
Immunosuppressive Tumor Microenvironment and Immunotherapy of Epstein–Barr Virus-Associated Malignancies


1
Department of Pathology, Collaborative Innovation Center for Cancer Medicine, State Key Laboratory of Oncology in South China, Sun Yat-sen University Cancer Center, Guangzhou 510060, China
2
Department of Liver Surgery, Collaborative Innovation Center for Cancer Medicine, State Key Laboratory of Oncology in South China, Sun Yat-sen University Cancer Center, Guangzhou 510060, China
*
Authors to whom correspondence should be addressed.
Viruses 2022, 14(5), 1017; https://0-doi-org.brum.beds.ac.uk/10.3390/v14051017
Submission received: 31 March 2022
/
Revised: 3 May 2022
/
Accepted: 9 May 2022
/
Published: 10 May 2022
(This article belongs to the Special Issue EBV and Cell Interactions in the Tumor Microenvironment)
Abstract
:The Epstein–Barr virus (EBV) can cause different types of cancer in human beings when the virus infects different cell types with various latent patterns. EBV shapes a distinct and immunosuppressive tumor microenvironment (TME) to its benefit by influencing and interacting with different components in the TME. Different EBV-associated malignancies adopt similar but slightly specific immunosuppressive mechanisms by encoding different EBV products to escape both innate and adaptive immune responses. Strategies reversing the immunosuppressive TME of EBV-associated malignancies have been under evaluation in clinical practice. As the interactions among EBV, tumor cells, and TME are intricate, in this review, we mainly discuss the epidemiology of EBV, the life cycle of EBV, the cellular and molecular composition of TME, and a landscape of different EBV-associated malignancies and immunotherapy by targeting the TME.
1. Introduction
Epstein–Barr virus (EBV), the first discovered human tumor virus, was isolated in Burkitt lymphoma samples by Michael Anthony Epstein and Yvonne Barr in 1964 [1,2]. This offers important insights into the natural infectious history of EBV and the mechanisms of how this virus drives the oncogenic process. EBV, also known as human herpesvirus 4, a member of the gammaherpesvirus subfamily [3], is characterized by the most common virus infection in humans, with a prevalence of more than 95% in the human population worldwide [4]. This virus normally persists as a lifelong infection without symptoms in humans, thus striking a subtle balance between the virus, the host cell, and the host immune system [5].
EBV, with a diameter of about 150–170 nm, consists of about 170 kb double-stranded DNA, which encodes more than 85 genes [6]. The viral genome controls EBV infectious mechanisms and contributes to the initiation and symptoms of EBV-associated human diseases. However, the particular function of 30% to 40% of the EBV genes remains unknown. There are two major EBV types, type 1 and type 2, which are mainly different in the genes encoding Epstein–Barr nuclear antigen 2 (EBNA2) and EBNA3 genes [7,8]. Type 1 is more prevalent worldwide and potentially transmits to B cells with a higher transforming efficiency while type 2 is mainly located in Africa [9].
Like other herpesviruses, EBV can build lifelong latency after its primary infection. EBV in teenagers usually causes symptomatic infectious mononucleosis but in adults the primary EBV infection is asymptomatic [10]. However, people with immunosuppressed conditions can also suffer severe acute diseases after EBV infection [11] and life-threatening EBV-associated malignancies originating from the lymphoid and epithelial cells [12,13]. It is reported that EBV causes more than 200,000 cases of cancer each year and 1.8% of cancer-related deaths [14,15]. Males encounter a higher prevalence of EBV-attributable malignancies compared to females [16]. Geographically, EBV-associated malignancy is present all over the world but is more prevalent in Asia and Africa than in the Western world [15]. It is well known that EBV can transform tumor precursor cells into EBV-associated malignancies [9,14].
In addition, EBV can also shape the supportive and immunosuppressed tumor microenvironment (TME) for its oncogenic process [17,18]. Available evidence indicates that the microenvironment plays an important role in the development of EBV-associated malignancies and the complex cross-talk among EBV and the microenvironment is not only relevant for establishing persistent EBV infection but also associated with the development of EBV-driven tumors and keeping the subtle EBV–host balance [19,20].
2. Viral Life Cycle and Gene Expression
The EBV life cycle in human beings often starts with its access to naive B cells in the oropharyngeal mucosa following transmission through the saliva. In the process of primary infection, EBV-infected naive B cells lead to a latency III growth program, which proliferates and expands the infected cells with no epigenetic repression of latent EBV genes [21]. Therefore, these cells express all the latent genes, including EBNAs, latent membrane protein (LMPs), EBV-encoded small RNAs (EBERs), and microRNAs (miRNAs) [22,23]. Being highly immunogenic, these cells can be recognized by the host immunity and rapidly eliminated by EBV-specific T cells [24]. Primary infection with EBV is followed by asymptomatic persistence of the virus in memory B cells where EBV gene expression is probably completely silenced. In this progress, the infected B cells epigenetically repress most genes to avoid the immune attack with a very high level of CpG methylation of its DNA [25]. The mechanism by which EBV gains access to the memory B cell pool is controversial. The most widely held model proposes that EBV-infected memory B cells are generated via a germinal center reaction. In this process, the virus enters the default program in the germinal center [26]. B cells in the default program are characterized by the expression of latency II proteins, including EBNA1, LMP1, and LMP2. These proteins are similar to the same signals as antigen-driven B cell responses in the normal germinal center [27,28]. Then, EBV-infected B cells leave the germinal center and gain entry into the memory B cell pool. These cells shut down all the viral genes (latency 0) that encode viral proteins and mimic the same signals inducing the proliferation of normal memory B cells. To allow the viral genome to replicate along with the cell, the EBV-infected memory B cells need to express EBNA1 (latency I), which can bind with the origin of plasmid replication (oriP), the viral plasmid origin of DNA replication [29,30]. Memory B cells can differentiate terminally into plasma cells, which is regarded to be associated with the switch to the EBV lytic program [31]. The lytic program can be further divided into three different lytic phases, including immediate early, early, and late phases. This process is characterized by the expression of up to 80 lytic genes [32]. During EBV lytic cycle, the viral chromatin opens up, which initiates the replication of the viral gene and synthesizes the full cascade of viral proteins [33,34]. The newly synthesized viral DNA, which lacks histones and methylated CpG dinucleotides, is packaged into viral capsid structures [35]. Then, the newly produced virions are released from the infected cell and, in the process, destroy the cells. Then, the virions may infect new target cells such as B cells and epithelial cells, where the virus can further replicate and then be transmitted to new hosts. Like the above replication program, the EBV life cycle oscillates between lytic phases and latent phases in infected cells [36].
