
Coagulopathy in Acute Puumala Hantavirus Infection

 , ,
, ,
Abstract
:1. Introduction
2. Puumala Hantavirus Infection
2.1. Clinical Features of the Disease
2.2. Acute Kidney Injury
2.3. Capillary Leakage
2.4. Hemorrhagic Manifestations
3. Thrombocytopenia in PUUV Infection
3.1. Thrombopoiesis and Platelet Activation
3.2. Decreased Platelet Count Association with Pentraxin-3, Cell-Free DNA, and Interleukin-6 Levels
4. Coagulopathy in PUUV Infection
4.1. Endothelial Activation
4.1.1. Neutrophil Activation
4.1.2. Vascular Endothelial Growth Factor, Bradykinin, and Nitric Oxide
4.2. Coagulation Activation
4.3. Fibrinolysis
4.4. Disseminated Intravascular Coagulation (DIC)
5. Complement Activation
Interactions between Coagulation, Predictive Biomarkers and the Complement System
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Vaheri, A.; Henttonen, H.; Voutilainen, L.; Mustonen, J.; Sironen, T.; Vapalahti, O. Hantavirus infections in Europe and their impact on public health. Rev. Med. Virol. 2013, 23, 35–49. [Google Scholar] [CrossRef]
- Vapalahti, O.; Mustonen, J.; Lundkvist, Å.; Henttonen, H.; Plyusnin, A.; Vaheri, A. Hantavirus infections in Europe. Lancet Infect. Dis. 2003, 3, 653–661. [Google Scholar] [CrossRef]
- Jonsson, C.B.; Figueiredo, L.T.; Vapalahti, O. A global perspective on hantavirus ecology, epidemiology, and disease. Clin. Microbiol. Rev. 2010, 23, 412–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makary, P.; Kanerva, M.; Ollgren, J.; Virtanen, M.J.; Vapalahti, O.; Lyytikäinen, O. Disease burden of Puumala virus infections, 1995–2008. Epidemiol. Infect. 2010, 138, 1484–1492. [Google Scholar] [CrossRef] [PubMed]
- Vaheri, A.; Strandin, T.; Hepojoki, J.; Sironen, T.; Henttonen, H.; Mäkelä, S.; Mustonen, J. Uncovering the mysteries of hantavirus infections. Nat. Rev. Microbiol. 2013, 11, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Mustonen, J.; Mäkelä, S.; Outinen, T.; Laine, O.; Jylhävä, J.; Arstila, P.T.; Hurme, M.; Vaheri, A. The pathogenesis of nephropathia epidemica: New knowledge and unanswered questions. Antivir. Res. 2013, 100, 589–604. [Google Scholar] [CrossRef]
- Hjertqvist, M.; Klein, S.l.; Ahlm, C.; Klingström, J. Mortality rate patterns for hemorrhagic fever with renal syndrome caused by Puumala virus. Emerg. Inf. Dis. 2010, 16, 1584–1586. [Google Scholar] [CrossRef]
- Connolly-Andersen, A.M.; Hammargren, E.; Whitaker, H.; Eliasson, M.; Holmgren, L.; Klingström, J.; Ahlm, C. Increased risk of acute myocardial infarction and stroke during hemorrhagic fever with renal syndrome: A self-controlled case series study. Circulation 2014, 129, 1295–1302. [Google Scholar] [CrossRef] [Green Version]
- Connolly-Andersen, A.M.; Whitaker, H.; Klingström, J.; Ahlm, C. Risk of venous thromboembolism following hemorrhagic fever with renal syndrome: A self-controlled case series study. Clin. Infect. Dis. 2018, 66, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Klingström, J.; Smed-Sörensen, A.; Maleki, K.T.; Solà-Riera, C.; Ahlm, C.; Björkström, N.K.; Ljunggren, H.G. Innate and adaptive immune responses against human Puumala virus infection: Immunopathogenesis and suggestions for novel treatment strategies for severe hantavirus-associated syndromes. J. Intern. Med. 2019, 285, 510–523. [Google Scholar] [CrossRef] [Green Version]
- Outinen, T.K.; Laine, O.K.; Mäkelä, S.; Pörsti, I.; Huhtala, H.; Vaheri, A.; Mustonen, J. Thrombocytopenia associates with the severity of inflammation and variables reflecting capillary leakage in Puumala hantavirus infection, an analysis of 546 Finnish patients. Infect. Dis. 2016, 48, 682–687. [Google Scholar] [CrossRef]
- Laine, O.; Mäkelä, S.; Mustonen, J.; Huhtala, H.; Szanto, T.; Vaheri, A.; Lassila, R.; Joutsi-Korhonen, L. Enhanced thrombin formation and fibrinolysis during acute Puumala hantavirus infection. Thromb. Res. 2010, 126, 154–158. [Google Scholar] [CrossRef]
