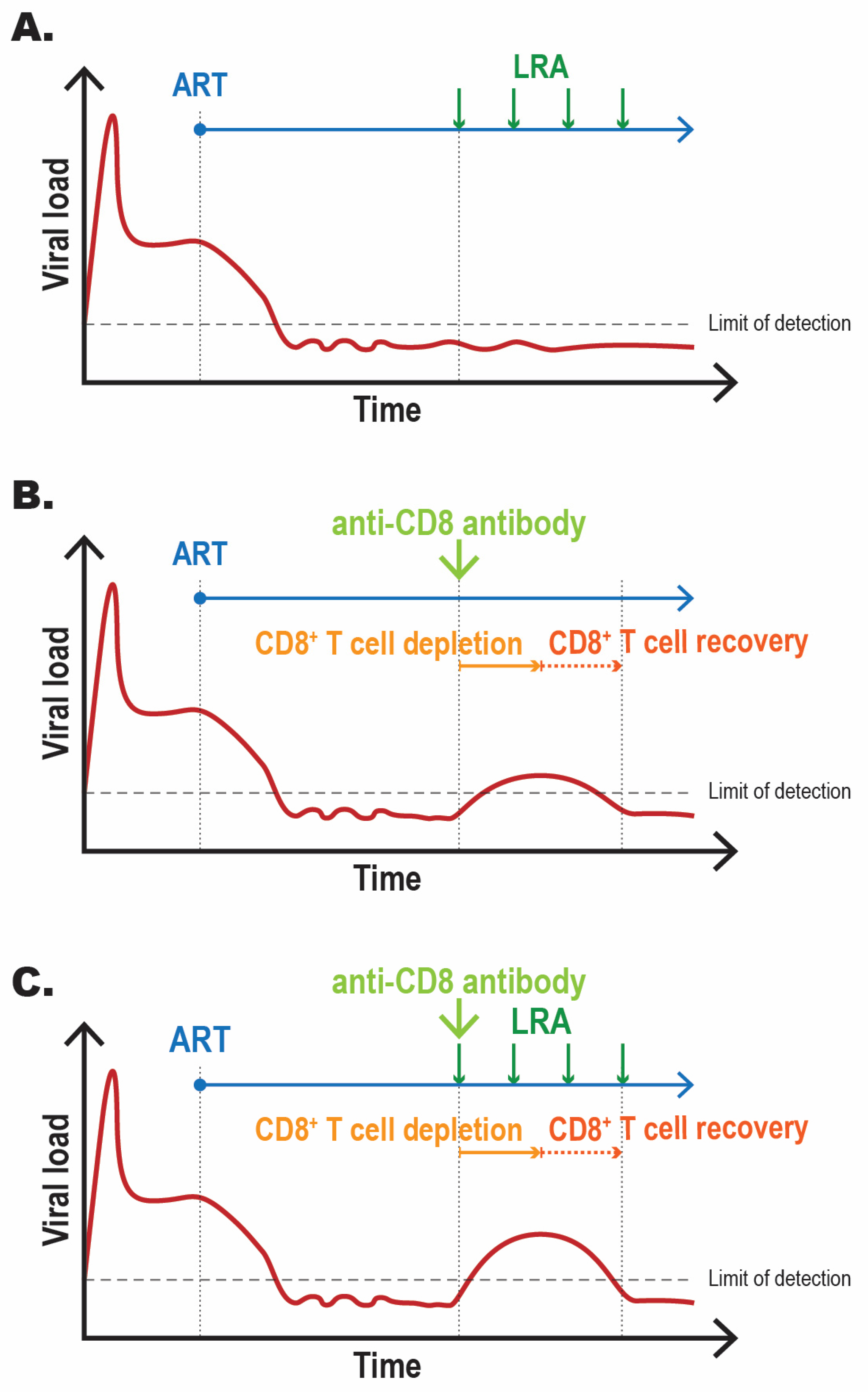
4.2. Impact of CD8+ T Cell Depletion on Viremia in a Nonhuman Primate Model of ART-Treated HIV-1 Infection
4.3. Impact of CD8+ T Cell Depletion on In Vivo LRA Activity in Animal Models of ART-Treated HIV-1 Infection
4.4. Deciphering the Mechanism(s) of CD8+ T Cell-Mediated Pro-Latency Function(s) Using In Vitro Assays
4.5. Outstanding Questions about the Role of NK Cells in Promoting Viral Latency
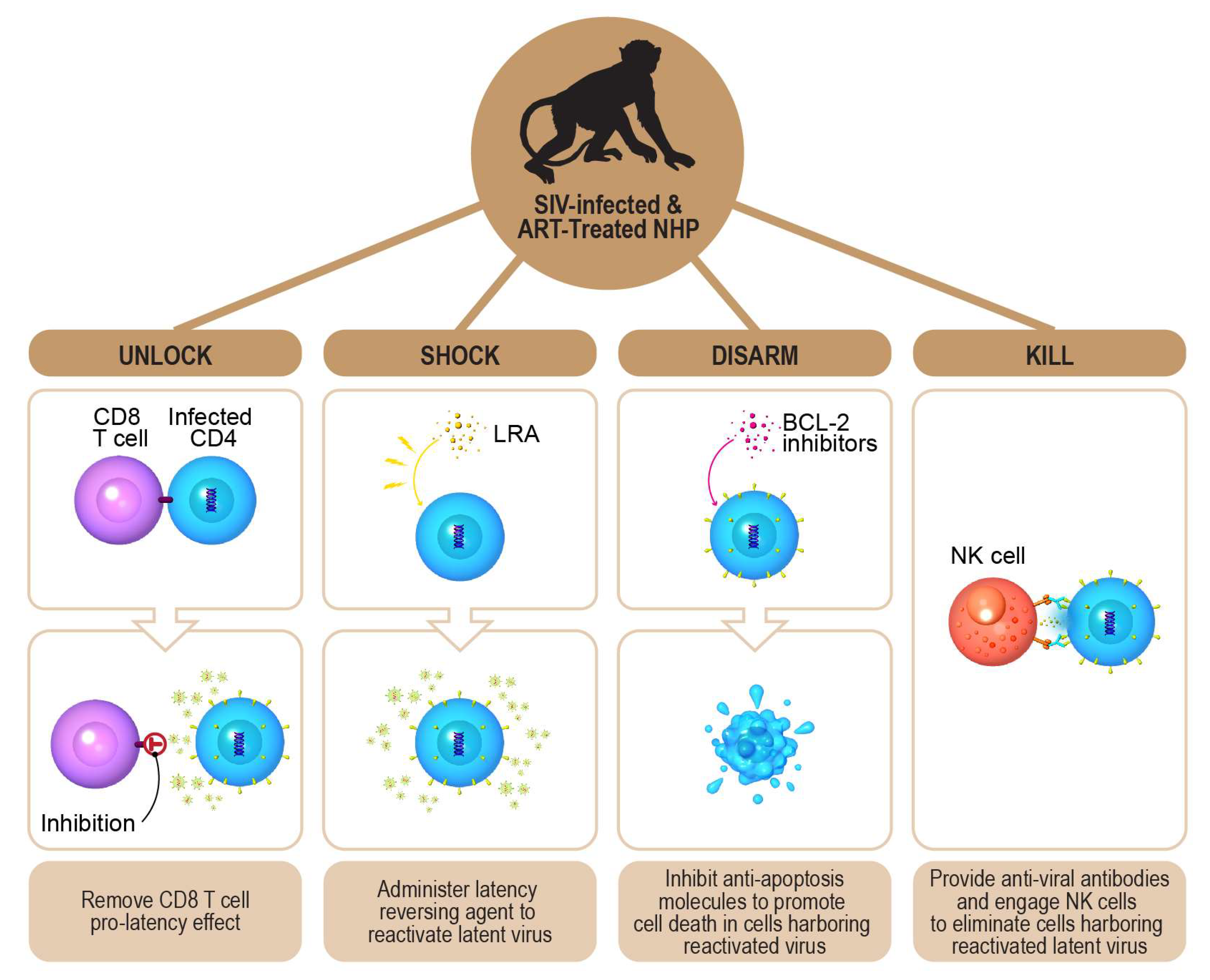
5. A Novel Multi-Pronged “Shock and Kill” HIV-1 Cure Strategy
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gulick, R.M.; Mellors, J.W.; Havlir, D.; Eron, J.J.; Gonzalez, C.; McMahon, D.; Richman, D.D.; Valentine, F.T.; Jonas, L.; Meibohm, A.; et al. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N. Engl. J. Med. 1997, 337, 734–739. [Google Scholar] [CrossRef] [Green Version]
- Palella, F.J., Jr.; Delaney, K.M.; Moorman, A.C.; Loveless, M.O.; Fuhrer, J.; Satten, G.A.; Aschman, D.J.; Holmberg, S.D. HIV Outpatient Study Investigators. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. N. Engl. J. Med. 1998, 338, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.K.; Hezareh, M.; Gunthard, H.F.; Havlir, D.V.; Ignacio, C.C.; Spina, C.A.; Richman, D.D. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 1997, 278, 1291–1295. [Google Scholar] [CrossRef]
- Colby, D.J.; Trautmann, L.; Pinyakorn, S.; Leyre, L.; Pagliuzza, A.; Kroon, E.; Rolland, M.; Takata, H.; Buranapraditkun, S.; Intasan, J.; et al. Rapid HIV RNA rebound after antiretroviral treatment interruption in persons durably suppressed in Fiebig I acute HIV infection. Nat. Med. 2018, 24, 923–926. [Google Scholar] [CrossRef] [PubMed]
- Joos, B.; Fischer, M.; Kuster, H.; Pillai, S.K.; Wong, J.K.; Boni, J.; Hirschel, B.; Weber, R.; Trkola, A.; Gunthard, H.F.; et al. HIV rebounds from latently infected cells, rather than from continuing low-level replication. Proc. Natl. Acad. Sci. USA 2008, 105, 16725–16730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeks, S.G. HIV: Shock and kill. Nature 2012, 487, 439–440. [Google Scholar] [CrossRef] [PubMed]
- Zerbato, J.M.; Purves, H.V.; Lewin, S.R.; Rasmussen, T.A. Between a shock and a hard place: Challenges and developments in HIV latency reversal. Curr. Opin. Virol. 2019, 38, 1–9. [Google Scholar] [CrossRef]
- Ait-Ammar, A.; Kula, A.; Darcis, G.; Verdikt, R.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Rohr, O.; Van Lint, C. Current Status of Latency Reversing Agents Facing the Heterogeneity of HIV-1 Cellular and Tissue Reservoirs. Front. Microbiol. 2020, 10, 3060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borducchi, E.N.; Liu, J.; Nkolola, J.P.; Cadena, A.M.; Yu, W.H.; Fischinger, S.; Broge, T.; Abbink, P.; Mercado, N.B.; Chandrashekar, A.; et al. Antibody and TLR7 agonist delay viral rebound in SHIV-infected monkeys. Nature 2018, 563, 360–364. [Google Scholar] [CrossRef]
- Elliott, J.H.; McMahon, J.H.; Chang, C.C.; Lee, S.A.; Hartogensis, W.; Bumpus, N.; Savic, R.; Roney, J.; Hoh, R.; Solomon, A.; et al. Short-term administration of disulfiram for reversal of latent HIV infection: A phase 2 dose-escalation study. Lancet HIV 2015, 2, e520–e529. [Google Scholar] [CrossRef] [Green Version]
- Elliott, J.H.; Wightman, F.; Solomon, A.; Ghneim, K.; Ahlers, J.; Cameron, M.J.; Smith, M.Z.; Spelman, T.; McMahon, J.; Velayudham, P.; et al. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog. 2014, 10, e1004473. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, C.; Serrano-Villar, S.; Madrid-Elena, N.; Perez-Elias, M.J.; Martin, M.E.; Barbas, C.; Ruiperez, J.; Munoz, E.; Munoz-Fernandez, M.A.; Castor, T.; et al. Bryostatin-1 for latent virus reactivation in HIV-infected patients on antiretroviral therapy. AIDS 2016, 30, 1385–1392. [Google Scholar] [CrossRef]
- Rasmussen, T.A.; Tolstrup, M.; Brinkmann, C.R.; Olesen, R.; Erikstrup, C.; Solomon, A.; Winckelmann, A.; Palmer, S.; Dinarello, C.; Buzon, M.; et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: A phase 1/2, single group, clinical trial. Lancet HIV 2014, 1, e13–e21. [Google Scholar] [CrossRef]
