
No Exchange of Picornaviruses in Vietnam between Humans and Animals in a High-Risk Cohort with Close Contact despite High Prevalence and Diversity

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Metagenomic Analysis
2.3. Genomic and Phylogenetic Analysis
3. Results
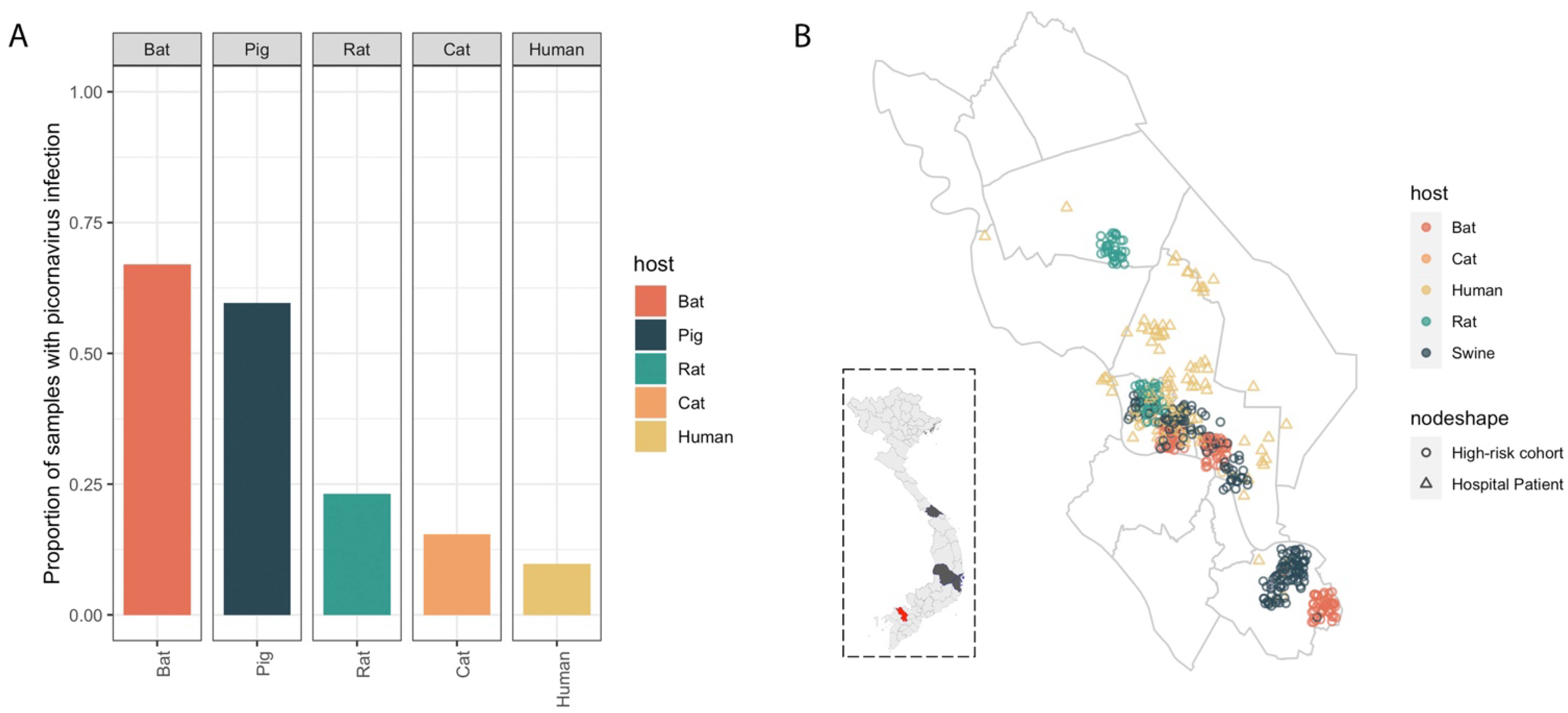
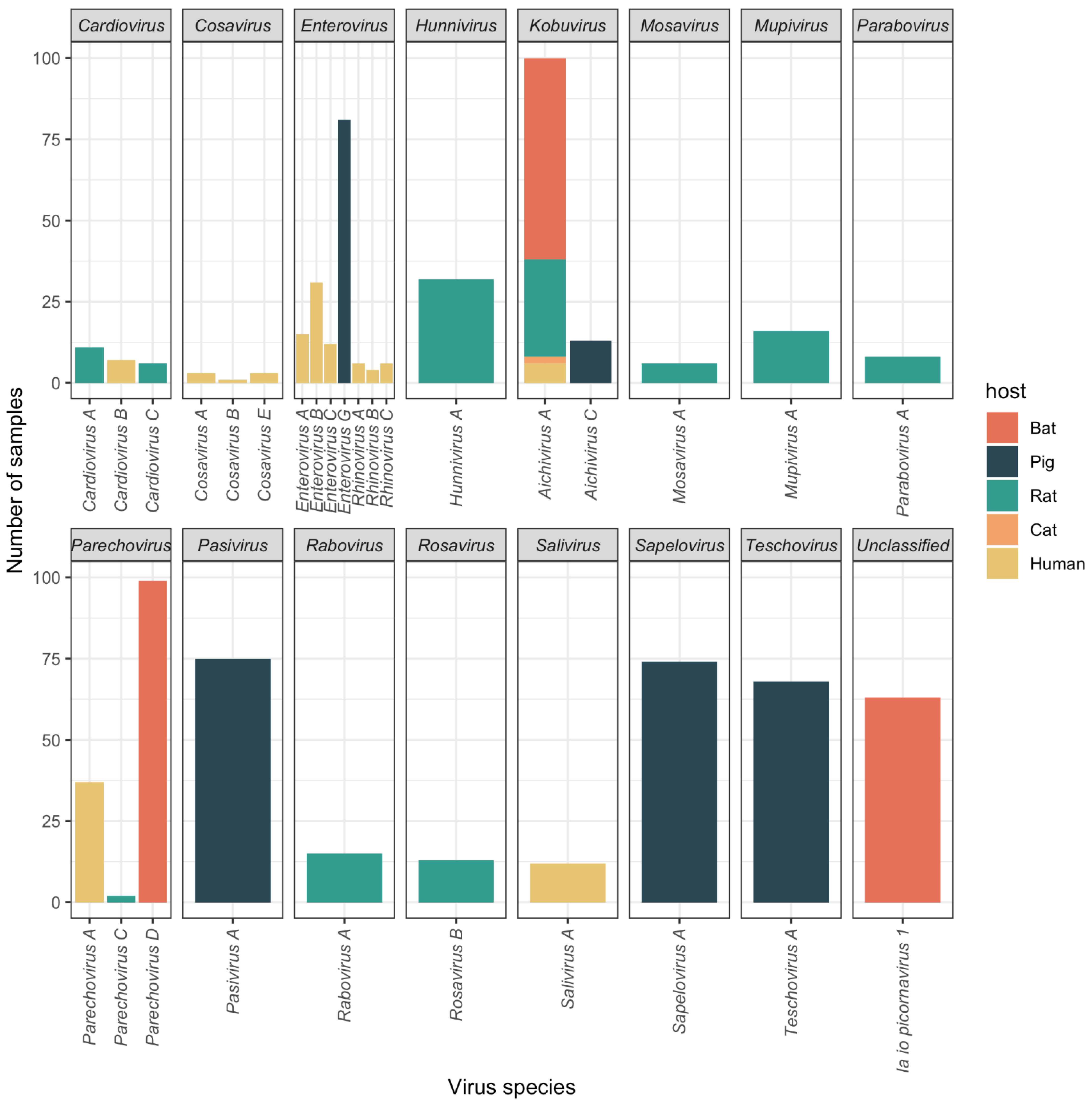
3.1. High Prevalence of Diverse Picornaviruses
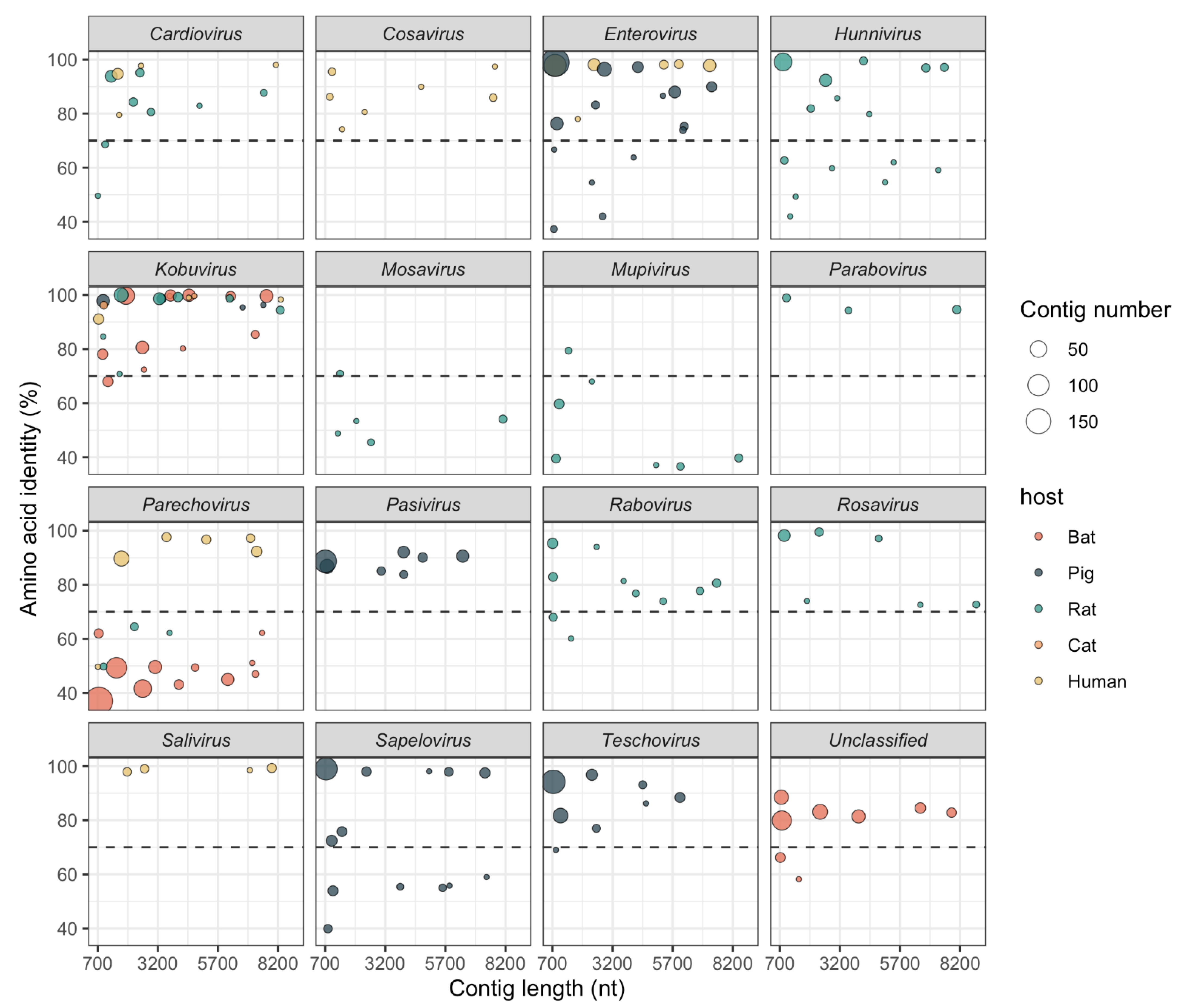
3.2. Putative Novel Picornaviruses
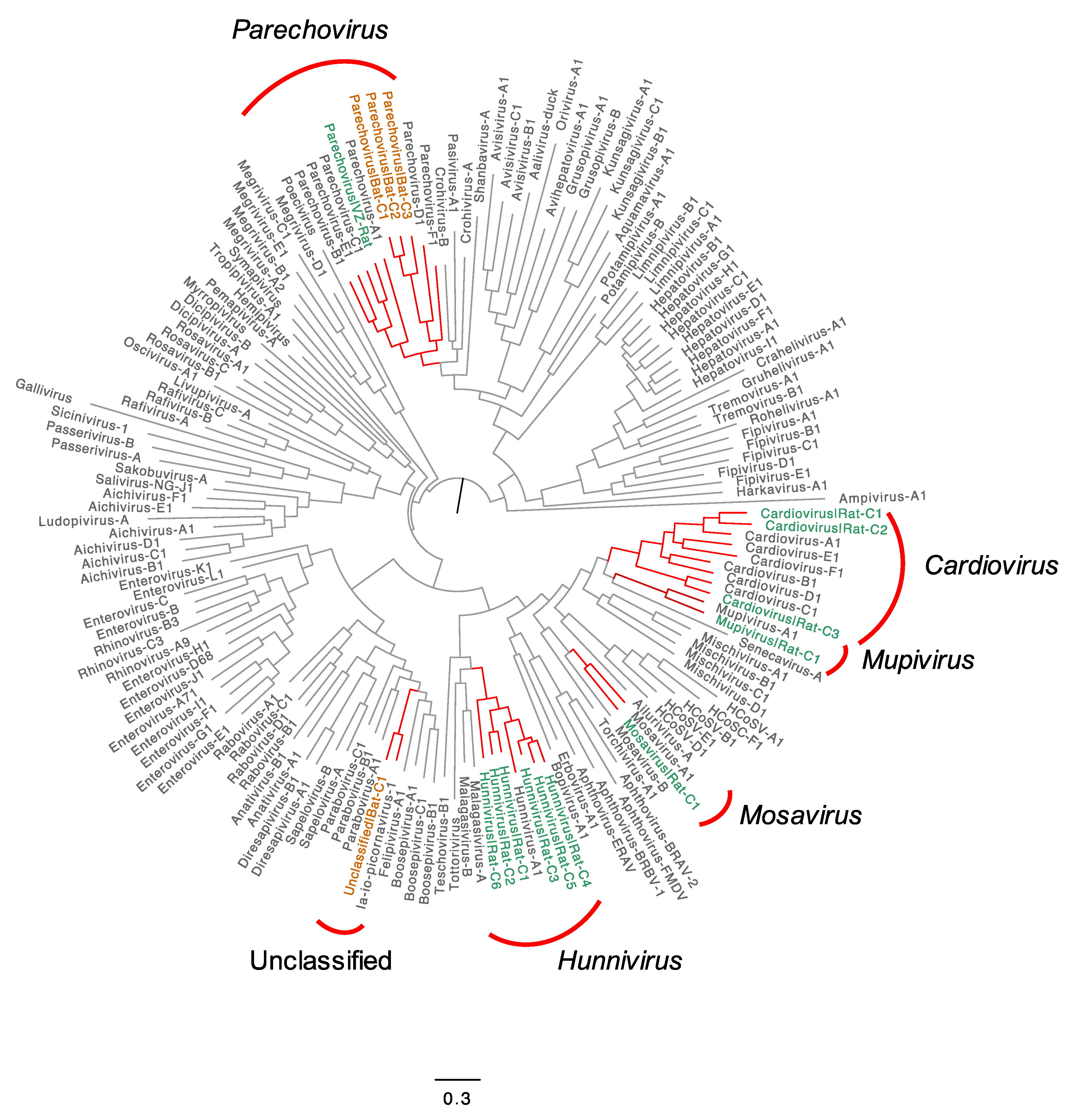
3.3. Evolutionary Characteristics of Putatively Novel Picornaviruses Found in Rats and Bats
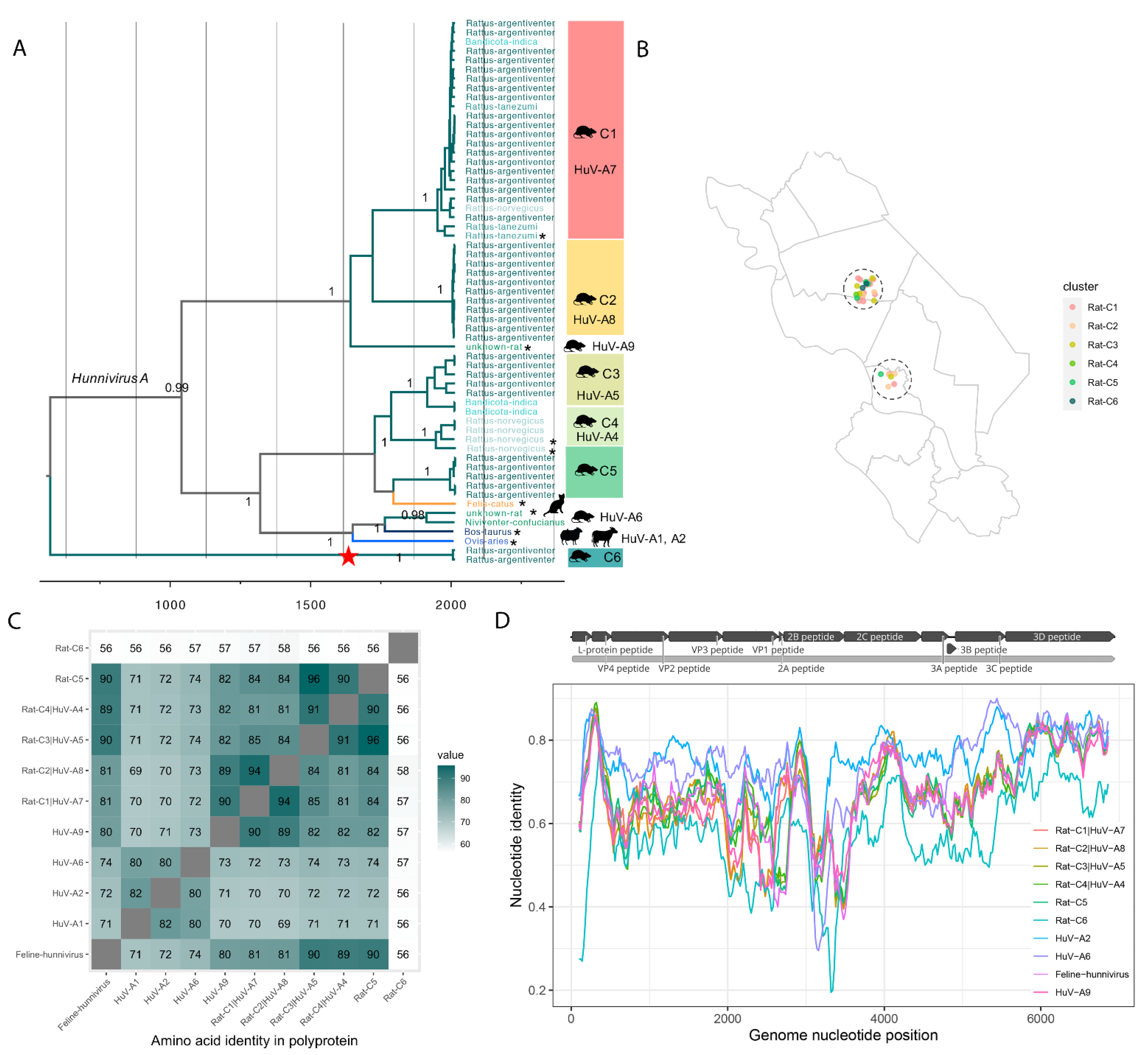
3.3.1. Diversity of Hunniviruses in Rats
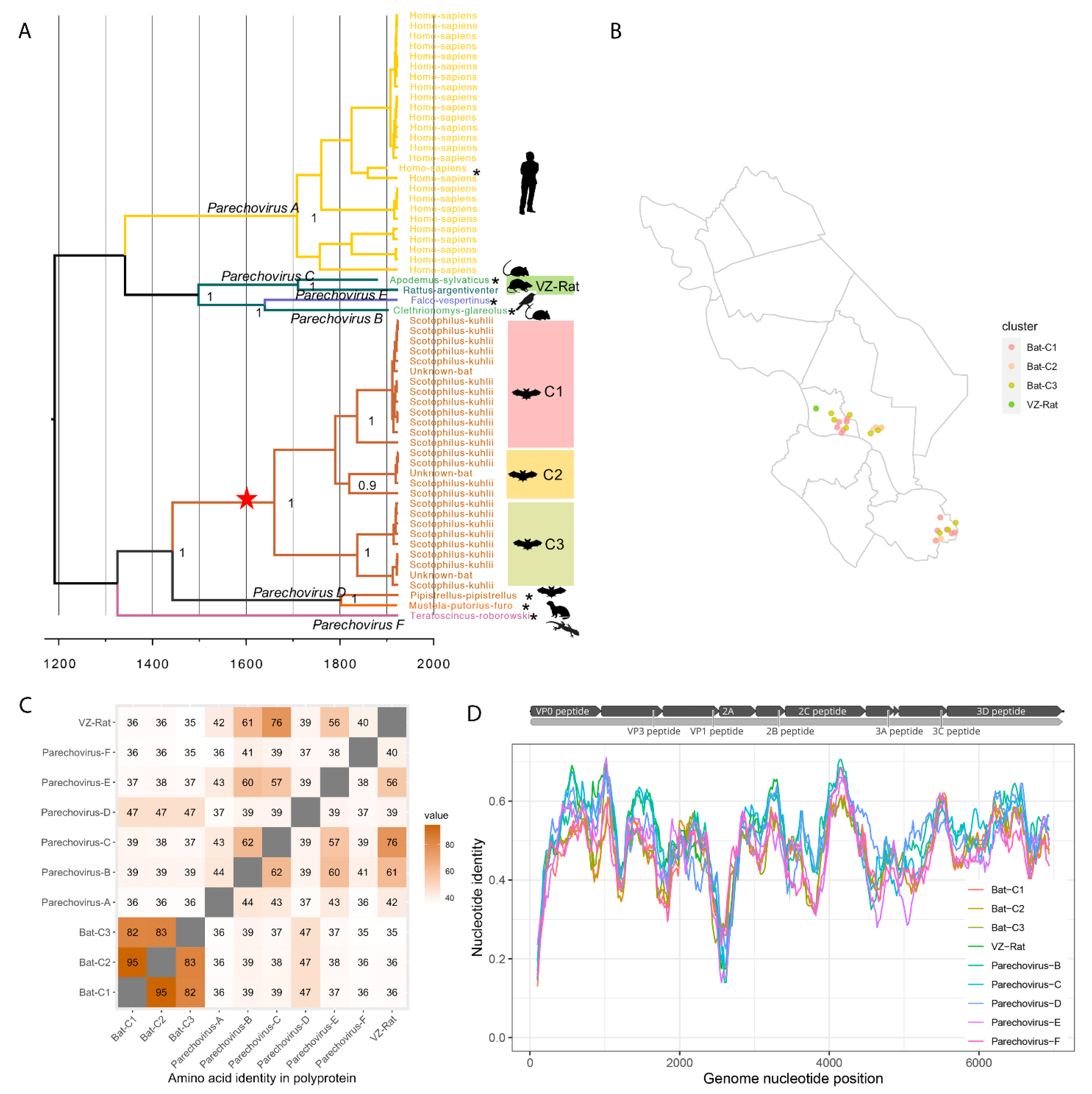
3.3.2. Diversity of Parechoviruses in Bats and Rats
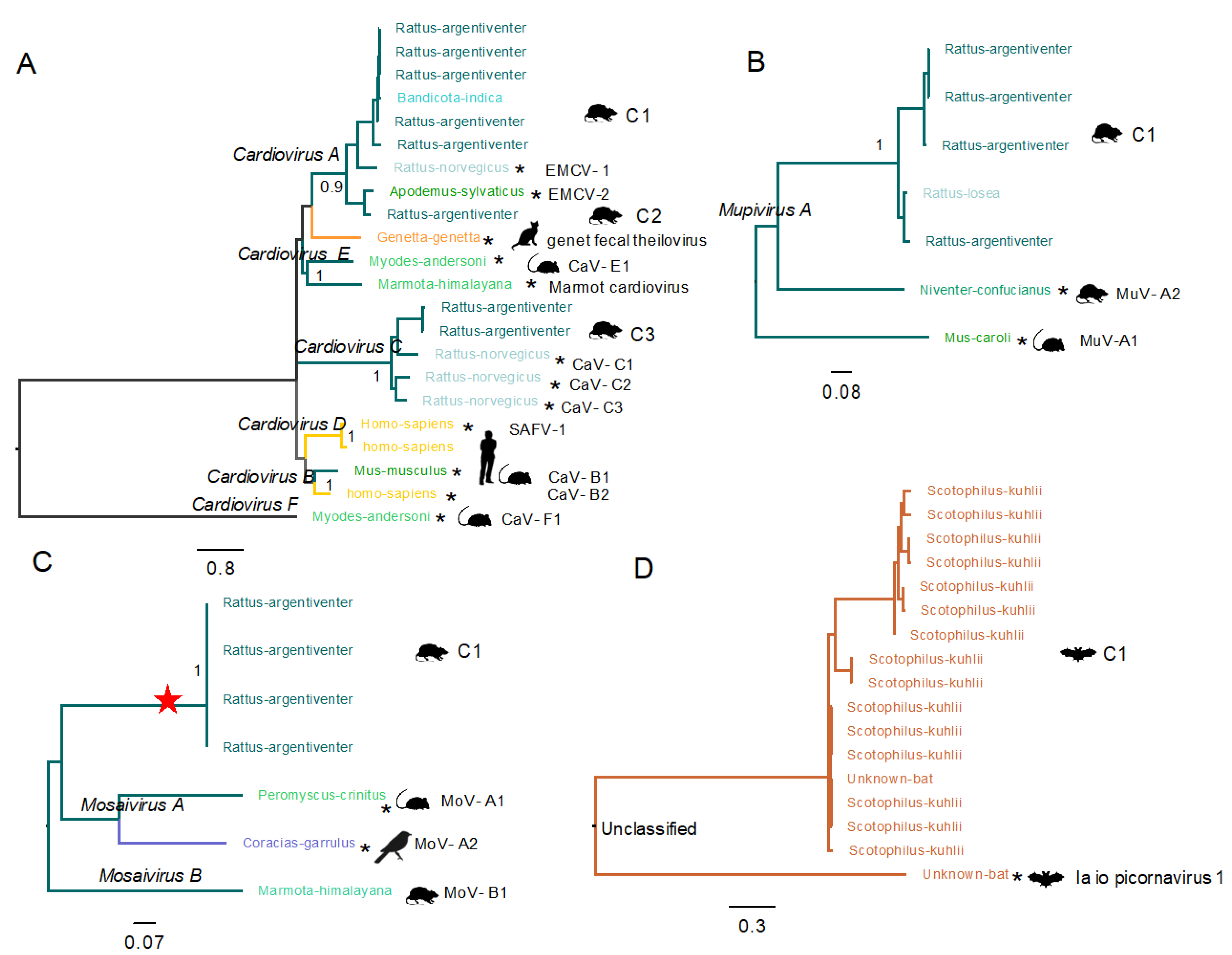
3.3.3. Diversity of Cardioviruses in Bats and Rats
3.3.4. A New Type of Mupivirus in Rats
3.3.5. Novel Species of Mosavirus in Rats
3.3.6. Unclassified Picornavirus in Bats
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boni, M.F.; Lemey, P.; Jiang, X.; Lam, T.T.; Perry, B.W.; Castoe, T.A.; Rambaut, A.; Robertson, D.L. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat. Microbiol. 2020, 5, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus-Infected Pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Magouras, I.; Brookes, V.J.; Jori, F.; Martin, A.; Pfeiffer, D.U.; Durr, S. Emerging Zoonotic Diseases: Should We Rethink the Animal-Human Interface? Front. Vet. Sci. 2020, 7, 582743. [Google Scholar] [CrossRef] [PubMed]