- Laine, O.; Mäkelä, S.; Mustonen, J.; Helminen, M.; Vaheri, A.; Lassila, R.; Joutsi-Korhonen, L. Platelet ligands and ADAMTS13 during Puumala hantavirus infection and associated thrombocytopenia. Blood Coagul. Fibrinolysis 2011, 22, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Laine, O.K.; Koskela, S.M.; Outinen, T.K.; Joutsi-Korhonen, L.; Huhtala, H.; Vaheri, A.; Hurme, M.A.; Jylhävä, J.; Mäkelä, S.M.; Mustonen, J.T. Plasma pentraxin-3 and coagulation and fibrinolysis variables during acute Puumala hantavirus infection and associated thrombocytopenia. Blood Coagul. Fibrinolysis 2014, 25, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Sane, J.; Laine, O.; Mäkelä, S.; Paakkala, A.; Jarva, H.; Mustonen, J.; Vapalahti, O.; Meri, S.; Vaheri, A. Complement activation in Puumala hantavirus infection correlates with disease severity. Ann. Med. 2012, 44, 468–475. [Google Scholar] [CrossRef]
- Cosgriff, T.M. Mechanisms of disease in Hantavirus infection: Pathophysiology of hemorrhagic fever with renal syndrome. Rev. Infect. Dis. 1991, 13, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Gavrilovskaya, I.; Gorbunova, E.; Mackow, E.R. Pathogenic hantaviruses direct the adherence of quiescent platelets to infected endothelial cells. J. Virol. 2010, 84, 4832–4839. [Google Scholar] [CrossRef] [Green Version]
- Mackow, E.R.; Gorbunova, E.E.; Gavrilovskaya, I.N. Endothelial cell dysfunction in viral hemorrhage and edema. Front. Microbiol. 2015, 5, 733. [Google Scholar] [CrossRef]
- Laine, O.; Joutsi-Korhonen, L.; Lassila, R.; Koski, T.; Huhtala, H.; Vaheri, A.; Mäkelä, S.; Mustonen, J. Hantavirus infection-induced thrombocytopenia triggers increased production but associates with impaired aggregation of platelets except for collagen. Thromb. Res. 2015, 136, 1126–1132. [Google Scholar] [CrossRef]
- Connolly-Andersen, A.M.; Sundberg, E.; Ahlm, C.; Hultdin, J.; Baudin, M.; Larsson, J.; Dunne, E.; Kenny, D.; Lindahl, T.L.; Ramström, S.; et al. Increased thrombopoiesis and platelet activation in hantavirus-infected patients. J. Infect. Dis. 2015, 212, 1061–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koskela, S.; Laine, O.; Mäkelä, S.; Pessi, T.; Tuomisto, S.; Huhtala, H.; Karhunen, P.J.; Pörsti, I.; Mustonen, J. Endothelial nitric oxide synthase G894T polymorphism associates with disease severity in Puumala hantavirus infection. PLoS ONE 2015, 10, e0142872. [Google Scholar] [CrossRef] [Green Version]
- Connolly-Andersen, A.M.; Thunberg, T.; Ahlm, C. Endothelial activation and repair during hantavirus infection: Association with disease outcome. Open Forum Inf. Dis. 2014, 1, ofu027. [Google Scholar] [CrossRef] [Green Version]
- Mustonen, J.; Outinen, T.; Laine, O.; Pörsti, I.; Vaheri, A.; Mäkelä, S. Kidney disease in Puumala hantavirus infection. Inf. Dis. 2017, 49, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Hautala, N.; Kauma, H.; Vapalahti, O.; Mähönen, S.M.; Vainio, O.; Vaheri, A.; Hautala, T. Prospective study on ocular findings in acute Puumala hantavirus infection in hospitalised patients. Br. J. Ophthalmol. 2011, 95, 559–562. [Google Scholar] [CrossRef]
- Hautala, T.; Mähönen, S.M.; Sironen, T.; Hautala, N.; Pääkkö, E.; Karttunen, A.; Salmela, P.T.; Ilonen, J.; Vainio, O.; Glaumoff, V.; et al. Central nervous system-related symptoms and findings are common in acute Puumala hantavirus infection. Ann. Med. 2010, 42, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Mäkelä, S.; Ala-Houhala, I.; Mustonen, J.; Koivisto, A.M.; Kouri, T.; Turjanmaa, V.; Vapalahti, O.; Vaheri, A.; Pasternack, A. Renal function and blood pressure five years after puumala virus-induced nephropathy. Kidney Int. 2000, 58, 1711–1718. [Google Scholar] [CrossRef] [Green Version]