- Riddler, S.A.; Para, M.; Benson, C.A.; Mills, A.; Ramgopal, M.; DeJesus, E.; Brinson, C.; Cyktor, J.; Jacobs, J.; Koontz, D.; et al. Vesatolimod, a toll-like receptor 7 agonist, induces immune activation in virally suppressed adults with HIV-1. Clin. Infect. Dis. 2021, 72, e815–e824. [Google Scholar] [CrossRef]
- Sogaard, O.S.; Graversen, M.E.; Leth, S.; Olesen, R.; Brinkmann, C.R.; Nissen, S.K.; Kjaer, A.S.; Schleimann, M.H.; Denton, P.W.; Hey-Cunningham, W.J.; et al. The Depsipeptide Romidepsin Reverses HIV-1 Latency In Vivo. PLoS Pathog. 2015, 11, e1005142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vibholm, L.; Schleimann, M.H.; Hojen, J.F.; Benfield, T.; Offersen, R.; Rasmussen, K.; Olesen, R.; Dige, A.; Agnholt, J.; Grau, J.; et al. Short-Course Toll-Like Receptor 9 Agonist Treatment Impacts Innate Immunity and Plasma Viremia in Individuals With Human Immunodeficiency Virus Infection. Clin. Infect. Dis. 2017, 64, 1686–1695. [Google Scholar] [CrossRef]
- Vibholm, L.K.; Konrad, C.V.; Schleimann, M.H.; Frattari, G.; Winckelmann, A.; Klastrup, V.; Jensen, N.M.; Jensen, S.S.; Schmidt, M.; Wittig, B.; et al. Effects of 24-week Toll-like receptor 9 agonist treatment in HIV type 1+ individuals. AIDS 2019, 33, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Bateson, R.; Tripathy, M.K.; Crooks, A.M.; Yang, K.H.; Dahl, N.P.; Kearney, M.F.; Anderson, E.M.; Coffin, J.M.; Strain, M.C.; et al. HIV-1 expression within resting CD4+ T cells after multiple doses of vorinostat. J. Infect. Dis. 2014, 210, 728–735. [Google Scholar] [CrossRef]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.; Choudhary, S.K.; Kuruc, J.D.; Crooks, A.M.; Parker, D.C.; Anderson, E.M.; Kearney, M.F.; Strain, M.C.; et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. [Google Scholar] [CrossRef] [Green Version]
- Madrid-Elena, N.; Garcia-Bermejo, M.L.; Serrano-Villar, S.; Diaz-de Santiago, A.; Sastre, B.; Gutierrez, C.; Dronda, F.; Coronel Diaz, M.; Dominguez, E.; Lopez-Huertas, M.R.; et al. Maraviroc Is Associated with Latent HIV-1 Reactivation through NF-κB Activation in Resting CD4(+) T Cells from HIV-Infected Individuals on Suppressive Antiretroviral Therapy. J. Virol. 2018, 92, e01931-17. [Google Scholar] [CrossRef] [Green Version]
- McBrien, J.B.; Mavigner, M.; Franchitti, L.; Smith, S.A.; White, E.; Tharp, G.K.; Walum, H.; Busman-Sahay, K.; Aguilera-Sandoval, C.R.; Thayer, W.O.; et al. Robust and persistent reactivation of SIV and HIV by N-803 and depletion of CD8(+) cells. Nature 2020, 578, 154–159. [Google Scholar] [CrossRef] [PubMed]
- McBrien, J.B.; Wong, A.K.H.; White, E.; Carnathan, D.G.; Lee, J.H.; Safrit, J.T.; Vanderford, T.H.; Paiardini, M.; Chahroudi, A.; Silvestri, G. Combination of CD8beta Depletion and Interleukin-15 Superagonist N-803 Induces Virus Reactivation in Simian-Human Immunodeficiency Virus-Infected, Long-Term ART-Treated Rhesus Macaques. J. Virol. 2020, 94, e00755-20. [Google Scholar] [CrossRef] [PubMed]
- Webb, G.M.; Molden, J.; Busman-Sahay, K.; Abdulhaqq, S.; Wu, H.L.; Weber, W.C.; Bateman, K.B.; Reed, J.S.; Northrup, M.; Maier, N.; et al. The human IL-15 superagonist N-803 promotes migration of virus-specific CD8+ T and NK cells to B cell follicles but does not reverse latency in ART-suppressed, SHIV-infected macaques. PLoS Pathog. 2020, 16, e1008339. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, E.K.; Spicer, L.; Smith, S.A.; Lee, D.; Fast, R.; Paganini, S.; Lawson, B.O.; Nega, M.; Easley, K.; Schmitz, J.E.; et al. CD8(+) Lymphocytes Are Required for Maintaining Viral Suppression in SIV-Infected Macaques Treated with Short-Term Antiretroviral Therapy. Immunity 2016, 45, 656–668. [Google Scholar] [CrossRef] [Green Version]
- Bruel, T.; Guivel-Benhassine, F.; Amraoui, S.; Malbec, M.; Richard, L.; Bourdic, K.; Donahue, D.A.; Lorin, V.; Casartelli, N.; Noel, N.; et al. Elimination of HIV-1-infected cells by broadly neutralizing antibodies. Nat. Commun. 2016, 7, 10844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, M.S.; Chung, A.W.; Kent, S.J. Importance of Fc-mediated functions of anti-HIV-1 broadly neutralizing antibodies. Retrovirology 2018, 15, 58. [Google Scholar] [CrossRef]