Although it is not well understood how the virus establishes its latency and switches to a lytic program in the target cells, the progress has been investigated intensively and is thought to be well regulated by epigenetic regulation. As a rule, the EBV’s genomic DNA in infected cells is presented as extrachromosomal plasmids. A powerful epigenetic repression of lytic genes ensures the latent pattern of the EBV-infected cells, whose expression of different latent viral genes depends on different cell types. Epigenetic regulation also controls the progress from latency III to latency I, which can be regulated by CpG methylation of viral DNA, polycomb group proteins, and high-density packaging of nucleosomes [25].
Disruption of this well-regulated B cell infection can result in EBV-associated B cell tumors in all latency types except latency 0 [22]. The role of EBV in the oncogenic process of epithelial tumors, such as undifferentiated nasopharyngeal carcinoma (NPC) and EBV-associated gastric carcinoma (EBVaGC), remains unknown but is considered as a result of the aberrance of viral latency in epithelial cells with the existence of premalignant gene changes [37,38]. The progress and distinguished gene expression profiles of B cells infected by EBV are summarized in Figure 1.
3. TME of EBV-Associated Malignancy
Some EBV-associated malignancies develop after chronic latent EBV infection. It has been well known that tumor cells can be recognized and eliminated by the innate and adaptive immune system. However, the development of EBV-associated malignancies is tightly associated with the TME shaped by tumor cells to suppress the hosts’ immune system and evade immune surveillance. Indeed, tumor cells can turn immune cells to a tolerogenic and exhausted state by different components in the TME, including cell–cell interactions, soluble molecules, physical molecules, and extracellular vesicles (EVs) (Figure 2).
3.1. Cellular Components
Immune cells are the major components in the TME of EBV-associated malignancies. These cells try to clear and kill the EBV-infected malignant cells on the one hand and establish an immunosuppressive environment to provide local support for the tumor on the other hand.
3.1.1. T Cells
Antivirus responses depend heavily on potent virus-specific T cells. The EBV resides in the host cell most of the time after the establishment of virus infection. T cells recognize and eliminate EBV-infected cells based on virus-derived peptides presented on the surface of HLA molecules. The numbers of different T cell subsets vary greatly between individuals.
CD8+ T cells are the most powerful immune cells targeting the EBV-infected cells and executing the antiviral immunity. As HLA class I molecules (HLA I) are expressed in all nucleated human cells, antiviral CD8+ T cells will detect and target the EBV-infected cells. Compared to EBV-negative tumors, the number of CD8+ T cells is increased in EBV-positive tumors. Higher numbers of CD8+ T cells are associated with a higher frequency of effector T cells expressing the cytotoxic molecules. To evade recognition by EBV-specific CD8+ T cells, EBV encodes products that independently interfere with antigen presentation on HLA I by shutting off the expression of HLA I molecules, downregulating the HLA I on the cellular surface, and blocking the process of antigen presentation in the cell [5]. A recent study showed high numbers of exhausted CD8+ T cells were present in both the TME and the peripheral blood of EBV-positive tumors, resulting in the reduced cytotoxic activity of CD8+ T cells [39]. The exhausted CD8+ T cells are induced by EBV in several ways, including increasing checkpoint molecules, higher levels of immunosuppressive cytokines, and downregulation of death receptors, e.g., FasL and TRAIL.
CD4+ T cells are an indispensable component of the adaptive immunity against EBV, especially when EBV-infected cells express a high level of HLA II. These CD4+ T helper (Th) cells, CD4+ Th1 cells, in particular, can specifically target and eliminate EBV-infected cells either directly through their cytotoxic capacity or indirectly through activating CD8+ cytotoxic T cells and B cells. In response, EBV adopts varying immune escape strategies interfering with CD4+ T cell immunity at different levels. Gp42 is initially described as an entry receptor for EBV to bind to HLA class II molecules present on B cells [40]. Furthermore, it also serves as an immunosuppressive molecule interacting with HLA class II/peptide complexes to block the function of T cell receptors and prevent the activation of CD4+ T cells [41]. The early host shutoff protein BGLF5 encoded by EBV decreases cell-surface HLA II by degradation of HLA II mRNAs [42]. EBV also encodes a viral IL-10 homolog BCRF1, acting as an anti-inflammatory cytokine that inhibits and modulates the function of CD4+ T cells [43].
In the peripheral blood and tumor tissues of patients with EBV-positive tumors, an increasing number of natural CD4+CD25+ regulatory T cells (Tregs) have been found [44,45]. The viral latent protein EBNA1 plays a direct role in recruiting natural Tregs by upregulating CCL20 chemokine [46], and LMP1 can also serve as a peptide to promote infiltration of Treg cells [47]. A recent study showed that EBV-infected cells increased the infiltration of regulatory Type 1 cells (Tr1), which upregulate the gene expression of Tr1-related markers (ITGA2, ITGB2, LAG3) and associated immunosuppressive cytokines (IL-10) [48]. This upregulation is associated with an overexpression of several chemokine markers, such as CCL17, CCL19, and CCL20, which are known to attract T-helper type 2 (Th2) and regulatory T cells, thus contributing to more powerful immunosuppression.