- Mäkelä, S.; Jaatinen, P.; Miettinen, M.; Salmi, J.; Ala-Houhala, I.; Huhtala, H.; Hurme, M.; Pörsti, I.; Vaheri, A.; Mustonen, J. Hormonal deficiencies during and after Puumala hantavirus infection. Eur. J. Clin. Microbiol. Infect. Dis. 2010, 29, 705–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krautkrämer, E.; Grouls, S.; Stein, N.; Reiser, J.; Zeier, M. Pathogenic old world hantaviruses infect renal glomerular and tubular cells and induce disassembling of cell-to-cell contacts. J. Virol. 2011, 85, 9811–9823. [Google Scholar] [CrossRef] [Green Version]
- Kanerva, M.; Mustonen, J.; Vaheri, A. Pathogenesis of Puumala and other hantavirus infections. Rev. Med. Virol. 1998, 8, 67–86. [Google Scholar] [CrossRef]
- Outinen, T.K.; Mäkelä, S.M.; Ala-Houhala, I.O.; Huhtala, H.S.; Hurme, M.; Paakkala, A.S.; Pörsti, I.H.; Syrjänen, J.T.; Mustonen, J.T. The severity of Puumala hantavirus induced nephropathia epidemica can be better evaluated using plasma interleukin-6 than C-reactive protein determinations. BMC Infect. Dis. 2010, 10, 132. [Google Scholar] [CrossRef] [Green Version]
- Outinen, T.K.; Mäkelä, S.; Huhtala, H.; Hurme, M.; Meri, S.; Pörsti, I.; Sane, J.; Vaheri, A.; Syrjänen, J.; Mustonen, J. High pentraxin-3 plasma levels associate with thrombocytopenia in acute Puumala hantavirus-induced nephropathia epidemica. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 957–963. [Google Scholar] [CrossRef]
- Outinen, T.K.; Mäkelä, S.M.; Ala-Houhala, I.O.; Huhtala, H.S.; Hurme, M.; Libraty, D.H.; Oja, S.S.; Pörsti, I.H.; Syrjänen, J.T.; Vaheri, A.; et al. High activity of indoleamine 2,3-dioxygenase is associated with renal insufficiency in Puumala hantavirus induced nephropathia epidemica. J. Med. Virol. 2011, 83, 731–737. [Google Scholar] [CrossRef] [Green Version]
- Outinen, T.K.; Tervo, L.; Mäkelä, S.; Huttunen, R.; Mäenpää, N.; Huhtala, H.; Vaheri, A.; Mustonen, J.; Aittoniemi, J. Plasma levels of soluble urokinase-type plasminogen activator receptor levels associate with the clinical severity of acute Puumala hantavirus infection. PLoS ONE 2013, 8, e71335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libraty, D.H.; Mäkelä, S.; Vlk, J.; Hurme, M.; Vaheri, A.; Ennis, F.A.; Mustonen, J. The degree of leukocytosis and urine GATA-3 mRNA levels are risk factors for severe acute kidney injury in Puumala virus nephropathia epidemica. PLoS ONE 2012, 7, e35402. [Google Scholar] [CrossRef] [PubMed]
- Hepojoki, J.; Vaheri, A.; Strandin, T. The fundamental role of endothelial cells in hantavirus pathogenesis. Front. Microbiol. 2014, 5, 727. [Google Scholar] [CrossRef] [Green Version]
- Nuutinen, H.; Vuoristo, M.; Färkkilä, M.; Kahri, A.; Seppälä, K.; Valtonen, V.; Joutsiniemi, T.; Miettinen, T. Hemorrhagic gastropathy in epidemic nephropathy. Gastrointest. Endosc. 1992, 38, 476–480. [Google Scholar] [CrossRef]
- Hautala, T.; Sironen, T.; Vapalahti, O.; Pääkkö, E.; Särkioja, T.; Salmela, P.I.; Vaheri, A.; Plyusnin, A.; Kauma, A. Hypophyseal hemorrhage and panhypopituitarism during Puumala virus infection: Magnetic resonance imaging and detection of viral antigen in the hypophysis. Clin. Infect. Dis. 2002, 35, 96–101. [Google Scholar] [CrossRef] [Green Version]
- Valtonen, M.; Kauppila, M.; Kotilainen, P.; Lähdevirta, J.; Svartbäck, C.M.; Kosunen, O.; Nurminen, J.; Sarkkinen, H.; Brummer-Korvenkontio, M. Four fatal cases of nephropathia epidemica. Scand. J. Infect. Dis. 1995, 27, 515–517. [Google Scholar] [CrossRef]
- Alexeyev, O.A.; Morozov, V.G.; Efremov, A.G.; Settegren, B. A case of haemorrhagic fever with renal syndrome complicated by spleen haemorrhage. Scand. J. Infect. Dis. 1994, 26, 491–492. [Google Scholar] [CrossRef] [PubMed]
- Lähdevirta, J. Nephropathia epidemica in Finland. A clinical, histological and epidemiological study. Ann. Clin. Res. 1971, 3, S1–S154. [Google Scholar]
- Laine, O.; Joutsi-Korhonen, L.; Lassila, R.; Huhtala, H.; Vaheri, A.; Mäkelä, S.; Mustonen, J. Elevated thrombopoietin and platelet indices confirm active thrombopoiesis but fail to predict clinical severity of puumala hantavirus infection. Medicine 2016, 95, e5689. [Google Scholar] [CrossRef] [PubMed]