- von Bredow, B.; Arias, J.F.; Heyer, L.N.; Moldt, B.; Le, K.; Robinson, J.E.; Zolla-Pazner, S.; Burton, D.R.; Evans, D.T. Comparison of Antibody-Dependent Cell-Mediated Cytotoxicity and Virus Neutralization by HIV-1 Env-Specific Monoclonal Antibodies. J. Virol. 2016, 90, 6127–6139. [Google Scholar] [CrossRef] [Green Version]
- Bonaparte, M.I.; Barker, E. Killing of human immunodeficiency virus-infected primary T-cell blasts by autologous natural killer cells is dependent on the ability of the virus to alter the expression of major histocompatibility complex class I molecules. Blood 2004, 104, 2087–2094. [Google Scholar] [CrossRef]
- Khoury, G.; Darcis, G.; Lee, M.Y.; Bouchat, S.; Van Driessche, B.; Purcell, D.F.J.; Van Lint, C. The Molecular Biology of HIV Latency. Adv. Exp. Med. Biol. 2018, 1075, 187–212. [Google Scholar]
- Kumar, A.; Darcis, G.; Van Lint, C.; Herbein, G. Epigenetic control of HIV-1 post integration latency: Implications for therapy. Clin. Epigenetics 2015, 7, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archin, N.M.; Espeseth, A.; Parker, D.; Cheema, M.; Hazuda, D.; Margolis, D.M. Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid. AIDS Res. Hum. Retrovir. 2009, 25, 207–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, S.; Wightman, F.; Ramanayake, S.; Alexander, M.; Kumar, N.; Khoury, G.; Pereira, C.; Purcell, D.; Cameron, P.U.; Lewin, S.R. Expression and reactivation of HIV in a chemokine induced model of HIV latency in primary resting CD4+ T cells. Retrovirology 2011, 8, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, D.G.; Chiang, V.; Fyne, E.; Balakrishnan, M.; Barnes, T.; Graupe, M.; Hesselgesser, J.; Irrinki, A.; Murry, J.P.; Stepan, G.; et al. Histone deacetylase inhibitor romidepsin induces HIV expression in CD4 T cells from patients on suppressive antiretroviral therapy at concentrations achieved by clinical dosing. PLoS Pathog. 2014, 10, e1004071. [Google Scholar] [CrossRef] [Green Version]
- Wightman, F.; Lu, H.K.; Solomon, A.E.; Saleh, S.; Harman, A.N.; Cunningham, A.L.; Gray, L.; Churchill, M.; Cameron, P.U.; Dear, A.E.; et al. Entinostat is a histone deacetylase inhibitor selective for class 1 histone deacetylases and activates HIV production from latently infected primary T cells. AIDS 2013, 27, 2853–2862. [Google Scholar] [CrossRef]
- Bernhard, W.; Barreto, K.; Saunders, A.; Dahabieh, M.S.; Johnson, P.; Sadowski, I. The Suv39H1 methyltransferase inhibitor chaetocin causes induction of integrated HIV-1 without producing a T cell response. FEBS Lett. 2011, 585, 3549–3554. [Google Scholar] [CrossRef]
- Bouchat, S.; Gatot, J.S.; Kabeya, K.; Cardona, C.; Colin, L.; Herbein, G.; De Wit, S.; Clumeck, N.; Lambotte, O.; Rouzioux, C.; et al. Histone methyltransferase inhibitors induce HIV-1 recovery in resting CD4(+) T cells from HIV-1-infected HAART-treated patients. AIDS 2012, 26, 1473–1482. [Google Scholar] [CrossRef]
- Fernandez, G.; Zeichner, S.L. Cell line-dependent variability in HIV activation employing DNMT inhibitors. Virol. J. 2010, 7, 266. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.; Cho, W.K.; Chu, C.K.; Keedy, K.S.; Archin, N.M.; Margolis, D.M.; Karn, J. Epigenetic silencing of HIV-1 by the histone H3 lysine 27 methyltransferase enhancer of Zeste 2. J. Virol. 2011, 85, 9078–9089. [Google Scholar] [CrossRef] [Green Version]
- Imai, K.; Togami, H.; Okamoto, T. Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J. Biol. Chem. 2010, 285, 16538–16545. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.L.; Hokello, J.; Sonti, S.; Zicari, S.; Sun, L.; Alqatawni, A.; Bukrinsky, M.; Simon, G.; Chauhan, A.; Daniel, R.; et al. CBF-1 Promotes the Establishment and Maintenance of HIV Latency by Recruiting Polycomb Repressive Complexes, PRC1 and PRC2, at HIV LTR. Viruses 2020, 12, 1040. [Google Scholar] [CrossRef]