3.1.2. Tumor-Associated Macrophages (TAMs)
The TME of EBV-positive tumors is also characterized by a more pronounced infiltration by TAMs as compared to EBV-negative cases. Interacting with the various signals in the TME, TAMs can show polarized activation into two functional states, including the M1 macrophages, which present a proinflammatory phenotype to promote Th1 responses and kill tumor cells, and the M2 macrophages, which can have an immunosuppressive role to promote tumor growth by remodeling and promoting Th2 responses. The macrophages are more likely to polarize into M1 macrophages to keep pace with a predominant Th1 microenvironment in EBV-positive tumors [49]. However, compared to EBV-negative tumors, EBV-positive tumors still have significantly higher numbers of M2 macrophages [50], which are mainly distributed in the stroma and involved in tumor progression [51]. This type of macrophage is attracted and recruited to the TME by chemokines such as CCL2 and CCL5, which can be induced by the EBV latent protein LMP1 in an NF-kappa B (NF-κB)-dependent manner [50].
3.1.3. Dendritic Cells (DCs)
DCs are the most potent antigen-presenting cells in the initiation of antiviral immune responses [52,53]. In EBV-associated malignancies, high levels of DCs are found to infiltrate into tumor regions [54,55]. However, the activation and maturation of DCs into different functional subsets depends largely on the cytokine milieu in the TME. EBERs within exosomes could promote DC activation and subsequent T cell activation, resulting in the systemic production of proinflammatory cytokines. EBV exosomes containing deoxyuridine triphosphate nucleotidohydrolase (dUTPase) can regulate the innate immune function of DCs through toll-like receptor (TLR)-2 leading to NF-κB activation and the production of proinflammatory cytokines, thus modulating the cellular microenvironment [56]. To evade the host immune responses, the EBV-infected cells suppress the function of DCs in several ways. The maturation of DCs is suppressed by galectin-1, a carbohydrate-binding protein within exosomes from EBV-infected cells [57]. Peripheral DCs infiltrate the tumor and are induced to differentiate into LAMP3+ DCs [58], which express PD-L1/PD-L2 interacting with PD-1 on CD8+ T cells and thus promote CD8+ T cell exhaustion [59].
3.1.4. Myeloid-Derived Suppressor Cells (MDSCs)
MDSCs originate from immature myeloid cells which are incapable of differentiating into mature myeloid cells, such as DCs, macrophages, and granulocytes [60]. MDSCs are expanded and induced by LMP1 in EBV-positive tumors of latency type II and III [61], exhibiting a strong immunosuppressive function. The LMP1 regulates the production of IL-1β, IL-6, and GM-CSF to enhance tumor-associated MDSC expansion [61]. MDSCs can support the naive CD4+ T cells to differentiate into Tregs by secreting retinoic acid and TGF-β and promote the transdifferentiation of Th17 cells into Foxp3+ Tregs [62]. Additionally, MDSCs can also trigger immunosuppressive functions of Tregs through mediating the release of IL-10 and IFN-γ [63]. Tumor-infiltrating MDSCs also present high levels of CCR5 ligands to recruit high numbers of Tregs into the TME [64], establishing an additional interaction between MDSCs and Tregs. Therefore, the MDSCs play an important role in the establishment of an immunosuppressive TME in EBV-positive tumors.
3.1.5. NK Cells
NK cells are the one arm of the innate immune response against EBV and have an important antiviral function during primary infection [65,66]. Although NK cells are highly infiltrated in EBV-positive malignancies, their functions can be impaired to various degrees when EBV-infected cells enter the latency phase [67]. LMP1 induces the expression of several antiapoptotic proteins such as Survivin, A20, Bcl2 [68], and a prosurvival receptor, 4-1BB [69,70], in EBV-infected cells to resist infected-cell death and apoptosis induced by NK cells via death receptors, which is regarded as the main mechanism of how NK cells kill the EBV-infected cells [71]. LMP2A and LMP2B can impair the antiviral response against EBV-infected cells by downregulating IFN signaling [72], and subsequently weaken cytokine-mediated antiviral effects. The EBV-encoded miRNAs, such as miRNA-BARTs, can dull NK cell immune recognition, and impair NK cell cytotoxic response by diminishing IL-12 release [73,74]. In such ways, the EBV weakens the antitumor function of NK cells and contributes to immune evasion in EBV-positive malignancies [75].
3.1.6. B Cells
Upon activation, B cells can differentiate into plasma cells and secrete antibodies to tag pathogens or infected cells for immune destruction. However, few studies have been focused on the function of B cells within the TME of EBV-associated malignancies. It is reported that B cells infiltrate into many EBV-positive tumors, but with a lower frequency than T cells do. Moreover, the number of infiltrated B cells varies greatly and they mostly reside in the stroma [76]. LMP1 can block the differentiation of B cells into antibody-secreting cells [77] through the upregulation of indoleamine 2,3-dioxygenase 1 (IDO1). IDO1 can also inhibit B cell function, thus further suppressing immune responses [77]. It is reported that EBV-infected cells can express miRNA-21 and then induce the expression of IL-10 in naive B cells, which is capable of suppressing CD8+ T cell activities [78].
3.1.7. Cancer-Associated Fibroblasts (CAFs)
CAFs, a major component of the cancer stroma derived from normal fibroblasts, can be induced and activated by LMP1 in EBV-infected cells through the NF-κB or ERK-MAPK pathway [79,80]. These CAFs recruited to the TME can promote cancer progression [81], facilitate metastasis by secreting proteases to degrade the extracellular matrix, provide support, fuel tumors’ insatiable growth by releasing paracrine cytokine [82], and recruit other stromal cell types with immunomodulatory properties [83]. The interplay between EBV-infected cells and CAFs forms the immunosuppressive microenvironment.