- Koskela, S.; Laine, O.; Paakkala, A.; Mäkelä, S.; Mustonen, J. Spleen enlargement is a common finding in acute Puumala hantavirus infection and it does not associate with thrombocytopenia. Scand. J. Infect. Dis. 2014, 46, 723–726. [Google Scholar] [CrossRef] [PubMed]
- Gavrilovskaya, I.N.; Brown, E.J.; Ginsberg, M.H.; Mackow, E.R. Cellular entry of Hantaviruses which cause hemorrhagic fever with renal syndrome is mediated by B3 integrins. J. Virol. 1999, 73, 3951–3959. [Google Scholar] [CrossRef] [Green Version]
- Bennet, J.S. Structure and function of the platelet integrin alphaIIbbeta3. J. Clin. Investig. 2005, 115, 3363–3369. [Google Scholar] [CrossRef] [Green Version]
- Sadler, J.E. Biochemistry and genetics of von Willebrand factor. Ann. Rev. Biochem. 1998, 67, 395–424. [Google Scholar] [CrossRef]
- Tietäväinen, J.; Mantula, P.; Outinen, T.; Huhtala, H.; Pörsti, I.H.; Niemelä, O.; Vaheri, A.; Mäkelä, S.; Mustonen, J. Glucosuria predicts the severity of Puumala hantavirus infection. Kidney Int. Rep. 2019, 4, 1296–1303. [Google Scholar] [CrossRef] [Green Version]
- Outinen, T.K.; Kuparinen, T.; Jylhävä, J.; Leppänen, S.; Mustonen, J.; Mäkelä, S.; Pörsti, I.; Syrjänen, J.; Vaheri, A.; Hurme, M. Plasma cell-free DNA levels are elevated in acute Puumala hantavirus infection. PLoS ONE 2012, 7, e31455. [Google Scholar] [CrossRef]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar] [PubMed]
- Kishimoto, T.; Akira, S.; Narazaki, M.; Taga, T. Interleukin-6 family of cytokines and gp130. Blood 1995, 86, 1243–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linderholm, M.; Groeneveld, P.H.; Tärnvik, A. Increased production of nitric oxide in patients with hemorrhagic fever with renal syndrome-relation to arterial hypotension and tumor necrosis factor. Infection 1996, 24, 337–340. [Google Scholar] [CrossRef]
- Takala, A.; Lähdevirta, J.; Jansson, S.E.; Vapalahti, O.; Orpana, A.; Karonen, S.L.; Repo, H. Systemic inflammation in hemorrhagic fever with renal syndrome correlates with hypotension and thrombocytopenia but not with renal injury. J. Infect. Dis. 2000, 181, 1964–1970. [Google Scholar] [CrossRef]
- Bottazzi, B.; Garlanda, C.; Cotena, A.; Moalli, F.; Jaillon, S.; Deban, L.; Mantovani, A. The long pentraxin PTX3 as a prototypic humoral pattern recognition receptor: Interplay with cellular innate immunity. Immunol. Rev. 2009, 227, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Deban, L.; Jarva, H.; Lehtinen, M.J.; Bottazzi, B.; Bastone, A.; Doni, A.; Jokiranta, T.S.; Mantovani, A.; Meri, S. Binding of the long pentraxin PTX3 to factor H: Interacting domains and function in the regulation of complement activation. J. Immunol. 2008, 181, 8433–8440. [Google Scholar] [CrossRef] [Green Version]
- Rasche, F.M.; Uhel, B.; Krüger, D.H.; Karges, W.; Czock, D.; Hampl, W.; Keller, F.; Meisel, H.; von Müller, L. Thrombocytopenia and acute renal failure in Puumala hantavirus infections. Emerg. Infect. Dis. 2004, 10, 1420–1425. [Google Scholar] [CrossRef] [Green Version]
- Latus, J.; Kitterer, D.; Segerer, S.; Artunc, F.; Alscher, M.D.; Braun, N. Severe thrombocytopenia in hantavirus-induced nephropathia epidemica. Infection 2015, 43, 83–87. [Google Scholar] [CrossRef]
- Latus, J.; Schwab, M.; Tacconelli, E.; Pieper, F.M.; Wegener, D.; Rettenmaier, B.; Schwab, A.; Hoffmann, L.; Dippon, J.; Müller, S.; et al. Acute kidney injury and tools for risk-stratification in 456 patients with hantavirus-induced nephropathia epidemica. Nephrol. Dial. Transplant. 2015, 30, 245–251. [Google Scholar] [CrossRef] [Green Version]
- Mackow, E.R.; Gavrilovskaya, I.N. Hantavirus regulation of endothelial cell functions. Thromb. Haemost. 2009, 102, 1030–1041. [Google Scholar] [CrossRef] [PubMed]
