
Complete Genome Characterization of Reticuloendotheliosis Virus Detected in Chickens with Multiple Viral Coinfections

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Cases and Samples
2.2. Detection of REV and Concomitant Viruses
2.3. Genome Sequencing
2.4. Sequence and Phylogenetic Analyses
2.5. Polymorphism Analysis and Protein Modeling
3. Results
3.1. Detection of REV and Concomitant Viruses
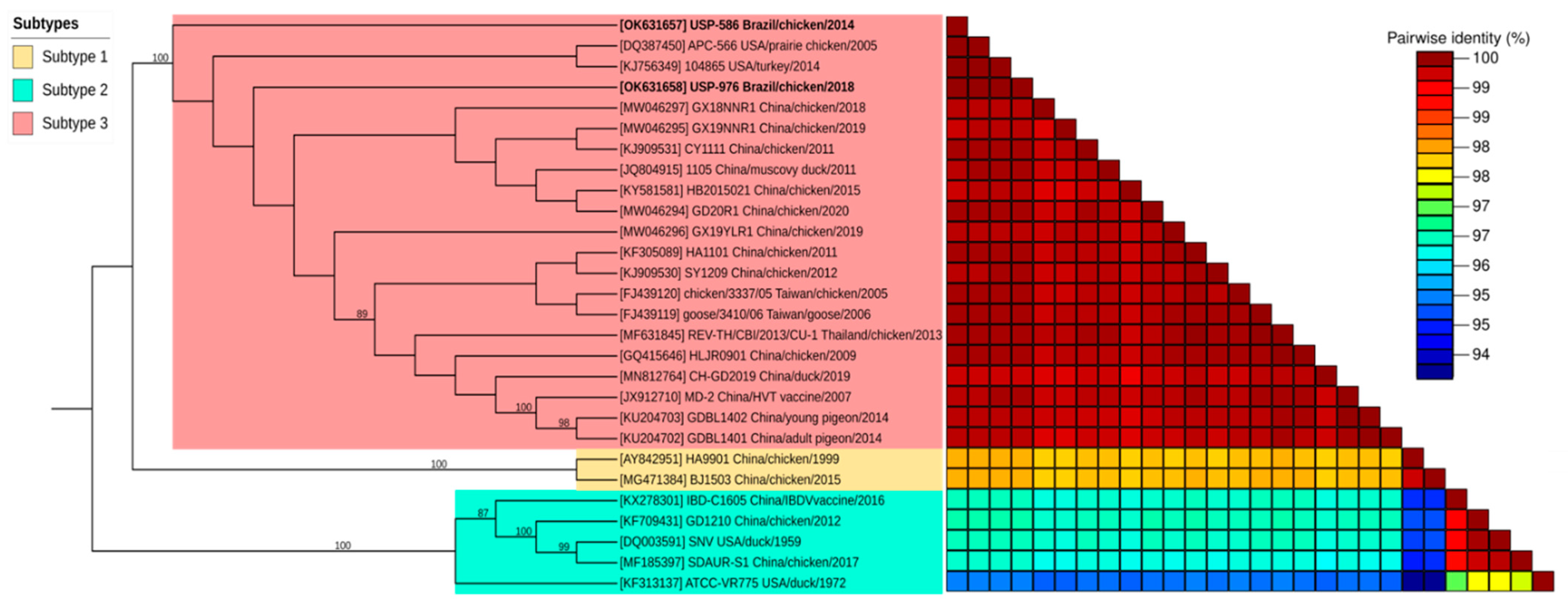
3.2. Genome Sequence and Phylogenetic Analyses
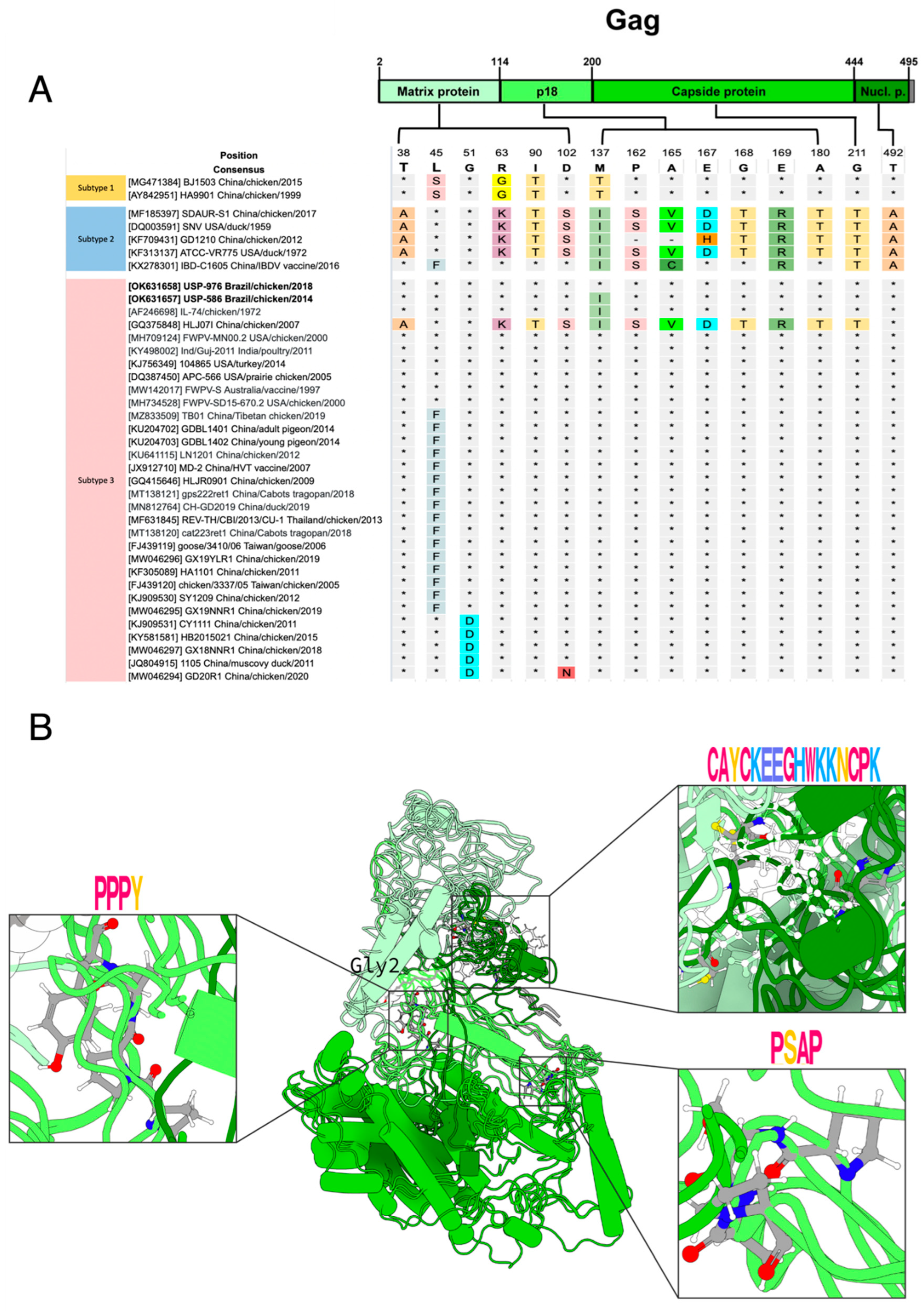
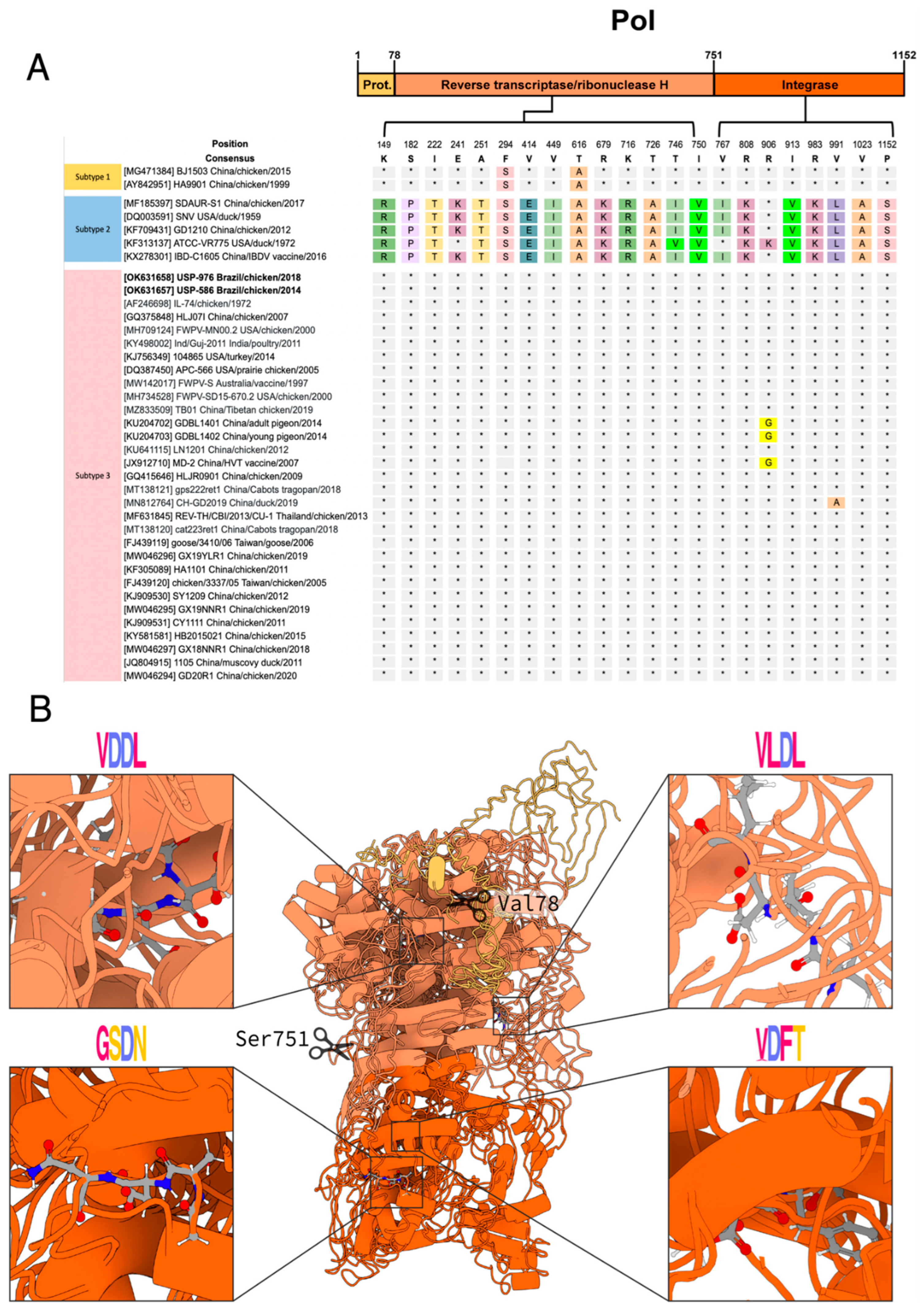
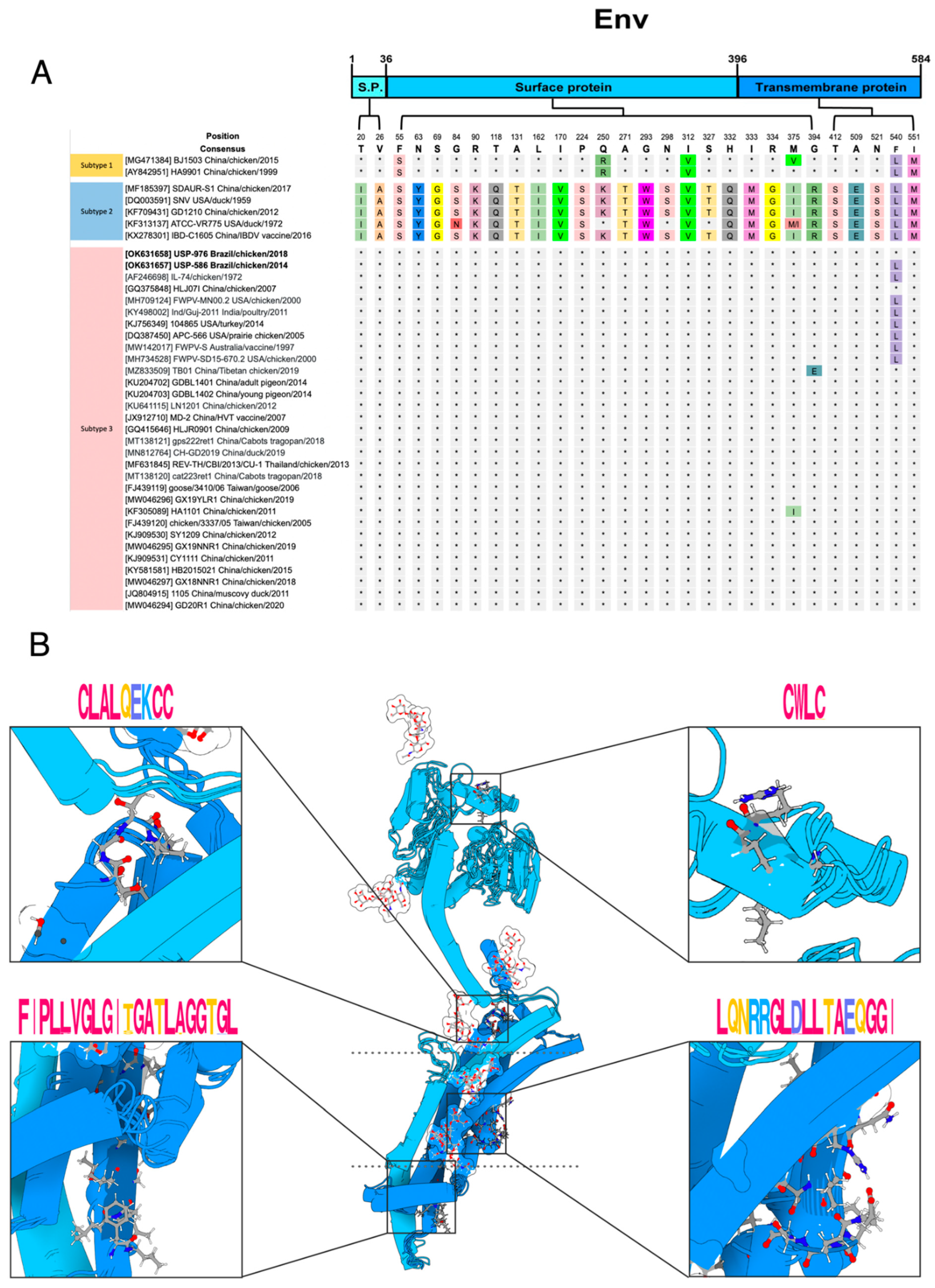
3.3. Polymorphism Analysis and Protein Modeling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nair, V.; Gimeno, I.; Dunn, J.; Zavala, G.; Williams, S.M.; Reece, R.L.; Hafner, S. Neoplastic Diseases. In Diseases of Poultry; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2020; pp. 548–715. ISBN 978-1-119-37119-9. [Google Scholar]
- Albritton, L.M. Chapter 1—Retrovirus Receptor Interactions and Entry. In Retrovirus-Cell Interactions; Parent, L.J., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 1–49. ISBN 978-0-12-811185-7. [Google Scholar]
- Robinson, F.R.; Twiehaus, M.J. Isolation of Tha Avian Reticuloendothelial Virus (Strain T). Avian Dis. 1974, 18, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Hoelzer, J.D.; Franklin, R.B.; Bose, H.R. Transformation by Reticuloendotheliosis Virus: Development of a Focus Assay and Isolation of a Nontransforming Virus. Virology 1979, 93, 20–30. [Google Scholar] [CrossRef]
- Drew, M.L. Retroviral Infections. In Infectious Diseases of Wild Birds; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2007; pp. 216–235. ISBN 978-0-470-34466-8. [Google Scholar]
- Wang, G.; Wang, Y.; Yu, L.; Jiang, Y.; Liu, J.; Cheng, Z. New Pathogenetic Characters of Reticuloendotheliosis Virus Isolated from Chinese Partridge in Specific-Pathogen-Free Chickens. Microb. Pathog. 2012, 53, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Cui, Z.; Lee, L.F.; Witter, R.L. Serologic Differences among Nondefective Reticuloendotheliosis Viruses. Arch. Virol. 1987, 93, 233–245. [Google Scholar] [CrossRef]
- Hertig, C.; Coupar, B.E.; Gould, A.R.; Boyle, D.B. Field and Vaccine Strains of Fowlpox Virus Carry Integrated Sequences from the Avian Retrovirus, Reticuloendotheliosis Virus. Virology 1997, 235, 367–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isfort, R.; Jones, D.; Kost, R.; Witter, R.; Kung, H.J. Retrovirus Insertion into Herpesvirus in Vitro and in Vivo. Proc. Natl. Acad. Sci. USA 1992, 89, 991–995. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Zhao, P.; Chang, S.; Ju, S.; Li, Y.; Meng, F.; Sun, P.; Cui, Z. Synergistic Pathogenic Effects of Co-Infection of Subgroup J Avian Leukosis Virus and Reticuloendotheliosis Virus in Broiler Chickens. Avian Pathol. 2015, 44, 43–49. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, Z.; Lan, X.; Zhang, F.; Wang, Q.; Li, K.; Pan, Q.; Gao, Y.; Qi, X.; Cui, H.-Y.; et al. A High Frequency of Gallid Herpesvirus-2 Co-Infection with Reticuloendotheliosis Virus is Associated with High Tumor Rates in Chinese Chicken Farms. Vet. Microbiol. 2019, 237, 108418. [Google Scholar] [CrossRef]
- Liang, M.; Zhao, Q.; Liu, G.; Yang, S.; Zuo, X.; Cui, G.; Zhong, S.; Sun, J.; Liu, J.; Zhu, R. Pathogenicity of Bordetella Avium under Immunosuppression Induced by Reticuloendotheliosis Virus in Specific-Pathogen-Free Chickens. Microb. Pathog. 2013, 54, 40–45. [Google Scholar] [CrossRef]
- Sun, G.-R.; Zhang, Y.-P.; Zhou, L.-Y.; Lv, H.-C.; Zhang, F.; Li, K.; Gao, Y.-L.; Qi, X.-L.; Cui, H.-Y.; Wang, Y.-Q.; et al. Co-Infection with Marek’s Disease Virus and Reticuloendotheliosis Virus Increases Illness Severity and Reduces Marek’s Disease Vaccine Efficacy. Viruses 2017, 9, 158. [Google Scholar] [CrossRef]
- Chacón, R.D.; Astolfi-Ferreira, C.S.; Guimarães, M.B.; Torres, L.N.; De la Torre, D.I.; Sá, L.R.M.D.; Piantino Ferreira, A.J. Detection and Molecular Characterization of a Natural Coinfection of Marek’s Disease Virus and Reticuloendotheliosis Virus in Brazilian Backyard Chicken Flock. Vet. Sci. 2019, 6, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chacón, R.D.; Astolfi-Ferreira, C.S.; De la Torre, D.I.; de Sá, L.R.M.; Piantino Ferreira, A.J. An Atypical Clinicopathological Manifestation of Fowlpox Virus Associated with Reticuloendotheliosis Virus in Commercial Laying Hen Flocks in Brazil. Transbound. Emerg. Dis. 2020, 67, 2923–2935. [Google Scholar] [CrossRef] [PubMed]
- Caleiro, G.S.; Nunes, C.F.; Urbano, P.R.; Kirchgatter, K.; de Araujo, J.; Durigon, E.L.; Thomazelli, L.M.; Stewart, B.M.; Edwards, D.C.; Romano, C.M. Detection of Reticuloendotheliosis Virus in Muscovy Ducks, Wild Turkeys, and Chickens in Brazil. J. Wildl. Dis. 2020, 56, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Carranza, C.; Astolfi-Ferreira, C.S.; Santander Parra, S.H.; Nuñez, L.F.N.; Penzes, Z.; Chacón, J.L.; Sesti, L.; Chacón, R.D.; Piantino Ferreira, A.J. Genetic Characterisation and Analysis of Infectious Bronchitis Virus Isolated from Brazilian Flocks between 2010 and 2015. Br. Poult. Sci. 2017, 58, 610–623. [Google Scholar] [CrossRef]