3.1.8. Endothelial Cells
Endothelial cells are involved in cancer pathogenesis and progression in various processes, including inflammation, fibrinolysis, and angiogenesis [84]. In EBV-positive tumors, LMP1 can induce the expression of fibroblast growth factor 2 (FGF2) and vascular endothelial growth factor (VEGF) to activate the endothelial cells to support tumor neoangiogenesis [85]. Furthermore, EBERs can be detected in endothelial cells in EBV-infected tissues, implicating that EBERs released from EBV-infected cells can act on endothelial cells and promote angiogenesis by stimulating VCAM-1 expression [86]. Innate immune modulation induced by EBV lytic infection can also promote endothelial cell inflammation and vascular injury [87].
3.2. Molecular Components
The local secretion of proinflammatory and immunosuppressive cytokines and chemokines is induced by EBV-infected cells. In turn, these aberrant soluble components also affect the function of immune cells and thus shape a TME where EBV-infected cells can proliferate, escape from apoptosis, and survive host antitumor defense [88].
3.2.1. Soluble Molecules
As for proinflammatory function, IFN-γ is upregulated in EBV-positive tumors. The cytokine IFN-γ, released by immune cells, such as T cells and DCs which can be recruited by EBV-infected cells, plays a central role in the resistance of the host to EBV infection via direct antiviral effects as well as modulation of the immune response. IFN-γ can directly enhance the macrophage’s differentiation into M1-like macrophages to establish an inflammatory TME. The BLZF1 encoded by EBV-infected cells seems to inhibit the function of the IFN-γ signaling pathway by downregulating the IFN-γ receptor and the downstream effector of IFN-γ. IP-10, a 10 kDa IFN-γ-inducible protein, is upregulated in many EBV-positive tumors. IP-10 is a member of the CXC chemokine family which is involved in chemotaxis, induction of apoptosis, regulation of cell growth, and mediation of angiostatic effects [89]. It is reported that the expression of IP-10 can be also mediated by LMP1, independent of IFN-γ, not only through transcriptional but also post-transcriptional mechanisms [90]. IL-1β is found to be upregulated in EBV-positive tumors [91]. The upregulation of IL-1β is induced by EBV genomic DNA and EBERs [92]. Tumor-derived IL-1β inhibits tumor growth and enhances survival through host responses. Mechanistically, IL-1β-mediated antitumor effects depend on infiltrated immunostimulatory neutrophils, which are significantly associated with better survival in patients with EBV-positive tumors [92]. TNF-α can be upregulated by LMP1 through TRAF2,5 and the NF-κB pathway in EBV-infected cells [93]. TNF-α may induce apoptosis and cell injuries via binding to TNF-α receptor-1 (TNFR1) [94]. However, these LMP1-expressing EBV-infected cells are resistant to TNF-α-induced apoptosis by downregulating TNFR1 and suppressing the downstream activities of apoptotic caspases 3, 8, and 9 [93].
Besides proinflammatory soluble molecules, immunosuppressive cytokines are found more frequently in EBV-positive tumors than in EBV-negative tumors. IL-10 is a potent immunosuppressive cytokine that induces Tregs and inhibits Th1 cells and cytotoxic T lymphocytes (CTLs) [95,96]. The expression of IL-10 is higher in EBV-positive tumors compared to EBV-negative tumors [96,97]. Higher levels of IL-10 are associated with lower numbers of CTLs in EBV-associated malignancies [98]. In EBV-infected cells, LMP2A can induce the expression of IL-10 [99,100]. IL-10 can also be produced by DCs, Tr1 cells, TAMs, and MDSCs to play the immunosuppressive role in EBV-associated malignancies. It is reported that higher levels of IL-4, IL-6, and IL-13 are present in EBV-positive tumors acting as a growth factor [101] and induce expression of the EBV-encoded protein LMP1 [102]. In turn, the induced LMP1 may directly enhance the production of immunosuppressive cytokines such as IL-6 and IL-8 via an NF-κB pathway [103,104]. The interplay between EBV-infected cells and immunosuppressive cytokines contributes to establishing a beneficial environment for tumor growth. Moreover, the expression of galectin-1 can be enhanced by both LMP1 and LMP2 proteins to induce tolerogenic DCs and cause the selective apoptosis of CD4+ T cells and CTLs [105].
3.2.2. Checkpoint Molecules
Immunomodulatory molecules such as PD-1 and its ligands PD-L1 and PD-L2 have been carefully investigated in EBV-associated malignancies. They play an indispensable role in assisting tumor cells to escape the host immune system [106]. Anti-PD-1 antibodies have been tested and applied in clinical practice and showed a remarkable response in EBV-associated malignancies [107,108]. Indeed, LMP1 can upregulate the expression of PD-L1 in EBV-infected cells through activation of TLR signaling and STAT3 or the engagement of AP-1-associated enhancers [109,110]. The induced expression of PD-L1 can be further enhanced by stimulation with IFN-γ [111]. Moreover, EBV-specific T cells can be also induced to express higher levels of PD-1 to further suppress immune responses against EBV-associated malignancies [112]. T-lymphocyte antigen-4 (CTLA-4) is another important immune checkpoint that is upregulated in T cells of EBV-infected tissues. High expression of CTLA-4 is significantly associated with disease progression and worse overall survival in patients with EBV-positive tumors [113]. Furthermore, other immunomodulatory molecules, such as TIM-3, LAG-3, and VISTA, are upregulated in EBV-specific T cells, which can further impair the function of T cell targeting the EBV [114,115].