- Zaki, S.R.; Greer, P.W.; Coffield, L.M.; Goldsmith, C.S.; Nolte, K.B.; Foucar, K.; Feddersen, R.M.; Zumwalt, R.E.; Miller, G.L.; Khan, A.S.; et al. Hantavirus pulmonary syndrome. Pathogenesis of an emerging infectious disease. Am. J. Pathol. 1995, 146, 552–579. [Google Scholar]
- Nusshag, C.; Osberghaus, A.; Baumann, A.; Schnitzler, P.; Zeier, M.; Krautkrämer, E. Deregulation of levels of angiopoietin-1 and angiopoietin-2 associated with severe courses of hantavirus infection. J. Clin. Virol. 2017, 94, 33–36. [Google Scholar] [CrossRef]
- Sadeghi, M.; Eckerle, I.; Daniel, V.; Burkhardt, U.; Opelz, G.; Schnitzler, P. Cytokine expression during early and late phase of acute Puumala hantavirus infection. BMC Immunol. 2011, 12, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Q.; Zhang, L.; Liu, Z.; Kang, W.; Lou, S.; Qiu, J.; Li, Z.; Zhang, G.; Wang, Y.; Li, M.; et al. Elevated sICAM-1 levels in patients with hemorrhagic fever with renal syndrome caused by Hantaan virus. Eur. J. Clin. Microbiol. Infect. Dis. 2010, 29, 1507–1511. [Google Scholar] [CrossRef]
- Kyriakidis, I.; Papa, A. Serum TNF-α, sTNFR1, IL-6, IL-8 and IL-10 levels in hemorrhagic fever with renal syndrome. Virus Res. 2013, 175, 91–94. [Google Scholar] [CrossRef]
- Klingström, J.; Plyusnin, A.; Vaheri, A.; Lundkvist, A. Wild-type Puumala hantavirus infection induces cytokines, C-reactive protein, creatinine, and nitric oxide in cynomolgus macaques. J. Virol. 2002, 76, 444–449. [Google Scholar] [CrossRef] [Green Version]
- Sironen, T.; Klingström, J.; Vaheri, A.; Andersson, L.C.; Lundkvist, A.; Plyusnin, A. Pathology of Puumala hantavirus infection in macaques. PLoS ONE 2008, 3, e3035. [Google Scholar] [CrossRef]
- Odobasic, D.; Kitching, A.R.; Holdsworth, S.R. Neutrophil-mediated regulation of innate and adaptive immunity: The role of myeloperoxidase. J. Immunol. Res. 2016, 2016, 2349817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yipp, B.G.; Kubes, P. NETosis: How vital is it? Blood 2013, 122, 2784–2794. [Google Scholar] [CrossRef]
- Raftery, M.J.; Lalwani, P.; Krautkrämer, E.; Peters, T.; Scharffetter-Kochanek, K.; Krüger, R.; Hofmann, J.; Seeger, K.; Krüger, D.H.; Schönrich, G. Beta2 integrin mediates Hantavirus-induced release of neutrophil extracellular traps. J. Exp. Med. 2014, 211, 1485–1497. [Google Scholar] [CrossRef]
- Vaheri, A.; Strandin, T.; Jääskeläinen, A.J.; Vapalahti, O.; Jarva, H.; Lokki, M.L.; Antonen, J.; Leppänen, I.; Mäkelä, S.; Meri, S.; et al. Pathophysiology of a severe case of Puumala Hantavirus infection successfully treated with bradykinin receptor antagonist icatibant. Antiviral. Res. 2014, 111, 23–25. [Google Scholar] [CrossRef]
- Strandin, T.; Mäkelä, S.; Mustonen, J.; Vaheri, A. Neutrophil activation in acute hemorrhagic fever is mediated by hantavirus-infected microvascular endothelial cells. Front. Immunol. 2018, 9, 2098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delabranche, X.; Helms, J.; Meziani, F. Immunohaemostasis: A new view on haemostasis during sepsis. Ann. Intensiv. Care 2017, 7, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, T.A.; Bhandari, A.A.; Wagner, D.D. Histones induce rapid and profound thrombocytopenia in mice. Blood 2011, 118, 3708–3714. [Google Scholar] [CrossRef] [Green Version]
- Massberg, S.; Grahl, L.; von Bruehl, M.L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Resiprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef]
- Ammollo, C.T.; Semeraro, F.; Xu, J.; Esmon, N.L.; Esmon, C.T. Extracellular histones increase plasma thrombin generation by impairing thrombomodulin-dependent protein C activation. J. Thromb. Haemost. 2011, 9, 1795–1803. [Google Scholar] [CrossRef] [PubMed]