- Sun, F.; Ferro, P.J.; Lupiani, B.; Kahl, J.; Morrow, M.E.; Flanagan, J.P.; Estevez, C.; Clavijo, A. A Duplex Real-Time Polymerase Chain Reaction Assay for the Simultaneous Detection of Long Terminal Repeat Regions and Envelope Protein Gene Sequences of Reticuloendotheliosis Virus in Avian Blood Samples. J. Vet. Diagn. Investig. 2011, 23, 937–941. [Google Scholar] [CrossRef] [Green Version]
- Hauck, R.; Prusas, C.; Hafez, H.M.; Lüschow, D. Quantitative PCR as a Tool to Determine the Reticuloendotheliosis Virus-Proviral Load of Fowl Poxvirus. Avian Dis. 2009, 53, 211–215. [Google Scholar] [CrossRef]
- Luan, H.; Wang, Y.; Li, Y.; Cui, Z.; Chang, S.; Zhao, P. Development of a Real-Time Quantitative RT-PCR to Detect REV Contamination in Live Vaccine. Poult. Sci. 2016, 95, 2023–2029. [Google Scholar] [CrossRef]
- Li, K.; Gao, H.; Gao, L.; Qi, X.; Qin, L.; Gao, Y.; Xu, Y.; Wang, X. Development of TaqMan Real-Time PCR Assay for Detection and Quantitation of Reticuloendotheliosis Virus. J. Virol. Methods 2012, 179, 402–408. [Google Scholar] [CrossRef]
- Todd, D.; Trudgett, J.; McNeilly, F.; McBride, N.; Donnelly, B.; Smyth, V.J.; Jewhurst, H.L.; Adair, B.M. Development and Application of an RT-PCR Test for Detecting Avian Nephritis Virus. Avian Pathol. 2010, 39, 207–213. [Google Scholar] [CrossRef]
- De la Torre, D.; Astolfi-Ferreira, C.S.; Chacón, R.D.; Puga, B.; Piantino Ferreira, A.J. Emerging New Avian Reovirus Variants from Cases of Enteric Disorders and Arthritis/Tenosynovitis in Brazilian Poultry Flocks. Br. Poult. Sci. 2021, 62, 361–372. [Google Scholar] [CrossRef]
- De la Torre, D.; Astolfi-Ferreira, C.S.; Chacon, R.D.; Piantino Ferreira, A.J. Sensitive SYBR Green-Real Time PCR for the Detection and Quantitation of Avian Rotavirus A. Vet. Sci. 2018, 6, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Techera, C.; Tomás, G.; Panzera, Y.; Banda, A.; Perbolianachis, P.; Pérez, R.; Marandino, A. Development of Real-Time PCR Assays for Single and Simultaneous Detection of Infectious Bursal Disease Virus and Chicken Anemia Virus. Mol. Cell. Probes 2019, 43, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Smyth, V.J.; Jewhurst, H.L.; Wilkinson, D.S.; Adair, B.M.; Gordon, A.W.; Todd, D. Development and Evaluation of Real-Time TaqMan® RT-PCR Assays for the Detection of Avian Nephritis Virus and Chicken Astrovirus in Chickens. Avian Pathol. 2010, 39, 467–474. [Google Scholar] [CrossRef]