3.3. EVs
Oncogenic viruses shape a beneficial protumoral milieu to promote the development and progression of malignancies [116,117]. EVs are a major component to facilitate intercellular communications in the TME. EVs can be divided into three major groups, including exosomes (30–150 nm in diameter, inward budding from the endosomal membrane), microvesicles (50–1000 nm in diameter, outward budding directly from plasma membranes), and apoptotic bodies (800–5000 nm in diameter, released directly from the plasma membrane of apoptotic cells), according to their production mode [118]. However, due to the limitations of methods to purify the EVs, it is almost impossible to isolate the pure subtypes and study their distinct functions. Since exosomes are the most frequently studied EVs in the EBV research field, we will mainly discuss the exosomes in the TME of EBV-associated malignancies.
EVs can transmit bioactive molecules enclosed in a lipid bilayer [119] to the TME to contribute to physiological and pathological processes by influencing recipient cells [117]. EBV is capable of modifying EVs’ content and influencing their release, thus enhancing the interplay between EBV-infected cells and immune cells in the TME. EBV can package various viral products, such as viral proteins and RNAs into the EVs [120] to aid in the evasion of the host immune system and promote tumor progression within the TME [120,121] (Figure 3).
LMP1 has been identified in exosomes. This latent EBV protein, when taken up by recipient cells, enables the induction of cellular responses by inhibiting apoptosis and promoting cell growth. LMP1 also supports tumor progression by mediating the function of immune cells in the TME. LMP1 can act on B cells and subsequently suppress humoral immune responses by preventing B cells’ differentiation into antibody-secreting cells [77]. LMP1 can upregulate the expression of IDO1 and then inhibit B cell function in the surrounding cells, thus suppressing immune effects [77]. Interestingly, LMP1 is found to regulate PD-L1 [122] and increase the packaging of PD-L1 into exosomes [123] in EBV-infected cells, which is involved in immune suppression. Moreover, exosomes containing LMP1 can induce the expression of angiogenic factors FGF2 and VEGF [124,125], which can enhance the proliferation of blood vessel endothelial cells and promote angiogenesis in tumor development. Therefore, the LMP1 released in EVs can modulate the immune responses and contribute to an immunosuppressive TME.
LMP2 is found to be secreted from EBV-infected cells in exosomes [126]. Though the biological function of LMP2 extracellular vesicles targeting recipient cells is not well understood, exosomal LMP2A may mimic the B cell receptor signal and inhibit B cell activity. LMP2A can promote the EBV life cycle by providing growth signals in the recipient cells while LMP2B has a distinct function in recipient cells and activates B cells [127].
EBERs are noncoding RNAs that can be found in all EBV-infected cells, thus regarded as the gold standard to identify the EBV infection in clinical practice. EBERs can be packaged with the EBER binding protein La into exosomes [128]. Evidence showed that EBERs can inhibit apoptosis, increase cell proliferation, and induce tumor formation [129,130]. EBERs are found to promote the release of IFN-I and inflammatory cytokines through TLR-3-mediated signaling [131]. Therefore, EBERs circulating within exosomes could trigger DC activation and subsequent T cell activation, leading to the systemic production of proinflammatory cytokines.
The miRNAs can be packaged into EVs and secreted into the TME for interaction with other cells [132]. There are 44 mature EBV miRNAs, including four of BamHI fragment H rightward open reading frame 1 (BHRF1), and forty BamHI-A rightward transcripts (BARTs) [133]. EBV miRNAs can suppress the secretion of proinflammatory cytokines, which can further suppress CD4+ T cell differentiation and reduce the activation of cytotoxic EBV-specific CD4+ effector T cells [134]. EBV-infected cells also reduce the functions of CD8+ T cells by releasing miRNAs that directly influence the process of antigen presentation and reduce MHC class 1 molecules present on the cellular surface. The miRNAs can also reduce EBNA1 levels, which is a target of CD8+ T cells, and therefore reduce the recognition and cytotoxic effects of CD8+ T cells [134].
EBV exosomes also contain dUTPase, a protein encoded by the EBV lytic gene BLLF3, which can influence the TME by modulating both innate and adaptive immune responses [56]. In particular, exosomes containing EBV dUTPase were shown to enhance the production of inflammatory cytokines and induce NF-κB activation by interacting with DCs through TLR-2 [56].
Galectin-1 is found to be packaged in EBV exosomes. Galectin-1 can affect local immune responses by promoting the apoptosis of Th1, Th17, and CTLs [135] and inducing the tolerant state of DCs [136].
Galectin-9, a β-galactoside-binding protein, can be derived from EBV exosomes in NPC patients. It can be induced to release by EBV products and interact with LMP1 [137]. These galectin-9-containing exosomes are shown to have an immunoregulatory function, by inducing apoptosis in EBV-specific CD4+ T cells through galectin-9/Tim-3 interactions [138] and expanding the number of Tregs and enhancing their suppressive activities [139].
Although the immunosuppressive role of EVs creates obstacles in controlling the tumor, EVs released by EBV-infected cells are now becoming more universally accepted as a superior source of biomarkers for diagnosing cancer due to their greater stability and higher diagnostic performances. In the case of EBV-associated malignancies, EVs containing EBV miRNAs have been investigated and proposed as early diagnostic markers in NPC and gastric cancer [140,141]. In addition, EVs are also considered as a promising immunotherapy target against EBV-associated malignancies for their immunosuppressive role by carrying different EBV products. A recent study showed that exosomes derived from Vδ2-T cells could promote the expansion of EBV-specific CD4+ and CD8+ T cells and control EBV-associated tumor cells through FasL and TRAIL pathways [142]. However, further studies need to be done to investigate the potential role of exosomes as specific therapeutic targets against EBV-associated malignancies. For example, preventing EBV-encoded products from loading into exosomes or inhibiting the release of such exosomes might serve as novel strategies [143].