- Rabaa, M.A.; Tue, N.T.; Phuc, T.M.; Carrique-Mas, J.; Saylors, K.; Cotten, M.; Bryant, J.E.; Nghia, H.D.; Cuong, N.V.; Pham, H.A.; et al. The Vietnam Initiative on Zoonotic Infections (VIZIONS): A Strategic Approach to Studying Emerging Zoonotic Infectious Diseases. Ecohealth 2015, 12, 726–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zell, R.; Delwart, E.; Gorbalenya, A.E.; Hovi, T.; King, A.M.Q.; Knowles, N.J.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Reuter, G.; et al. ICTV Virus Taxonomy Profile: Picornaviridae. J. Gen. Virol. 2017, 98, 2421–2422. [Google Scholar] [CrossRef]
- Zell, R. Picornaviridae-the ever-growing virus family. Arch. Virol. 2018, 163, 299–317. [Google Scholar] [CrossRef]
- Woolhouse, M.E.J.; Brierley, L. Epidemiological characteristics of human-infective RNA viruses. Sci. Data 2018, 5, 180017. [Google Scholar] [CrossRef]
- Lu, L.; Van Dung, N.; Bryant, J.E.; Carrique-Mas, J.; Van Cuong, N.; Anh, P.H.; Rabaa, M.A.; Baker, S.; Simmonds, P.; Woolhouse, M.E. Evolution and phylogeographic dissemination of endemic porcine picornaviruses in Vietnam. Virus Evol. 2016, 2, vew001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Van Dung, N.; Ivens, A.; Bogaardt, C.; O’Toole, A.; Bryant, J.E.; Carrique-Mas, J.; Van Cuong, N.; Anh, P.H.; Rabaa, M.A.; et al. Genetic diversity and cross-species transmission of kobuviruses in Vietnam. Virus Evol. 2018, 4, vey002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotten, M.; Oude