- Pandelo Jose, D.; Bartholomeeusen, K.; da Cunha, R.D.; Abreu, C.M.; Glinski, J.; da Costa, T.B.; Bacchi Rabay, A.F.; Pianowski Filho, L.F.; Dudycz, L.W.; Ranga, U.; et al. Reactivation of latent HIV-1 by new semi-synthetic ingenol esters. Virology 2014, 462–463, 328–339. [Google Scholar] [CrossRef] [Green Version]
- DeChristopher, B.A.; Loy, B.A.; Marsden, M.D.; Schrier, A.J.; Zack, J.A.; Wender, P.A. Designed, synthetically accessible bryostatin analogues potently induce activation of latent HIV reservoirs in vitro. Nat. Chem. 2012, 4, 705–710. [Google Scholar] [CrossRef]
- Mehla, R.; Bivalkar-Mehla, S.; Zhang, R.; Handy, I.; Albrecht, H.; Giri, S.; Nagarkatti, P.; Nagarkatti, M.; Chauhan, A. Bryostatin modulates latent HIV-1 infection via PKC and AMPK signaling but inhibits acute infection in a receptor independent manner. PLoS ONE 2010, 5, e11160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkosky, J.; Culnan, D.M.; Roman, J.; Dornadula, G.; Schnell, M.; Boyd, M.R.; Pomerantz, R.J. Prostratin: Activation of latent HIV-1 expression suggests a potential inductive adjuvant therapy for HAART. Blood 2001, 98, 3006–3015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spivak, A.M.; Bosque, A.; Balch, A.H.; Smyth, D.; Martins, L.; Planelles, V. Ex Vivo Bioactivity and HIV-1 Latency Reversal by Ingenol Dibenzoate and Panobinostat in Resting CD4(+) T Cells from Aviremic Patients. Antimicrob. Agents Chemother. 2015, 59, 5984–5991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Huertas, M.R.; Jimenez-Tormo, L.; Madrid-Elena, N.; Gutierrez, C.; Rodriguez-Mora, S.; Coiras, M.; Alcami, J.; Moreno, S. The CCR5-antagonist Maraviroc reverses HIV-1 latency in vitro alone or in combination with the PKC-agonist Bryostatin-1. Sci. Rep. 2017, 7, 2385. [Google Scholar] [CrossRef]
- Pache, L.; Dutra, M.S.; Spivak, A.M.; Marlett, J.M.; Murry, J.P.; Hwang, Y.; Maestre, A.M.; Manganaro, L.; Vamos, M.; Teriete, P.; et al. BIRC2/cIAP1 Is a Negative Regulator of HIV-1 Transcription and Can Be Targeted by Smac Mimetics to Promote Reversal of Viral Latency. Cell Host Microb. 2015, 18, 345–353. [Google Scholar] [CrossRef] [Green Version]
- Contreras, X.; Barboric, M.; Lenasi, T.; Peterlin, B.M. HMBA releases P-TEFb from HEXIM1 and 7SK snRNA via PI3K/Akt and activates HIV transcription. PLoS Pathog. 2007, 3, 1459–1469. [Google Scholar] [CrossRef]
- Banerjee, C.; Archin, N.; Michaels, D.; Belkina, A.C.; Denis, G.V.; Bradner, J.; Sebastiani, P.; Margolis, D.M.; Montano, M. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J. Leukoc. Biol. 2012, 92, 1147–1154. [Google Scholar] [CrossRef]
- Darcis, G.; Kula, A.; Bouchat, S.; Fujinaga, K.; Corazza, F.; Ait-Ammar, A.; Delacourt, N.; Melard, A.; Kabeya, K.; Vanhulle, C.; et al. An In-Depth Comparison of Latency-Reversing Agent Combinations in Various In Vitro and Ex Vivo HIV-1 Latency Models Identified Bryostatin-1+JQ1 and Ingenol-B+JQ1 to Potently Reactivate Viral Gene Expression. PLoS Pathog. 2015, 11, e1005063. [Google Scholar] [CrossRef] [PubMed]
- Doyon, G.; Zerbato, J.; Mellors, J.W.; Sluis-Cremer, N. Disulfiram reactivates latent HIV-1 expression through depletion of the phosphatase and tensin homolog. AIDS 2013, 27, F7–F11. [Google Scholar] [CrossRef] [PubMed]
- Kula, A.; Delacourt, N.; Bouchat, S.; Darcis, G.; Avettand-Fenoel, V.; Verdikt, R.; Corazza, F.; Necsoi, C.; Vanhulle, C.; Bendoumou, M.; et al. Heterogeneous HIV-1 Reactivation Patterns of Disulfiram and Combined Disulfiram+Romidepsin Treatments. J. Acquir. Immune Defic. Syndr. 2019, 80, 605–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, S.; Bullen, C.K.; Shroff, N.S.; Shan, L.; Yang, H.C.; Manucci, J.L.; Bhat, S.; Zhang, H.; Margolick, J.B.; Quinn, T.C.; et al. Disulfiram reactivates latent HIV-1 in a Bcl-2-transduced primary CD4+ T cell model without inducing global T cell activation. J. Virol. 2011, 85, 6060–6064. [Google Scholar] [CrossRef] [Green Version]