4. Landscape of Different EBV-Associated Malignancies
All EBV-associated malignancies can be regarded as rare accidents of the normal EBV life cycle. Although these diseases have similar oncogenic routes, different EBV-associated malignancies are distinguished from each other in epidemiology, histologic appearance, EBV positive rates, latency pattern, and TME. The TME of EBV-associated malignancies is characterized by the coexistence and subtle balance of proinflammatory and immunosuppressive factors. These malignancies adopt similar but slightly specific immunosuppressive mechanisms to evade the host immune responses. Here, we describe the epidemiology, TME, immune evasion strategies, and immunotherapy of different EBV-associated malignancies (Table 1).
4.1. EBV-Associated Lymphomas
4.1.1. Post-transplant Lymphoproliferative Disorder (PTLD)
PTLD, a wide range of heterogeneous lymphoproliferative diseases from benign lymphoproliferations to malignant lymphomas, originates from B cells and is involved in 2%-10% of solid organ and hematopoietic stem cell transplants. These diseases are associated with EBV and are mainly characterized by a latency III pattern [144].
In the setting of acquired immune deficiency, EBV can fully exploit all its capabilities to promote the growth of the infected B cells and resist apoptosis. LMP1 is the main driver for B cell growth and survival by increasing the expression and secretion of cytokines and chemokines, such as IFN-γ, IL-6, and IL-10 [145,146]. LMP1 can also modulate the expression of some genes resisting apoptosis, including c-FLIP and BCL-2 [147,148], and upregulate the antiapoptotic Bcl-2 family member Mcl-1 through miRNA-155 expression [148]. Higher levels of IL-10 can also be induced by LMP-2A in B cells [149]. EBNA2, the main driver of type III latency, also enhances the growth and survival of B cells by reducing the expression of the proapoptotic BIK protein [150]. Besides latent proteins, uncontrolled EBV lytic replication may also contribute to the development of EBV-associated lymphoproliferations. A viral IL-10 homolog encoded by EBV lytic genes can also be expressed by EBV-infected cells to interfere with the host cytokine milieu. BZLF-1, the main EBV lytic transactivator, can upregulate the production of cytokines, such as IL-6, IL-10, and IL-13, to promote the growth of B cells [151]. PTLD contains sizeable T cell populations, mainly consisting of the memory/helper type while cytotoxic T cells are strikingly low in all samples [152].
Adoptive T cell therapy has been successfully applied for treating patients with EBV-associated PTLD by reconstituting host cellular immunity against the disease. The first clinical trial of adoptive T cell therapy showed that infusion of donor-derived EBV-specific T cells (EBVSTs) into patients with allogeneic hematopoietic stem cell transplantation (HSCT) induced complete regression in all five patients, but graft-versus-host disease developed [153]. Later, in vitro expanded donor-derived or autologous EBVSTs in phase I clinical trials were proven effective in the prevention and treatment of PTLD in HSCT or solid organ transplantation patients with minimal alloreactivity [154,155].
4.1.2. Hodgkin Lymphoma
The EBV-positive classical Hodgkin lymphoma, involved in approximately 50% of classical Hodgkin lymphoma, is a particular type of lymphoma with a specific TME, which is made up of a small proportion of Hodgkin and Reed–Sternberg (HRS) cells (<1%) and a large number of infiltrating immune cells (>90%) [156,157]. Therefore, the HRS cells greatly depend on interactions with immune cells in the TME by recruiting and communicating with various cellular components through autocrine and paracrine signals [18].
Classical Hodgkin lymphoma is distinguished by the expression of TNFR family members and their ligands. In addition, this lymphoma produces unbalanced Th2 cytokines and chemokines [158], thus leading to the reactive infiltration of eosinophils, Th2 cells, and fibroblasts [159]. These reactive cells also promote the growth of HRS cells. Such Th2 cytokines and chemokines can also attract Treg cells to suppress the EBV-specific immune responses [44]. The EBV-positive Hodgkin lymphoma is a tumor of type II latency pattern, expressing a series of latent genes, including EBNA1, LMP1, LMP2, EBER, and BART RNAs [160]. ENBA1 can further upregulate the expression of the chemokine CCL20 in the HRS cells and thus increase the migration and recruitment of Tregs to the TME. LMP1 epitopes are also found to promote infiltration of Treg cells into EBV-positive Hodgkin lymphoma [46,47]. In contrast, activated CD8+ T cells are scarcely found in areas that are abundant in tumor cells, suggesting that they may be ineffective when targeting the EBV-positive HRS cells. The HRS cells can induce the exhaustion of CD8+ T cells through the secretion of IL-10, TGF-β, and gelatin-1. In addition, PD-L1 is highly expressed in EBV-positive HRS cells, further inhibiting the function of CD8+ T cells by inducing their exhaustion [161].
Since PD-L1 plays an important role in the immunosuppressive TME of classical Hodgkin lymphoma, PD-1 inhibitors, such as nivolumab and pembrolizumab, have been tested in clinical trials and have shown to be effective in patients with classical Hodgkin lymphoma [107,108]. Clinical trials with in vitro expanded EBVSTs targeting type II latency antigens significantly increased response rates as well as overall survival in HL patients [162].
4.1.3. Burkitt Lymphoma (BL)
BL was the first human tumor found to be associated with a particular virus, EBV. Based on geographic distribution, BL is classified into three subtypes: endemic BL, sporadic BL, and immunodeficiency-associated BL. About 95% of endemic BL are EBV positive. In contrast, only less than 15% of sporadic BL and 40% of immunodeficiency-associated BL are associated with EBV.