Munnink, B.; Canuti, M.; Deijs, M.; Watson, S.J.; Kellam, P.; van der Hoek, L. Full genome virus detection in fecal samples using sensitive nucleic acid preparation, deep sequencing, and a novel iterative sequence classification algorithm. PLoS ONE 2014, 9, e93269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woolhouse, M.; Ashworth, J.; Bogaardt, C.; Tue, N.T.; Baker, S.; Thwaites, G.; Phuc, T.M. Sample descriptors linked to metagenomic sequencing data from human and animal enteric samples from Vietnam. Sci. Data 2019, 6, 202. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 3. [Google Scholar] [CrossRef]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchfink, B.; Reuter, K.; Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT: Iterative refinement and additional methods. Methods Mol. Biol. 2014, 1079, 131–146. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Hill, V.; Baele, G. Bayesian estimation of past population dynamics in BEAST 1.10 using the Skygrid coalescent model. Mol. Biol. Evol. 2019, 36, 2620–2628. [Google Scholar] [CrossRef] [Green Version]
- Oberste, M.S.; Maher, K.; Pallansch, M.A. Molecular phylogeny and proposed classification of the simian picornaviruses. J. Virol. 2002, 76, 1244–1251. [Google Scholar] [CrossRef] [Green Version]
- Muir, P.; Kammerer, U.; Korn, K.; Mulders, M.N.; Poyry, T.; Weissbrich, B.; Kandolf, R.; Cleator, G.M.; van Loon, A.M. Molecular typing of enteroviruses: Current status and future requirements. The European Union Concerted Action