- Nuñez, L.F.; Santander-Parra, S.H.; Chaible, L.; De la Torre, D.I.; Buim, M.R.; Murakami, A.; Zaidan Dagli, M.L.; Astolfi-Ferreira, C.S.; Piantino Ferreira, A.J. Development of a Sensitive Real-Time Fast-QPCR Based on SYBR® Green for Detection and Quantification of Chicken Parvovirus (ChPV). Vet. Sci. 2018, 5, 69. [Google Scholar] [CrossRef] [Green Version]
- Günes, A.; Marek, A.; Grafl, B.; Berger, E.; Hess, M. Real-Time PCR Assay for Universal Detection and Quantitation of All Five Species of Fowl Adenoviruses (FAdV-A to FAdV-E). J. Virol. Methods 2012, 183, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Nuñez, L.F.N.; Parra, S.H.S.; De la Torre, D.; Catroxo, M.H.; Buim, M.R.; Chacon, R.V.; Ferreira, C.S.A.; Piantino Ferreira, A.J. Isolation of Avian Nephritis Virus from Chickens Showing Enteric Disorders. Poult. Sci. 2018, 97, 3478–3488. [Google Scholar] [CrossRef]
- De la Torre, D.I.; Nuñez, L.F.; Astolfi-Ferreira, C.S.; Piantino Ferreira, A.J. Enteric Virus Diversity Examined by Molecular Methods in Brazilian Poultry Flocks. Vet. Sci. 2018, 5, 38. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT Online Service: Multiple Sequence Alignment, Interactive Sequence Choice and Visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Chacón, R.D.; Astolfi-Ferreira, C.S.; Chacón, J.L.; Nuñez, L.F.N.; De la Torre, D.I.; Piantino Ferreira, A.J. A Seminested RT-PCR for Molecular Genotyping of the Brazilian BR-I Infectious Bronchitis Virus Strain (GI-11). Mol. Cell. Probes 2019, 47, 101426. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. JModelTest 2: More Models, New Heuristics and Parallel Computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A Virus Classification Tool Based on Pairwise Sequence Alignment and Identity Calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]
- Blum, M.; Chang, H.-Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro Protein Families and Domains Database: 20 Years On. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Zheng, W.; Zhang, C.; Li, Y.; Pearce, R.; Bell, E.W.; Zhang, Y. Folding Non-Homologous Proteins by Coupling Deep-Learning Contact Maps with I-TASSER Assembly Simulations. Cell Rep. Methods 2021, 1, 100014. [Google Scholar] [CrossRef]
- Gupta, R.; Brunak, S. Prediction of Glycosylation across the Human Proteome and the Correlation to Protein Function. Pac. Symp. Biocomput. 2002, 7, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Cumberbatch, J.A.; Brewer, D.; Vidavsky, I.; Sharif, S. Chicken Major Histocompatibility Complex Class II Molecules of the B Haplotype Present Self and Foreign Peptides. Anim. Genet. 2006, 37, 393–396. [Google Scholar] [CrossRef]