- De Bont, C.M.; Boelens, W.C.; Pruijn, G.J.M. NETosis, complement, and coagulation: A triangular relationship. Cell. Mol. Immunol. 2019, 16, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Discovery factor of vascular permeability factor (VPF). Exp. Cell Res. 2006, 312, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Gavrilovskaya, I.N.; Gorbunova, E.E.; Mackow, N.A.; Mackow, E.R. Hantaviruses direct endothelial cell permeability by sensitizing cells to the vascular permeability factor VEGF, while angiopoietin 1 and sphingosine 1-phosphate inhibit Hantavirus-directed permeability. J. Virol. 2008, 82, 5797–5806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorbunova, E.; Gavrilovskaya, I.N.; Mackow, E.R. Pathogenic hantaviruses Andes virus and Hantaan virus induce adherens junction disassembly by directing vascular endothelial cadherin internalization in human endothelial cells. J. Virol. 2010, 84, 7405–7411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Wang, W.; Wang, J.P.; Pan, L.; Zhang, Y.; Yu, H.T.; Jiang, W.; Wang, P.Z.; Bai, X.F. Elevated vascular endothelial growth factor levels induce hyperpermeability of endothelial cells in hantavirus infection. J. Int. Med. Res. 2012, 40, 1812–1821. [Google Scholar] [CrossRef]
- Vestweber, D. VE-cadherin: The major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. Arterioscler. Thromb. Vasc. Biol. 2010, 28, 223–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorbunova, E.E.; Gavrilovskaya, I.N.; Pepini, T.; Mackow, E.R. VEGFR2 and Src kinase inhibitors suppress Andes virus-induced endothelial cell permeability. J. Virol. 2011, 85, 2296–2303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavrilovskaya, I.; Gorbunova, E.; Koster, F.; Mackow, E. Elevated VEGF levels in pulmonary edema fluid and PBMCs from patients with acute hantavirus pulmonary syndrome. Adv. Virol. 2012, 2012, 674360. [Google Scholar] [CrossRef]
- Maurer, M.; Bader, M.; Bas, M.; Bossi, F.; Cicardi, F.; Cugno, M.; Howarth, P.; Kaplan, A.; Kojda, G.; Leeb-Lundberg, F. New topics in bradykinin research. Allergy 2011, 66, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.L.; Wahl-Jensen, V.; Copeland, A.M.; Jahrling, P.B.; Schmaljohn, C.S. Endothelial cell permeability during hantavirus infection involves factor XII-dependent increased activation of the kallikrein-kinin system. PLoS Pathog. 2013, 9, e1003470. [Google Scholar] [CrossRef]
- Antonen, J.; Leppänen, I.; Tenhunen, J.; Arvola, P.; Mäkelä, S.; Vaheri, A.; Mustonen, J. A severe case of Puumala hantavirus infection successfully treated with bradykinin receptor antagonist icatibant. Scand. J. Infect. 2013, 45, 494–496. [Google Scholar] [CrossRef]
- Laine, O.; Leppänen, I.; Koskela, S.; Antonen, J.; Mäkelä, S.; Sinisalo, M.; Vaheri, A.; Mustonen, J. Severe Puumala virus infection in a patient with lymphoproliferative disease treated with icatibant. Infect. Dis. 2015, 47, 107–111. [Google Scholar] [CrossRef]
- Groeneveld, P.H.; Colson, P.; Kwappenberg, K.M.; Clement, J. Increased production of nitric oxide in patients infected with the European variant of hantavirus. Scand. J. Infect. Dis. 1995, 27, 453–456. [Google Scholar] [CrossRef]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Klingström, J.; Akerström, S.; Hardestam, J.; Stoltz, M.; Simon, M.; Falk, K.I.; Mirazimi, A.; Rottenberg, M.; Lundkvist, A. Nitric oxide and peroxynitrite have different antiviral effects against hantavirus replication and fee mature virions. Eur. J. Immunol. 2006, 36, 2649–2657. [Google Scholar] [CrossRef] [PubMed]
- Veldman, B.A.; Spiering, W.; Doevendans, P.A.; Vervoort, G.; Kroon, A.A.; de Leeuw, P.W.; Smits, P. The Glu298Asp polymorphism of the NOS 3 gene as a determinant of the baseline production of nitric oxide. J. Hypertens. 2002, 20, 2023–2027. [Google Scholar] [CrossRef]
- Erbs, S.; Möbius-Winkler, S.; Linke, A.; Adams, V.; Doll, N.; Gielen, S.; Gummert, J.F.; Mohr, F.W.; Schuler, G.; Hambrecht, R. Both T-786C and G894T polymorphism of endothelial nitric oxide synthase affect in-vitro endothelium-dependent relaxation of internal mammary artery rings from patients with coronary artery disease. Eur. J. Cardiovasc. Prev. Rehabil. 2006, 13, 826–831. [Google Scholar] [CrossRef]