on Virus Meningitis and Encephalitis. Clin. Microbiol. Rev. 1998, 11, 202–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIntyre, C.L.; Knowles, N.J.; Simmonds, P. Proposals for the classification of human rhinovirus species A, B and C into genotypically assigned types. J. Gen. Virol. 2013, 94, 1791–1806. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; McIntyre, C.; Savolainen-Kopra, C.; Tapparel, C.; Mackay, I.M.; Hovi, T. Proposals for the classification of human rhinovirus species C into genotypically assigned types. J. Gen. Virol. 2010, 91, 2409–2419. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Sheng, J.; Zhao, Z.; Yan, C.; Tu, C.; He, B. Genomic Characterization of the First Parechovirus in Bats. Virol. Sin. 2019, 34, 471–473. [Google Scholar] [CrossRef] [PubMed]
- Elmekki, A.A.; Nieuwenhuysen, P.; Vandergroen, G.; Pattyn, S.R. Characterization of Some Ungrouped Viruses. Trans. R. Soc. Trop. Med. Hyg. 1981, 75, 799–806. [Google Scholar]
- Firth, C.; Bhat, M.; Firth, M.A.; Williams, S.H.; Frye, M.J.; Simmonds, P.; Conte, J.M.; Ng, J.; Garcia, J.; Bhuva, N.P.; et al. Detection of zoonotic pathogens and characterization of novel viruses carried by commensal Rattus norvegicus in New York City. mBio 2014, 5, e01933-14. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Lu, L.; Du, J.; Yang, L.; Ren, X.; Liu, B.; Jiang, J.; Yang, J.; Dong, J.; Sun, L.; et al. Comparative analysis of rodent and small mammal viromes to better understand the wildlife origin of emerging infectious diseases. Microbiome 2018, 6, 178. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.G.; Kapusinszky, B.; Wang, C.; Rose, R.K.; Lipton, H.L.; Delwart, E.L. The fecal viral flora of wild rodents. PLoS Pathog. 2011, 7, e1002218. [Google Scholar] [CrossRef] [Green Version]
- Reuter, G.; Boros, A.; Kiss, T.; Delwart, E.; Pankovics, P. Complete genome characterization of mosavirus (family Picornaviridae) identified in droppings of a European roller (Coracias garrulus) in Hungary. Arch. Virol. 2014, 159, 2723–2729. [Google Scholar] [CrossRef]
- Luo, X.L.; Lu, S.; Jin, D.; Yang, J.; Wu, S.S.; Xu, J. Marmota himalayana in the Qinghai-Tibetan plateau as a special host for bi-segmented and unsegmented picobirnaviruses. Emerg. Microbes Infect. 2018, 7, 20. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Yang, L.; Ren, X.; He, G.; Zhang, J.; Yang, J.; Qian, Z.; Dong, J.; Sun, L.; Zhu, Y.; et al. Deciphering the bat virome catalog to better understand the ecological diversity of bat viruses and the bat origin of emerging infectious diseases. ISME J. 2016, 10, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Huong, N.Q.; Nga, N.T.T.; Long, N.V.; Luu, B.D.; Latinne, A.; Pruvot, M.; Phuong, N.T.; Quang, L.T.V.; Hung, V.V.; Lan, N.T.; et al. Coronavirus testing indicates transmission risk increases along wildlife supply chains for human consumption in Viet Nam, 2013–2014. PLoS ONE 2020, 15, e0237129. [Google Scholar] [CrossRef]