- Xue, M.; Shi, X.; Zhang, J.; Zhao, Y.; Cui, H.; Hu, S.; Gao, H.; Cui, X.; Wang, Y.-F. Identification of a Conserved B-Cell Epitope on Reticuloendotheliosis Virus Envelope Protein by Screening a Phage-Displayed Random Peptide Library. PLoS ONE 2012, 7, e49842. [Google Scholar] [CrossRef]
- Khairy, W.O.A.; Qian, K.; Shao, H.; Ye, J.; Qin, A. Identification of Two Conserved B-Cell Epitopes in the Gp90 of Reticuloendothelial Virus Using Peptide Microarray. Vet. Microbiol. 2017, 211, 107–111. [Google Scholar] [CrossRef]
- Barbosa, T.; Zavala, G.; Cheng, S.; Villegas, P. Full Genome Sequence and Some Biological Properties of Reticuloendotheliosis Virus Strain APC-566 Isolated from Endangered Attwater’s Prairie Chickens. Virus Res. 2007, 124, 68–77. [Google Scholar] [CrossRef]
- McNulty, M.S. Chicken Anaemia Agent: A Review. Avian Pathol. 1991, 20, 187–203. [Google Scholar] [CrossRef] [PubMed]
- Niewiadomska, A.M.; Gifford, R.J. The Extraordinary Evolutionary History of the Reticuloendotheliosis Viruses. PLoS Biol. 2013, 11, e1001642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathy, D.N.; Hanson, L.E.; Killinger, A.H. Atypical Fowlpox in a Poultry Farm in Illinois. Avian Dis. 1974, 18, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Schnitzlein, W.M.; Tripathy, D.N. Reticuloendotheliosis Virus Sequences within the Genomes of Field Strains of Fowlpox Virus Display Variability. J. Virol. 2003, 77, 5855–5862. [Google Scholar] [CrossRef] [Green Version]
- Joshi, L.R.; Bauermann, F.V.; Hain, K.S.; Kutish, G.F.; Armién, A.G.; Lehman, C.P.; Neiger, R.; Afonso, C.L.; Tripathy, D.N.; Diel, D.G. Detection of Fowlpox Virus Carrying Distinct Genome Segments of Reticuloendotheliosis Virus. Virus Res. 2019, 260, 53–59. [Google Scholar] [CrossRef]
- Sarker, S.; Athukorala, A.; Bowden, T.R.; Boyle, D.B. Characterisation of an Australian Fowlpox Virus Carrying a Near-Full-Length Provirus of Reticuloendotheliosis Virus. Arch. Virol. 2021, 166, 1485–1488. [Google Scholar] [CrossRef]
- Kim, T.J.; Tripathy, D.N. Reticuloendotheliosis Virus Integration in the Fowl Poxvirus Genome: Not a Recent Event. Avian Dis. 2001, 45, 663–669. [Google Scholar] [CrossRef]
- Riffel, N.; Harlos, K.; Iourin, O.; Rao, Z.; Kingsman, A.; Stuart, D.; Fry, E. Atomic Resolution Structure of Moloney Murine Leukemia Virus Matrix Protein and Its Relationship to Other Retroviral Matrix Proteins. Structure 2002, 10, 1627–1636. [Google Scholar] [CrossRef]