- Modlinger, P.S.; Wilcox, C.S.; Aslam, S. Nitric oxide, oxidative stress, and progression of chronic renal failure. Semin. Nephrol. 2004, 24, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Van Der Meijden, P.E.; Van Schilfgaarde, M.; Van Oerle, R.; Renné, T.; ten Cate, H.; Spronk, H.M. Platelet- and erythrocyte-derived microparticles trigger thrombin generation via factor XIIa. J. Thromb. Haemost. 2012, 10, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Goeijenbier, M.; Meijers, J.C.; Anfasa, F.; Roose, J.M.; van de Weg, C.A.; Bakhtiari, K.; Henttonen, H.; Vaheri, A.; Osterhaus, A.D.; van Gorp, E.C.; et al. Effect of Puumala hantavirus on human umbilical vein endothelial cell hemostatic function: Platelet interactions, tissue factor expression and fibrinolysis regulator release. Front. Microbiol. 2015, 6, 220. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y. Contact pathway of coagulation and inflammation. Thromb. J. 2015, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Lähdevirta, J. The minor problem of hemostatic impairment in nephropathia epidemica, the mild Scandinavian form of hemorrhagic fever with renal syndrome. Rev. Infect. Dis. 1989, 11, S860–S863. [Google Scholar] [CrossRef] [PubMed]
- Sundberg, E.; Hultdin, J.; Nilsson, S.; Ahlm, C. Evidence of disseminated intravascular coagulation in a hemorrhagic fever with renal syndrome—Scoring models and severe illness. PLoS ONE 2011, 6, e21134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owens, A.P., 3rd; Mackman, N. Microparticles in hemostasis and thrombosis. Circ. Res. 2011, 108, 1284–1297. [Google Scholar] [CrossRef]
- Schmedes, C.M.; Grover, S.P.; Hisada, Y.M.; Goeijenbier, M.; Hultdin, J.; Nilsson, S.; Thunberg, T.; Ahlm, C.; Mackman, N.; Fors Connolly, A.M. Circulating extracellular vesicle tissue factor activity during orthohantavirus infection is associated with intravascular coagulation. J. Infect. Dis. 2020, 222, 1392–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koskela, S.M.; Joutsi-Korhonen, L.; Mäkelä, S.; Huhtala, H.; Vaheri, A.; Pörsti, I.; Mustonen, J.; Laine, O. Diminished coagulation capacity assessed by calibrated automated thrombography during acute Puumala hantavirus infection. Blood Coagul. Fibrinolysis 2018, 29, 55–60. [Google Scholar] [CrossRef] [Green Version]
- Strandin, T.; Hepojoki, J.; Laine, O.; Mäkelä, S.; Klingström, J.; Lundkvist, Å.; Julkunen, I.; Mustonen, J.; Vaheri, A. Interferons induce STAT1-dependent expression of tissue plasminogen activator, a pathogenicity factor in Puumala hantavirus disease. J. Infect. Dis. 2016, 213, 1632–1641. [Google Scholar] [CrossRef] [Green Version]
- Bellomo, C.; Korva, M.; Papa, A.; Mäkelä, S.; Mustonen, J.; Avšič-Županc, T.; Vaheri, A.; Martinez, V.P.; Strandin, T. Differential regulation of PAI-1 in hantavirus cardiopulmonary syndrome and hemorrhagic fever with renal syndrome. Open Forum Infect. Dis. 2018, 5, ofy021. [Google Scholar] [CrossRef] [Green Version]
- Laine, O.; Joutsi-Korhonen, L.; Mäkelä, S.; Mikkelsson, J.; Pessi, T.; Tuomisto, S.; Huhtala, H.; Libraty, D.; Vaheri, A.; Karhunen, P.; et al. Polymorphisms of PAI-1 and platelet GP Ia may associate with impairment of renal function and thrombocytopenia in Puumala hantavirus infection. Thromb. Res. 2012, 129, 611–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M. Coagulopathy in patients with hemorrhagic fever with renal syndrome. J. Korean Med. Sci. 1987, 2, 201–211. [Google Scholar] [CrossRef] [Green Version]
- Dunkelberger, J.R.; Song, W.C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef] [Green Version]