- Bosque, A.; Nilson, K.A.; Macedo, A.B.; Spivak, A.M.; Archin, N.M.; Van Wagoner, R.M.; Martins, L.J.; Novis, C.L.; Szaniawski, M.A.; Ireland, C.M.; et al. Benzotriazoles Reactivate Latent HIV-1 through Inactivation of STAT5 SUMOylation. Cell Rep. 2017, 18, 1324–1334. [Google Scholar] [CrossRef]
- Sorensen, E.S.; Macedo, A.B.; Resop, R.S.; Howard, J.N.; Nell, R.; Sarabia, I.; Newman, D.; Ren, Y.; Jones, R.B.; Planelles, V.; et al. Structure-Activity Relationship Analysis of Benzotriazine Analogues as HIV-1 Latency-Reversing Agents. Antimicrob. Agents Chemother. 2020, 64, e00888-20. [Google Scholar] [CrossRef]
- Offersen, R.; Nissen, S.K.; Rasmussen, T.A.; Ostergaard, L.; Denton, P.W.; Sogaard, O.S.; Tolstrup, M. A Novel Toll-Like Receptor 9 Agonist, MGN1703, Enhances HIV-1 Transcription and NK Cell-Mediated Inhibition of HIV-1-Infected Autologous CD4+ T Cells. J. Virol. 2016, 90, 4441–4453. [Google Scholar] [CrossRef] [Green Version]
- Tsai, A.; Irrinki, A.; Kaur, J.; Cihlar, T.; Kukolj, G.; Sloan, D.D.; Murry, J.P. Toll-Like Receptor 7 Agonist GS-9620 Induces HIV Expression and HIV-Specific Immunity in Cells from HIV-Infected Individuals on Suppressive Antiretroviral Therapy. J. Virol. 2017, 91, e02166-16. [Google Scholar] [CrossRef] [Green Version]
- Evans, V.A.; van der Sluis, R.M.; Solomon, A.; Dantanarayana, A.; McNeil, C.; Garsia, R.; Palmer, S.; Fromentin, R.; Chomont, N.; Sekaly, R.P.; et al. Programmed cell death-1 contributes to the establishment and maintenance of HIV-1 latency. AIDS 2018, 32, 1491–1497. [Google Scholar] [CrossRef] [PubMed]
- Wightman, F.; Solomon, A.; Kumar, S.S.; Urriola, N.; Gallagher, K.; Hiener, B.; Palmer, S.; McNeil, C.; Garsia, R.; Lewin, S.R. Effect of ipilimumab on the HIV reservoir in an HIV-infected individual with metastatic melanoma. AIDS 2015, 29, 504–506. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.B.; Mueller, S.; O’Connor, R.; Rimpel, K.; Sloan, D.D.; Karel, D.; Wong, H.C.; Jeng, E.K.; Thomas, A.S.; Whitney, J.B.; et al. A Subset of Latency-Reversing Agents Expose HIV-Infected Resting CD4+ T-Cells to Recognition by Cytotoxic T-Lymphocytes. PLoS Pathog. 2016, 12, e1005545. [Google Scholar] [CrossRef] [Green Version]
- Battivelli, E.; Dahabieh, M.S.; Abdel-Mohsen, M.; Svensson, J.P.; Tojal Da Silva, I.; Cohn, L.B.; Gramatica, A.; Deeks, S.; Greene, W.C.; Pillai, S.K.; et al. Distinct chromatin functional states correlate with HIV latency reactivation in infected primary CD4(+) T cells. eLife 2018, 7, e34655. [Google Scholar] [CrossRef]
- Grau-Exposito, J.; Luque-Ballesteros, L.; Navarro, J.; Curran, A.; Burgos, J.; Ribera, E.; Torrella, A.; Planas, B.; Badia, R.; Martin-Castillo, M.; et al. Latency reversal agents affect differently the latent reservoir present in distinct CD4+ T subpopulations. PLoS Pathog. 2019, 15, e1007991. [Google Scholar] [CrossRef] [Green Version]
- Spina, C.A.; Anderson, J.; Archin, N.M.; Bosque, A.; Chan, J.; Famiglietti, M.; Greene, W.C.; Kashuba, A.; Lewin, S.R.; Margolis, D.M.; et al. An in-depth comparison of latent HIV-1 reactivation in multiple cell model systems and resting CD4+ T cells from aviremic patients. PLoS Pathog. 2013, 9, e1003834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laird, G.M.; Bullen, C.K.; Rosenbloom, D.I.; Martin, A.R.; Hill, A.L.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. Ex vivo analysis identifies effective HIV-1 latency-reversing drug combinations. J. Clin. Investig. 2015, 125, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Borrow, P.; Lewicki, H.; Hahn, B.H.; Shaw, G.M.; Oldstone, M.B. Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J. Virol. 1994, 68, 6103–6110. [Google Scholar] [CrossRef] [Green Version]
- Koup, R.A.; Safrit, J.T.; Cao, Y.; Andrews, C.A.; McLeod, G.; Borkowsky, W.; Farthing, C.; Ho, D.D. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J. Virol. 1994, 68, 4650–4655. [Google Scholar] [CrossRef] [Green Version]
- Borrow, P.; Lewicki, H.; Wei, X.; Horwitz, M.S.; Peffer, N.; Meyers, H.; Nelson, J.A.; Gairin, J.E.; Hahn, B.H.; Oldstone, M.B.; et al. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat. Med. 1997, 3, 205–211. [Google Scholar] [CrossRef]