- Segura-Morales, C.; Pescia, C.; Chatellard-Causse, C.; Sadoul, R.; Bertrand, E.; Basyuk, E. Tsg101 and Alix Interact with Murine Leukemia Virus Gag and Cooperate with Nedd4 Ubiquitin Ligases during Budding. J. Biol. Chem. 2005, 280, 27004–27012. [Google Scholar] [CrossRef] [Green Version]
- Kingston, R.L.; Fitzon-Ostendorp, T.; Eisenmesser, E.Z.; Schatz, G.W.; Vogt, V.M.; Post, C.B.; Rossmann, M.G. Structure and Self-Association of the Rous Sarcoma Virus Capsid Protein. Structure 2000, 8, 617–628. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Barklis, E. Nucleocapsid Protein Effects on the Specificity of Retrovirus RNA Encapsidation. J. Virol. 1995, 69, 5716–5722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorelick, R.J.; Nigida, S.M.; Bess, J.W.; Arthur, L.O.; Henderson, L.E.; Rein, A. Noninfectious Human Immunodeficiency Virus Type 1 Mutants Deficient in Genomic RNA. J. Virol. 1990, 64, 3207–3211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oroszlan, S.; Luftig, R.B. Retroviral Proteinases. Curr. Top Microbiol. Immunol. 1990, 157, 153–185. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Zhao, Y.; Qi, X.; Cui, H.; Gao, Y.; Gao, H.; Liu, C.; Wang, Y.; Zhang, Y.; Li, K.; et al. Soluble Expression and Enzymatic Activity Evaluation of Protease from Reticuloendotheliosis Virus. Protein Expr. Purif. 2015, 114, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Jacobo-Molina, A.; Arnold, E. HIV Reverse Transcriptase Structure-Function Relationships. Biochemistry 1991, 30, 6351–6356. [Google Scholar] [CrossRef]
- Svarovskaia, E.S.; Cheslock, S.R.; Zhang, W.-H.; Hu, W.-S.; Pathak, V.K. Retroviral Mutation Rates and Reverse Transcriptase Fidelity. Front. Biosci. 2003, 8, d117–d134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, M.; Wilhelm, F.X. Reverse Transcription of Retroviruses and LTR Retrotransposons. Cell. Mol. Life Sci. 2001, 58, 1246–1262. [Google Scholar] [CrossRef]
- Craigie, R.; Bushman, F.D. HIV DNA Integration. Cold Spring Harb. Perspect. Med. 2012, 2, a006890. [Google Scholar] [CrossRef] [Green Version]
- Aydin, H.; Cook, J.D.; Lee, J.E. Crystal Structures of Beta- and Gammaretrovirus Fusion Proteins Reveal a Role for Electrostatic Stapling in Viral Entry. J. Virol. 2014, 88, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Denner, J. Immunising with the Transmembrane Envelope Proteins of Different Retroviruses Including HIV-1: A Comparative Study. Hum. Vaccin. Immunother. 2013, 9, 462–470. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.D.; Griffin, K.; Brewster, C.D.; Kapuscinski, M.L.; Stenglein, M.D.; Tripp, D.W.; Quackenbush, S.L.; Fox, K.A. A Novel Retrovirus (Gunnison’s Prairie Dog Retrovirus) Associated with Thymic Lymphoma in Gunnison’s Prairie Dogs in Colorado, USA. Viruses 2020, 12, 606. [Google Scholar] [CrossRef] [PubMed]