Different from Hodgkin lymphoma containing at least 90% immune cells, BL seems to lack such a supportive TME. EBV-associated BL cells usually show the restricted type I latency pattern. EBERs can induce the secretion of the anti-inflammatory cytokine IL-10 by directly interacting with RIG-I and IRF3 activation [163]. To promote B cell growth, EBER2 can also induce the expression of cytokine IL-6 and EBNA1 can upregulate IL-2 receptors and enhance the antiapoptotic effects of IL-2 [164]. Higher levels of TAMs, M2-TAMs in particular, are the most prominent in the TME in BL [165]. They might play an important role in tumor progression through the secretion of chemokines, cytokines, and the expression of immune checkpoint-associated proteins such as PD-L1 [165].
With its rapid growth, BL is a highly chemotherapy-sensitive disease and was one of the early cancers in which cures were achieved with chemotherapy alone [166]. Therefore, fewer studies have been focused on immunotherapy against BL. It has been reported in a study that PD-1 antibody showed no objective response in patients with BL [167].
4.1.4. Diffuse Large B Cell Lymphoma (DLBCL)
DLBCL accounts for 30% of all lymphomas and is the most common B cell lymphoma. Approximately 10% of cases of DLBCL are associated with EBV [168]. Compared to the EBV-negative DLBCL, EBV-positive DLBCL has a more unfavorable prognosis. DLBCL, with the tumor cell content ranging from 60% to 80% [169], is characterized by a diffused growth pattern of large CD20+ B cells [170]. Cases arising in elderly patients tend to display latency III infection and cases in younger patients more frequently present a latency II infection pattern.
EBV-associated DLBCL expresses higher levels of PD-L1 compared to EBV-negative tumors, implicating an immune-tolerant mechanism of this tumor [171]. Increased PD-L1 expression in EBV-associated DLBCL cell lines can further induce PD-1 expression on T cells in vitro, subsequently exhausting CD8+T cells [172]. The expression of immunosuppressive cytokine IL-10 is increased in EBV-associated DLBCL [173], which can be induced by LMP1 [172] or Treg cells [17]. Although CD8+ T cells are increased [173], they might not be sufficient to eliminate EBV-infected cells in EBV-associated DLBCL because fewer central and effector memory CD8+ T cells are found among patients with EBV-positive DLBCL [174]. The immunosuppressive TME is also associated with the increased expression of other immune checkpoints, such as LAG3 and TIM3, and a greater protumoral M2 TAM polarization pattern [175]. Therefore, although characterized by inflammatory infiltration, EBV-associated DLBCL may still represent an ineffective immune attempt to control EBV-infected tumor cells [18].
Although the majority of patients with DLBCL can be cured with the standard immunochemotherapy R-CHOP, one-third of them relapse with a dismal outcome in most cases [176]. Chimeric antigen receptor (CAR) T cell therapy has emerged as a treatment option to treat these refractory DLBCLs. From early reports to multicenter studies, the clinical efficacy of CAR T cell therapy has been proven in these refractory patients [177]. As the PD-1/PD-L1 pathway is upregulated in EBV-associated DLBCL, it is expected that PD-1/PD-L1 blockade may improve the potency of CAR T cell therapies [178].
4.1.5. Extranodal NK/T Cell Lymphomas
Extranodal NK/T cell lymphomas are frequently EBV positive, usually affecting immunocompetent patients, although NK cells or T cells are the noncanonical cellular target of EBV. With a type II latency pattern, extranodal NK/T cell lymphomas are characterized by obvious inflammatory infiltration, implicating the supportive role of the inflammatory environment in the development of this tumor.
Extranodal NK/T cell lymphomas are often localized in the nasal area, where inflammation often occurs and B cells, T cells, and NK cells are thus recruited. The NK cells and T cells might acquire EBV infection when they kill EBV-infected B cells, which might produce the virus in the process. LMP1 expressed by the EBV-infected cells promotes the proliferation of the tumor, which is further potentiated by the cytokine IL-2 secreted by immune cells in the TME [179]. In turn, LMP1 can also increase the sensitivity of the cell to IL-2. Furthermore, activated T cells and macrophages produce IL-10 to enhance the tumor-growing function of IL-2. The sera levels of CD27 are increased in patients with NK/T cell lymphomas, which can interact with the CD70 receptor expressed in the EBV-infected NK/T cells to mediate tumor growth [180]. The EBV-infected T cells can upregulate the expression of TNF-α and promote the proinflammatory process. To keep an immunosuppressive TME, the LMP1 expressed in EBV-infected T cells can confer resistance to TNF-α-induced apoptosis through downregulating TNFR-1 [181]. NK/T cell lymphoma cells express PD-L1, which can be upregulated by LMP1 through the MAPK/NF-κB pathway [182]. Ligation of PD-1 on T cells with PD-L1 on lymphoma cells inhibits the function of T cells, providing a potential mechanism for NK/T cell lymphoma cells to evade immunosurveillance.
In stage I/II NK/T cell lymphoma, combined chemotherapy and radiotherapy is the best approach [183]. In relapsed/refractory cases, blockade of PD-1 has recently shown promising results with high response rates [184]. Further clinical studies remain to be done to prove the efficacy of immunotherapy in NK/T cell lymphoma.
4.2. NPC
The association between EBV and the undifferentiated NPC was found accidentally and was the first discovery that EBV infection is not restricted to B cells [185]. This tumor is characterized by a massive lymphocytic infiltration giving the tumor with a lymphoepithelial-like (LEL) appearance and its unique geographical distribution with high incidence throughout South-East Asia. Regardless of geographical distribution, all cases of undifferentiated NPC worldwide are EBV positive, with every malignant cell harboring EBV and presenting the viral gene [186].