- Amara, U.; Flierl, M.A.; Rittirsch, D.; Klos, A.; Chen, H.; Acker, B.; Brückner, U.B.; Nilsson, B.; Gebhard, F.; Lambris, J.D.; et al. Molecular intercommunications between the complement and coagulation systems. J. Immunol. 2010, 18, 5628–5636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paakkala, A.; Mustonen, J.; Viander, M.; Huhtala, H.; Pasternack, A. Complement activation in nephropathia epidemica caused by Puumala hantavirus. Clin. Nephrol. 2010, 536, 424–431. [Google Scholar]
- Kerr, H.; Richards, A. Complement-mediated injury and protection of endothelium: Lessons from atypical haemolytic uraemic syndrome. Immunobiology 2012, 217, 195–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukada, H.; Ying, X.; Fu, C.; Ishikawa, S.; McKeown-Longo, P.; Albelda, S.; Bhattacharya, S.; Bray, B.A.; Bhattacharya, J. Ligation of endothelial alpha v beta 3 integrin increases capillary hydraulic conductivity of rat lung. Circ. Res. 1995, 77, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Bossi, F.; Fischetti, F.; Pellis, V.; Bulla, R.; Ferrero, E.; Mollnes, T.E.; Regoli, D.; Tedesco, F. Platelet-activating factor and kinin-dependent vascular leakage as a novel functional activity of the soluble terminal complement complex. J. Immunol. 2004, 173, 6921–6927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bottazzi, B.; Doni, A.; Garlanda, C.; Mantovani, A. An integrated view of humoral innate immunity: Pentraxins as a paradigm. Annu. Rev. Immunol. 2010, 28, 157–183. [Google Scholar] [CrossRef]
- Deban, L.; Russo, R.C.; Sironi, M.; Moalli, F.; Scanziani, M.; Zambelli, V.; Cuccovillo, I.; Bastone, A.; Gobbi, M.; Valentino, S. Regulation of leukocyte recruitment by the long pentraxin PTX3. Nat. Immunol. 2010, 11, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.H.; Sniecinski, R.M.; Welsby, I.J.; Levi, M. Antithrombin: Anti-inflammatory properties and clinical applications. Thromb. Haemost. 2016, 115, 712–728. [Google Scholar] [CrossRef]
- Levi, M.; Schultz, M.; Van Der Poll, T. Sepsis and thrombosis. Semin. Thromb. Hemost. 2013, 39, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; Van Der Poll, T. The role of natural anticoagulants in the pathogenesis and management of systemic activation of coagulation and inflammation in critically ill patients. Semin. Thromb. Hemost. 2008, 34, 459–468. [Google Scholar] [CrossRef] [PubMed]


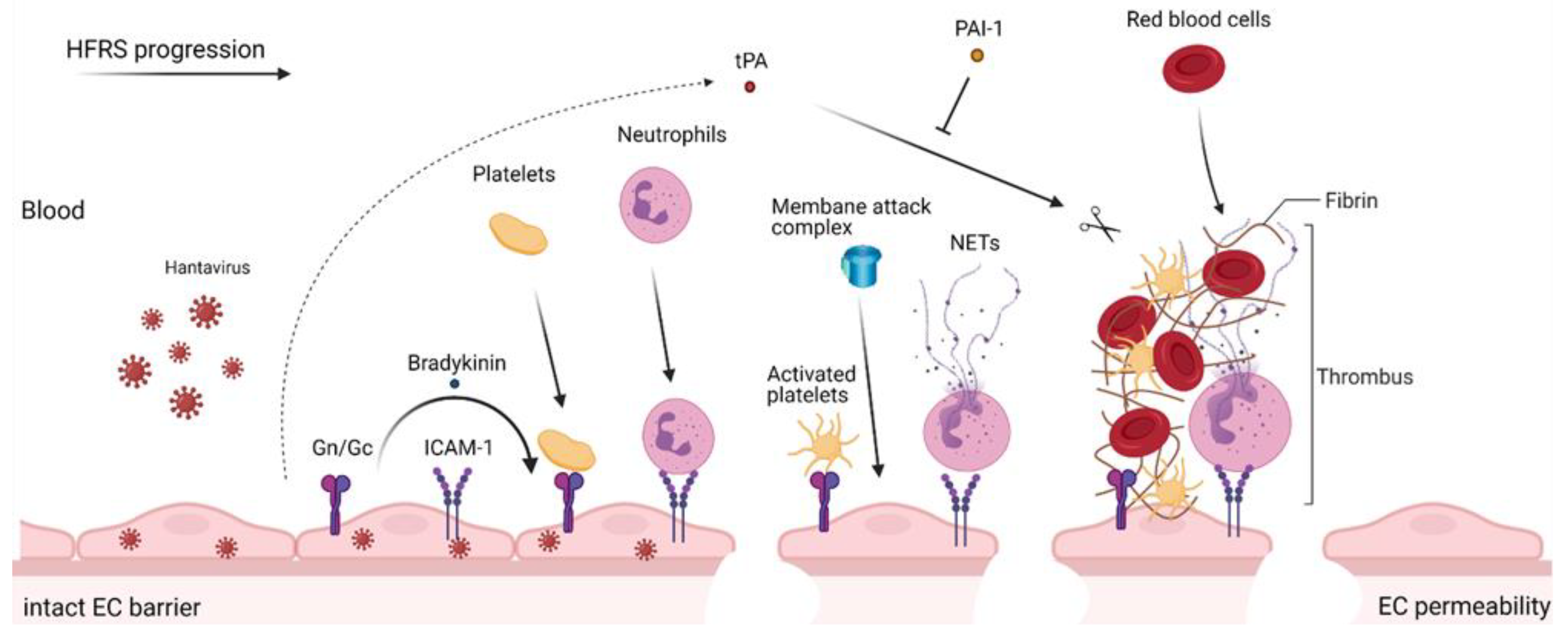{kind=link}
| Coagulation Markers | Acute-Phase | References |
| APTT | Prolonged | [12] |
| Prothrombin time | Shortened | [12] |
| Thrombin time | Shortened | [12] |
| Fibrinogen | Increased | [12,13] |
| F1+2 | Increased | [12] |
| D-dimer | Increased | [12] |
| Antithrombin activity | Decreased | [12] |
| Protein C activity | Decreased | [12] |
| Protein S free antigen | Decreased | [12] |
| Plasma PTX-3 | Increased | [31] |
| Plasma cf-DNA | Increased | [47] |
| tPA activity | Increased | [69,93,100] |
| PAI-1 | Not altered | [100,101] |
| Platelet Ligands and Markers of Endothelial Cell Activation | Acute Phase | References |
| vWF:Ag | Increased | [13] |
| vWF:RCo | Increased | [13] |
| Fibronectin | Decreased | [13] |
| ADAMTS13 activity | Decreased | [13] |
| Complement Activation | Acute Phase | References |
| SC5b-9 | Increased | [15,106] |
| C3 | Decreased | [15,106] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koskela, S.; Mäkelä, S.; Strandin, T.; Vaheri, A.; Outinen, T.; Joutsi-Korhonen, L.; Pörsti, I.; Mustonen, J.; Laine, O. Coagulopathy in Acute Puumala Hantavirus Infection. Viruses 2021, 13, 1553. https://0-doi-org.brum.beds.ac.uk/10.3390/v13081553
Koskela S, Mäkelä S, Strandin T, Vaheri A, Outinen T, Joutsi-Korhonen L, Pörsti I, Mustonen J, Laine O. Coagulopathy in Acute Puumala Hantavirus Infection. Viruses. 2021; 13(8):1553. https://0-doi-org.brum.beds.ac.uk/10.3390/v13081553
Chicago/Turabian StyleKoskela, Sirpa, Satu Mäkelä, Tomas Strandin, Antti Vaheri, Tuula Outinen, Lotta Joutsi-Korhonen, Ilkka Pörsti, Jukka Mustonen, and Outi Laine. 2021. "Coagulopathy in Acute Puumala Hantavirus Infection" Viruses 13, no. 8: 1553. https://0-doi-org.brum.beds.ac.uk/10.3390/v13081553