- Shalev, Z.; Duffy, S.P.; Adema, K.W.; Prasad, R.; Hussain, N.; Willett, B.J.; Tailor, C.S. Identification of a Feline Leukemia Virus Variant That Can Use THTR1, FLVCR1, and FLVCR2 for Infection. J. Virol. 2009, 83, 6706–6716. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Gene Target | Primer Name | Primer Sequence | Reference |
|---|---|---|---|---|
| REV | LTR | LTR-F | AGGCTCATAAACCATAAAAGGAAATGT | [18] |
| LTR-R | CCTTTACAACCATTGGCTCAGTATG | |||
| gag | qR 5 | GTTTTCTATACACACCAGCCTACCT | [19] | |
| qR 6 | TCCTGACCTCCCGCCTACT | |||
| pol | pol F | CCCCATTCATGTCCAGCTAT | [20] | |
| pol R | AGGGAGGAGAGGAGTGTTCC | |||
| env | gp90F | AAGAATCTGTGCGTGAAAG | [21] | |
| gp90R | TAAGGACCTGGTGAGTAGC | |||
| ANV | UTR | ANV F | ACGGCGAGTACCATCGAG | [22] |
| ANV R | AATGAAAAGCCCACTTTCGG | |||
| ARV | S4 | qREO-S4-F | GCTTTTTGAGTCCTTGTGCAG | [23] |
| qREO-S4-R | GGATGTGTTGCGGGAAACT | |||
| ARtV-A | VP6 | qRtVA-VP6-F | TTGGACCAGTATTTCCTGCTG | [24] |
| qRtVA-VP6-R | TGGTATGAGCTGTTACCCTCAA | |||
| CAV | VP2/VP3 | CAV-F630 | ACGCTAAGATCTGCAACTG | [25] |
| CAV-R756 | TTACCCTGTACTCGGAGG | |||
| CAstV | ORF1b-ORF2 | CAstV F | GCYGCTGCTGAAGAWATACAG | [26] |
| CAstV R | CATCCCTCTACCAGATTTTCTGAAA | |||
| ChPV | NS | PVA-F | GCAACTAACCTGACCGTGTG | [27] |
| PVA-R | CCCGGATTCAGAACCAGTAT | |||
| CPNV | VP1 | qCPNV-F | CGTAGACCTCGTCCTTCTGCTTKG | This study |
| qCPNV-R | GGGCGGTAACCATTCAGATACAYCC | |||
| FAdV | 52K | 52K-fw | ATGGCKCAGATGGCYAAGG | [28] |
| 52K-rw | AGCGCCTGGGTCAAACCGA |
| Gene Target | Primer Name | Primer Sequence | Location A | Length (bp) |
|---|---|---|---|---|
| LTR 5′ | LTR5-F1 | AATGTGGGAGGGAGCTCYG | 1–744 | 744 |
| LTR5-R1 | CAMCAACAATCAGAWAYCACAGA | |||
| gag | Gag-F2 | GAGGRTTTGGGAGGATCGGAGTG | 642–1415 | 774 |
| Gag-R2 | GATATGGAGGTGGAGRRGCTG | |||
| Gag-F3 | TGTAAACCCACAGGACCCTC | 1253–1992 | 740 | |
| Gag-R3 | CCCTGCCGAACCTCAGTTAT | |||
| Gag-F4 | TGGGAYCCTAACACRGGGAGA | 1868–2616 | 749 | |
| Gag-R4 | TTCCGTATRTTCCCAGTAGCC | |||
| pol | Pol-F5 | ACTCGCCCAGGAGAGTAGAG | 2269–3087 | 819 |
| Pol-R5 | GTGTTCCAGGGGGAGTGGAC | |||
| Pol-F6 | AAGTACCGCCCTACCTGTGA | 2959–3827 | 869 | |
| Pol-R6 | CTTCCTCTTTTTCRCCCCAC | |||
| Pol-F7 | CGAAAACCAAAAGGCARGTGCG | 3680–4586 | 907 | |
| Pol-R7 | GGRCGTGTAGAGTRGCGAAT | |||
| Pol-F8 | ACAAAGGCCCTGGAATGGAG | 4505–5415 | 911 | |
| Pol-R8 | AGGGCCTCACACAACTGCTG | |||