- Goulder, P.J.; Phillips, R.E.; Colbert, R.A.; McAdam, S.; Ogg, G.; Nowak, M.A.; Giangrande, P.; Luzzi, G.; Morgan, B.; Edwards, A.; et al. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat. Med. 1997, 3, 212–217. [Google Scholar] [CrossRef]
- Goulder, P.J.; Watkins, D.I. Impact of MHC class I diversity on immune control of immunodeficiency virus replication. Nat. Rev. Immunol. 2008, 8, 619–630. [Google Scholar] [CrossRef]
- Chowdhury, A.; Hayes, T.L.; Bosinger, S.E.; Lawson, B.O.; Vanderford, T.; Schmitz, J.E.; Paiardini, M.; Betts, M.; Chahroudi, A.; Estes, J.D.; et al. Differential Impact of In Vivo CD8+ T Lymphocyte Depletion in Controller versus Progressor Simian Immunodeficiency Virus-Infected Macaques. J. Virol. 2015, 89, 8677–8686. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Bauer, D.E.; Tuttleton, S.E.; Lewin, S.; Gettie, A.; Blanchard, J.; Irwin, C.E.; Safrit, J.T.; Mittler, J.; Weinberger, L.; et al. Dramatic rise in plasma viremia after CD8(+) T cell depletion in simian immunodeficiency virus-infected macaques. J. Exp. Med. 1999, 189, 991–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matano, T.; Shibata, R.; Siemon, C.; Connors, M.; Lane, H.C.; Martin, M.A. Administration of an anti-CD8 monoclonal antibody interferes with the clearance of chimeric simian/human immunodeficiency virus during primary infections of rhesus macaques. J. Virol. 1998, 72, 164–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzner, K.J.; Jin, X.; Lee, F.V.; Gettie, A.; Bauer, D.E.; Di Mascio, M.; Perelson, A.S.; Marx, P.A.; Ho, D.D.; Kostrikis, L.G.; et al. Effects of in vivo CD8(+) T cell depletion on virus replication in rhesus macaques immunized with a live, attenuated simian immunodeficiency virus vaccine. J. Exp. Med. 2000, 191, 1921–1931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migueles, S.A.; Osborne, C.M.; Royce, C.; Compton, A.A.; Joshi, R.P.; Weeks, K.A.; Rood, J.E.; Berkley, A.M.; Sacha, J.B.; Cogliano-Shutta, N.A.; et al. Lytic granule loading of CD8+ T cells is required for HIV-infected cell elimination associated with immune control. Immunity 2008, 29, 1009–1021. [Google Scholar] [CrossRef] [Green Version]
- Cocchi, F.; DeVico, A.L.; Garzino-Demo, A.; Arya, S.K.; Gallo, R.C.; Lusso, P. Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science 1995, 270, 1811–1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dragic, T.; Litwin, V.; Allaway, G.P.; Martin, S.R.; Huang, Y.; Nagashima, K.A.; Cayanan, C.; Maddon, P.J.; Koup, R.A.; Moore, J.P.; et al. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 1996, 381, 667–673. [Google Scholar] [CrossRef]
- Cocchi, F.; DeVico, A.L.; Lu, W.; Popovic, M.; Latinovic, O.; Sajadi, M.M.; Redfield, R.R.; Lafferty, M.K.; Galli, M.; Garzino-Demo, A.; et al. Soluble factors from T cells inhibiting X4 strains of HIV are a mixture of beta chemokines and RNases. Proc. Natl. Acad. Sci. USA 2012, 109, 5411–5416. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.L.; Francois, F.; Mosoian, A.; Klotman, M.E. CAF-mediated human immunodeficiency virus (HIV) type 1 transcriptional inhibition is distinct from alpha-defensin-1 HIV inhibition. J. Virol. 2003, 77, 6777–6784. [Google Scholar] [CrossRef] [Green Version]
- Levy, J.A.; Mackewicz, C.E.; Barker, E. Controlling HIV pathogenesis: The role of the noncytotoxic anti-HIV response of CD8+ T cells. Immunol. Today 1996, 17, 217–224. [Google Scholar] [CrossRef]
- Mackewicz, C.; Levy, J.A. CD8+ cell anti-HIV activity: Nonlytic suppression of virus replication. AIDS Res. Hum. Retrovir. 1992, 8, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Mackewicz, C.E.; Patterson, B.K.; Lee, S.A.; Levy, J.A. CD8(+) cell noncytotoxic anti-human immunodeficiency virus response inhibits expression of viral RNA but not reverse transcription or provirus integration. J. Gen. Virol. 2000, 81, 1261–1264. [Google Scholar] [CrossRef] [PubMed]