- Du, J.; Lu, L.; Liu, F.; Su, H.; Dong, J.; Sun, L.; Zhu, Y.; Ren, X.; Yang, F.; Guo, F.; et al. Distribution and characteristics of rodent picornaviruses in China. Sci. Rep. 2016, 6, 34381. [Google Scholar] [CrossRef] [Green Version]
- McFerran, J.B.; Nelson, R.; McCracken, J.M.; Ross, J.G. Viruses isolated from sheep. Nature 1969, 221, 194–195. [Google Scholar] [CrossRef]
- Lu, G.; Huang, M.; Chen, X.; Sun, Y.; Huang, J.; Hu, R.; Li, S. Identification and genome characterization of a novel feline picornavirus proposed in the Hunnivirus genus. Infect. Genet. Evol. 2019, 71, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Oberste, M.S.; Gotuzzo, E.; Blair, P.; Nix, W.A.; Ksiazek, T.G.; Comer, J.A.; Rollin, P.; Goldsmith, C.S.; Olson, J.; Kochel, T.J. Human febrile illness caused by encephalomyocarditis virus infection, Peru. Emerg. Infect. Dis. 2009, 15, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Hauffe, H.C.; Niklasson, B.; Olsson, T.; Bianchi, A.; Rizzoli, A.; Klitz, W. Ljungan virus detected in bank voles (Myodes glareolus) and yellow-necked mice (Apodemus flavicollis) from Northern Italy. J. Wildl. Dis. 2010, 46, 262–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindberg, A.M.; Johansson, S. Phylogenetic analysis of Ljungan virus and A-2 plaque virus, new members of the Picornaviridae. Virus Res. 2002, 85, 61–70. [Google Scholar] [CrossRef]