| Pol-F9 | ATGRTAACAGCCAAAGGGGG | 5198–6086 | 889 | |
| Pol-R9 | AGTTGCTGCRAGGGGTRAC | |||
| env | Env-F10 | ACTGTTCCAACCTGGTGAYCT | 5785–6537 | 753 |
| Env-R10 | AATCATGTCAGTGGGACCGC | |||
| Env-F11 | CGTATGAAGAYGGGCCTAAT | 6413–7167 | 755 | |
| Env-R11 | GGGGATAAACTGGACTGCYC | |||
| Env-F12 | GTGCATACTGGCATCAATCG | 7050–7752 | 703 | |
| Env-R12 | CCACATTCCCCACYGCTCTT | |||
| LTR 3′ | LTR3-F13 | TATTGTTCCTGACCCTCGGC | 7577–8295 | 719 |
| LTR3-R13 | CCCCCAAATGTTGTACMGAART | |||
| pMiniT 2.0 vector | Flank-F | ACCTGCCAACCAAAGCGAGAA | - | Insert B + 309 |
| Flank-R | TCAGGGTTATTGTCTCATGAG |
| Case | ID | Year | State A | REV | ANV | ARV | CAV | CAstV | ChPV | FAdV | Total B |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Farm 1 | 586 | 2014 | PR | + | − | − | − | + | + | − | 3 |
| Farm 2 | 599 | 2015 | PR | + | − | − | − | − | − | − | 1 |
| Farm 3 | 976 | 2018 | SP | + | + | − | + | − | + | − | 4 |
| Farm 4 | 1005 | 2018 | SP | + | + | + | + | + | + | + | 7 |
| Farm 5 | 1006 | 2018 | SP | + | + | + | + | + | + | + | 7 |
| Farm 6 | 1007 | 2018 | SP | + | + | + | + | + | + | + | 7 |
| Farm 7 | 1270 | 2019 | PR | + | − | + | − | + | + | − | 4 |
| Farm 8 | 1313 | 2019 | PR | + | − | + | − | + | + | − | 4 |
| Farm 9 | 1314 | 2019 | PR | + | − | + | − | + | + | − | 4 |
| Farm 10 | 1315 | 2019 | PR | + | − | + | − | + | + | − | 4 |
| Total C | 10 | 4 | 7 | 4 | 8 | 9 | 3 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chacón, R.D.; Sedano-Herrera, B.; Alfaro-Espinoza, E.R.; Quispe, W.U.; Liñan-Torres, A.; De la Torre, D.; de Oliveira, A.; Astolfi-Ferreira, C.S.; Ferreira, A.J.P. Complete Genome Characterization of Reticuloendotheliosis Virus Detected in Chickens with Multiple Viral Coinfections. Viruses 2022, 14, 798. https://0-doi-org.brum.beds.ac.uk/10.3390/v14040798
Chacón RD, Sedano-Herrera B, Alfaro-Espinoza ER, Quispe WU, Liñan-Torres A, De la Torre D, de Oliveira A, Astolfi-Ferreira CS, Ferreira AJP. Complete Genome Characterization of Reticuloendotheliosis Virus Detected in Chickens with Multiple Viral Coinfections. Viruses. 2022; 14(4):798. https://0-doi-org.brum.beds.ac.uk/10.3390/v14040798
Chicago/Turabian StyleChacón, Ruy D., Benjy Sedano-Herrera, Elizabeth Regina Alfaro-Espinoza, Wilma Ursula Quispe, Arturo Liñan-Torres, David De la Torre, Anderson de Oliveira, Claudete S. Astolfi-Ferreira, and Antonio J. Piantino Ferreira. 2022. "Complete Genome Characterization of Reticuloendotheliosis Virus Detected in Chickens with Multiple Viral Coinfections" Viruses 14, no. 4: 798. https://0-doi-org.brum.beds.ac.uk/10.3390/v14040798