- Tomaras, G.D.; Lacey, S.F.; McDanal, C.B.; Ferrari, G.; Weinhold, K.J.; Greenberg, M.L. CD8+ T cell-mediated suppressive activity inhibits HIV-1 after virus entry with kinetics indicating effects on virus gene expression. Proc. Natl. Acad. Sci. USA 2000, 97, 3503–3508. [Google Scholar] [CrossRef]
- Walker, C.M.; Erickson, A.L.; Hsueh, F.C.; Levy, J.A. Inhibition of human immunodeficiency virus replication in acutely infected CD4+ cells by CD8+ cells involves a noncytotoxic mechanism. J. Virol. 1991, 65, 5921–5927. [Google Scholar] [CrossRef] [Green Version]
- Walker, C.M.; Levy, J.A. A diffusible lymphokine produced by CD8+ T lymphocytes suppresses HIV replication. Immunology 1989, 66, 628–630. [Google Scholar]
- Wiviott, L.D.; Walker, C.M.; Levy, J.A. CD8+ lymphocytes suppress HIV production by autologous CD4+ cells without eliminating the infected cells from culture. Cell Immunol. 1990, 128, 628–634. [Google Scholar] [CrossRef]
- Han, K.P.; Zhu, X.; Liu, B.; Jeng, E.; Kong, L.; Yovandich, J.L.; Vyas, V.V.; Marcus, W.D.; Chavaillaz, P.A.; Romero, C.A.; et al. IL-15:IL-15 receptor alpha superagonist complex: High-level co-expression in recombinant mammalian cells, purification and characterization. Cytokine 2011, 56, 804–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhode, P.R.; Egan, J.O.; Xu, W.; Hong, H.; Webb, G.M.; Chen, X.; Liu, B.; Zhu, X.; Wen, J.; You, L.; et al. Comparison of the Superagonist Complex, ALT-803, to IL15 as Cancer Immunotherapeutics in Animal Models. Cancer Immunol. Res. 2016, 4, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Webster, R.L.; Johnson, R.P. Delineation of multiple subpopulations of natural killer cells in rhesus macaques. Immunology 2005, 115, 206–214. [Google Scholar] [CrossRef]
- Le Borgne, S.; Fevrier, M.; Callebaut, C.; Lee, S.P.; Riviere, Y. CD8(+)-Cell antiviral factor activity is not restricted to human immunodeficiency virus (HIV)-specific T cells and can block HIV replication after initiation of reverse transcription. J. Virol. 2000, 74, 4456–4464. [Google Scholar] [CrossRef]
- Zanoni, M.; Palesch, D.; Pinacchio, C.; Statzu, M.; Tharp, G.K.; Paiardini, M.; Chahroudi, A.; Bosinger, S.E.; Yoon, J.; Cox, B.; et al. Innate, non-cytolytic CD8+ T cell-mediated suppression of HIV replication by MHC-independent inhibition of virus transcription. PLoS Pathog. 2020, 16, e1008821. [Google Scholar] [CrossRef]
- Kulpa, D.A.; Talla, A.; Brehm, J.H.; Ribeiro, S.P.; Yuan, S.; Bebin-Blackwell, A.G.; Miller, M.; Barnard, R.; Deeks, S.G.; Hazuda, D.; et al. Differentiation into an Effector Memory Phenotype Potentiates HIV-1 Latency Reversal in CD4(+) T Cells. J. Virol. 2019, 93, e00969-19. [Google Scholar] [CrossRef] [Green Version]
- Mavilio, D.; Benjamin, J.; Kim, D.; Lombardo, G.; Daucher, M.; Kinter, A.; Nies-Kraske, E.; Marcenaro, E.; Moretta, A.; Fauci, A.S. Identification of NKG2A and NKp80 as specific natural killer cell markers in rhesus and pigtailed monkeys. Blood 2005, 106, 1718–1725. [Google Scholar] [CrossRef] [Green Version]
- Hong, H.S.; Rajakumar, P.A.; Billingsley, J.M.; Reeves, R.K.; Johnson, R.P. No monkey business: Why studying NK cells in non-human primates pays off. Front. Immunol. 2013, 4, 32. [Google Scholar] [CrossRef] [Green Version]
- Reeves, R.K.; Gillis, J.; Wong, F.E.; Yu, Y.; Connole, M.; Johnson, R.P. CD16- natural killer cells: Enrichment in mucosal and secondary lymphoid tissues and altered function during chronic SIV infection. Blood 2010, 115, 4439–4446. [Google Scholar] [CrossRef]
- Choi, E.I.; Reimann, K.A.; Letvin, N.L. In vivo natural killer cell depletion during primary simian immunodeficiency virus infection in rhesus monkeys. J. Virol. 2008, 82, 6758–6761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, E.I.; Wang, R.; Peterson, L.; Letvin, N.L.; Reimann, K.A. Use of an anti-CD16 antibody for in vivo depletion of natural killer cells in rhesus macaques. Immunology 2008, 124, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Manickam, C.; Shah, S.V.; Nohara, J.; Ferrari, G.; Reeves, R.K. Monkeying Around: Using Non-human Primate Models to Study NK Cell Biology in HIV Infections. Front. Immunol. 2019, 10, 1124. [Google Scholar] [CrossRef] [PubMed]