- Niklasson, B.; Kinnunen, L.; Hornfeldt, B.; Horling, J.; Benemar, C.; Hedlund, K.O.; Matskova, L.; Hyypia, T.; Winberg, G. A new picornavirus isolated from bank voles (Clethrionomys glareolus). Virology 1999, 255, 86–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, E.S.; Niklasson, B.; Tesh, R.B.; Shafren, D.R.; Travassos da Rosa, A.P.A.; Lindberg, A.M. Molecular characterization of M1146, an American isolate of Ljungan virus (LV) reveals the presence of a new LV genotype. J. Gen. Virol. 2003, 84, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Salisbury, A.M.; Begon, M.; Dove, W.; Niklasson, B.; Stewart, J.P. Ljungan virus is endemic in rodents in the UK. Arch. Virol. 2014, 159, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Jaaskelainen, A.J.; Voutilainen, L.; Lehmusto, R.; Henttonen, H.; Lappalainen, M.; Kallio-Kokko, H.; Vaheri, A.; Vapalahti, O. Serological survey in the Finnish human population implies human-to-human transmission of Ljungan virus or antigenically related viruses. Epidemiol. Infect. 2016, 144, 1278–1285. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.K.; Woo, P.C.; Lai, K.K.; Huang, Y.; Yip, C.C.; Shek, C.T.; Lee, P.; Lam, C.S.; Chan, K.H.; Yuen, K.Y. Complete genome analysis of three novel picornaviruses from diverse bat species. J. Virol. 2011, 85, 8819–8828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregorio, S.B.; Nakao, J.C.; Beran, G.W. Human enteroviruses in animals and arthropods in the central Philippines. Southeast Asian J. Trop. Med. Public Health 1972, 3, 45–51. [Google Scholar] [PubMed]
- Grew, N.; Gohd, R.S.; Arguedas, J.; Kato, J.I. Enteroviruses in rural families and their domestic animals. Am. J. Epidemiol. 1970, 91, 518–526. [Google Scholar] [CrossRef]