- DeGottardi, M.Q.; Okoye, A.A.; Vaidya, M.; Talla, A.; Konfe, A.L.; Reyes, M.D.; Clock, J.A.; Duell, D.M.; Legasse, A.W.; Sabnis, A.; et al. Effect of Anti-IL-15 Administration on T Cell and NK Cell Homeostasis in Rhesus Macaques. J. Immunol. 2016, 197, 1183–1198. [Google Scholar] [CrossRef] [Green Version]
- Grzywacz, B.; Kataria, N.; Verneris, M.R. CD56(dim)CD16(+) NK cells downregulate CD16 following target cell induced activation of matrix metalloproteinases. Leukemia 2007, 21, 356–359. [Google Scholar] [CrossRef] [Green Version]
- Jost, S.; Reardon, J.; Peterson, E.; Poole, D.; Bosch, R.; Alter, G.; Altfeld, M. Expansion of 2B4+ natural killer (NK) cells and decrease in NKp46+ NK cells in response to influenza. Immunology 2011, 132, 516–526. [Google Scholar] [CrossRef]
- Parsons, M.S.; Tang, C.C.; Jegaskanda, S.; Center, R.J.; Brooks, A.G.; Stratov, I.; Kent, S.J. Anti-HIV antibody-dependent activation of NK cells impairs NKp46 expression. J. Immunol. 2014, 192, 308–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanier, L.L. NK cell receptors. Annu. Rev. Immunol. 1998, 16, 359–393. [Google Scholar] [CrossRef]
- Veillette, M.; Coutu, M.; Richard, J.; Batraville, L.A.; Dagher, O.; Bernard, N.; Tremblay, C.; Kaufmann, D.E.; Roger, M.; Finzi, A. The HIV-1 gp120 CD4-bound conformation is preferentially targeted by antibody-dependent cellular cytotoxicity-mediating antibodies in sera from HIV-1-infected individuals. J. Virol. 2015, 89, 545–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veillette, M.; Desormeaux, A.; Medjahed, H.; Gharsallah, N.E.; Coutu, M.; Baalwa, J.; Guan, Y.; Lewis, G.; Ferrari, G.; Hahn, B.H.; et al. Interaction with cellular CD4 exposes HIV-1 envelope epitopes targeted by antibody-dependent cell-mediated cytotoxicity. J. Virol. 2014, 88, 2633–2644. [Google Scholar] [CrossRef] [Green Version]
- Gomes, S.E.; Simoes, A.E.; Pereira, D.M.; Castro, R.E.; Rodrigues, C.M.; Borralho, P.M. miR-143 or miR-145 overexpression increases cetuximab-mediated antibody-dependent cellular cytotoxicity in human colon cancer cells. Oncotarget 2016, 7, 9368–9387. [Google Scholar] [CrossRef] [PubMed]
- Masood, A.; Chitta, K.; Paulus, A.; Khan, A.N.; Sher, T.; Ersing, N.; Miller, K.C.; Manfredi, D.; Ailawadhi, S.; Borrelo, I.; et al. Downregulation of BCL2 by AT-101 enhances the antileukaemic effect of lenalidomide both by an immune dependant and independent manner. Br. J. Haematol. 2012, 157, 59–66. [Google Scholar] [CrossRef]
- Ren, Y.; Huang, S.H.; Patel, S.; Alberto, W.D.C.; Magat, D.; Ahimovic, D.; Macedo, A.B.; Durga, R.; Chan, D.; Zale, E.; et al. BCL-2 antagonism sensitizes cytotoxic T cell-resistant HIV reservoirs to elimination ex vivo. J. Clin. Investig. 2020, 130, 2542–2559. [Google Scholar] [CrossRef]
- Chandrasekar, A.P.; Cummins, N.W.; Badley, A.D. The Role of the BCL-2 Family of Proteins in HIV-1 Pathogenesis and Persistence. Clin. Microbiol. Rev. 2019, 33, e00107-19. [Google Scholar] [CrossRef]
- Roberts, A.W. Therapeutic development and current uses of BCL-2 inhibition. Hematol. Am. Soc. Hematol. Educ. Program. 2020, 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khoury, G.; Kulpa, D.A.; Parsons, M.S. Potential Utility of Natural Killer Cells for Eliminating Cells Harboring Reactivated Latent HIV-1 Following the Removal of CD8+ T Cell-Mediated Pro-Latency Effect(s). Viruses 2021, 13, 1451. https://0-doi-org.brum.beds.ac.uk/10.3390/v13081451
Khoury G, Kulpa DA, Parsons MS. Potential Utility of Natural Killer Cells for Eliminating Cells Harboring Reactivated Latent HIV-1 Following the Removal of CD8+ T Cell-Mediated Pro-Latency Effect(s). Viruses. 2021; 13(8):1451. https://0-doi-org.brum.beds.ac.uk/10.3390/v13081451
Chicago/Turabian StyleKhoury, Georges, Deanna A. Kulpa, and Matthew S. Parsons. 2021. "Potential Utility of Natural Killer Cells for Eliminating Cells Harboring Reactivated Latent HIV-1 Following the Removal of CD8+ T Cell-Mediated Pro-Latency Effect(s)" Viruses 13, no. 8: 1451. https://0-doi-org.brum.beds.ac.uk/10.3390/v13081451