- Graves, I.L.; Oppenheimer, J.R. Human viruses in animals in West Bengal: An ecological analysis. Hum. Ecol. 1975, 3, 105–130. [Google Scholar] [CrossRef]
- Harvala, H.; Sharp, C.P.; Ngole, E.M.; Delaporte, E.; Peeters, M.; Simmonds, P. Detection and genetic characterization of enteroviruses circulating among wild populations of chimpanzees in Cameroon: Relationship with human and simian enteroviruses. J. Virol. 2011, 85, 4480–4486. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, L.; Ashworth, J.; Nguyen, D.; Li, K.; Smith, D.B.; Woolhouse, M.; on behalf of the VIZIONS Consortium. No Exchange of Picornaviruses in Vietnam between Humans and Animals in a High-Risk Cohort with Close Contact despite High Prevalence and Diversity. Viruses 2021, 13, 1709. https://0-doi-org.brum.beds.ac.uk/10.3390/v13091709
Lu L, Ashworth J, Nguyen D, Li K, Smith DB, Woolhouse M, on behalf of the VIZIONS Consortium. No Exchange of Picornaviruses in Vietnam between Humans and Animals in a High-Risk Cohort with Close Contact despite High Prevalence and Diversity. Viruses. 2021; 13(9):1709. https://0-doi-org.brum.beds.ac.uk/10.3390/v13091709
Chicago/Turabian StyleLu, Lu, Jordan Ashworth, Dung Nguyen, Kejin Li, Donald B. Smith, Mark Woolhouse, and on behalf of the VIZIONS Consortium. 2021. "No Exchange of Picornaviruses in Vietnam between Humans and Animals in a High-Risk Cohort with Close Contact despite High Prevalence and Diversity" Viruses 13, no. 9: 1709. https://0-doi-org.brum.beds.ac.uk/10.3390/v13091709