It has been investigated for a long time how EBV triggers the pathogenesis of NPC [187,188]. Whole-genome sequencing has been performed to show the complex genomic changes in NPC. The latent pattern of NPC is best described as latency I/II, an intermediate form with the expression of EBNA1, the noncoding EBERs and miRNAs, and LMP2 [188]. Besides diverse gene profiles through genome sequencing analysis [189], NPC also shows another dimension of heterogeneity, diverse immune cells shaping the beneficial and immunosuppressive TME for tumor growth. EBV-positive NPC cells release diverse cytokines and chemokines to recruit multiple immune cells from the peripheral blood [190]. Naive CD8+ T cells infiltrate into the tumor and then develop into the effector CD8+ T cells and further exhausted CD8+ T cells. EBV-positive NPC cells express HLA-G, inhibiting the cytotoxic function of T cells and NK cells [39]. Peripheral DCs infiltrate into the tumor and then differentiate into LAMP3+ DCs [58]. The mature LAMP3+ DCs expressing PD-L1/PD-L2 can interact with PD-1 on CD8+ T cells, restraining the activation of CD8+ T cells and promoting their exhaustion [59]. The antigen presentation process of DCs will be limited when LAMP3+ DCs interact with Treg cells through CTLA4-CD80/CD86. In the process, the DCs secret exosomes containing IDO1, which enables them to recruit Treg cells and induce their proliferation and activation [139]. The intensive cell–cell interactions in the TME foster an immunosuppressive niche for the tumor growth of NPC.
To activate the immune cells to eliminate the NPC cells, clinical trials have been carried out to evaluate the effect of the monoclonal antibody against human PD-1. Recently, a phase III trial showed that the addition of toripalimab to the standard first-line chemotherapy can provide superior progression-free survival in patients with recurrent or metastatic NPC [191]. In addition, most therapeutic EBV vaccines have been focused on patients with NPC, and early clinical trials were done with DC-based EBV vaccines and recombinant viral vector vaccines [192,193]. Although the therapeutic EBV vaccines have shown increasing T cell responses in patients with NPC, future studies remain to be done to overcome the limited antigen epitopes delivered by DC-based EBV vaccines and the decreased antiviral vector immune responses after repeated immunizations [153,194].
4.3. EBVaGC
EBV is associated with approximately 10% of gastric cancer worldwide [195]. Compared to their EBV-negative counterparts, EBVaGC is more common in males and more likely to occur earlier in life, but have a relatively favorable prognosis [196]. When EBV infects normal gastric epithelial cells, EBV-infected cells grow clonally, resulting in the development of EBVaGC. The developmental processes of EBVaGC often carry out the sequence of gastritis–infection–cancer [197,198]. EBV infection occurs in gastric epithelial cells in the setting of the mucosa of atrophic gastritis. EBV-infected lymphocytes are recruited to the gastric mucosa and infect the epithelial cells. During this process, EBV establishes latent infection in the epithelial cells and these inflammatory signals play an important role in clonal growth in infected cells. In the setting of an inflammatory environment, the infected epithelial cells induce epithelial–mesenchymal transition and resistance against apoptosis of founder clones [199,200].
Gastric cancer can be divided into LEL-type tumors and conventional-type tumors based on histological appearance. The LEL-like gastric cancer resembles NPC, which is rich in lymphocytic LEL-like infiltration. These LEL-like tumors, accounting for only 1–4% of all gastric cancer, are associated with EBV in more than 90% of cases [201]. The remaining conventional-type tumors are EBV positive in 5–15% of cases. Viral gene expression in tumor cells is consistently either latency type I or an intermediate latency I/II in more than 50% of EBV-positive cases where LMP2 can be detected [202]. The TME plays a critical role in the pathogenesis of EBVaGC. The microenvironment of EBVaGC is characterized by the massive infiltration of CD8+ T cells [203,204]. These tumor-infiltrating lymphocytes (TILs) create an immune-active TME by eliminating EBV-infected malignant cells. However, there are also specific mechanisms that help EBV escape the host immune responses. The EBV-infected malignant cells express PD-L1 and then recruit PD-L1-positive immune cells to escape the host immune system [205]. In parallel with the immunosuppressive function of PD-L1, Tregs and TAMs are also recruited to remodel the TME of EBVaGC [206,207]. IDO1 is upregulated in EBVaGC, serving as a powerful inhibitory immune molecule reducing the immune responses [208]. In addition, EBVaGC cells release exosomes that contain some EBV-specific molecules and incorporate them with immune cells, contributing to the immunosuppressive TME [125].
To invert the immunosuppressive TME of EBVaGC, phase II clinical trials are underway to recruit patients who are stratified based on EBV status to assess the response to PD-1 inhibitors [209]. A phase I trial is assessing the safety of CRISPR-Cas9-mediated PD-1 knockout in EBV-specific CTL cells in stage IV GCs [209].
4.4. EBV-Associated Intrahepatic Cholangiocarcinoma (EBVaICC)
Studies of EBVaICC are so rare that the TME of EBVaICC has not been well characterized. A recent study reported that 6.6% of ICC are EBV positive [210]. EBVaICC is positive for EBNA1, but negative for LMP1 and EBNA2, suggesting that EBVaICC is a type I latency pattern. ICC can be divided into the LEL subtype and the conventional subtype. The LEL subtype of ICC has a close relationship with EBV infection, with 45% of cases of EBVaICC being the LEL subtype, while only 0.7% of non-EBVaICC belong to the LEL subtype. In terms of TME, the densities of tumor-infiltrating immune cells were significantly increased in EBValCC compared with non-EBVaICC. The TILs in EBVaICC mainly consist of CD3+ T cells, CD20+ B cells, and CD68+ TAMs. Among the T cell population, CD8+ T cells accounted for the predominant components. As for TAMs, M1 TAMs are more predominant than M2 TAMs in EBValCC [210]. Based on the PD-L1 expression and tumor-infiltrating CD8+ T cell densities, it is recommended to categorize the ICCs into four TME types: Type I (PD-L1+/CD8-High), Type II (PD-L1−/CD8-Low), Type III (PD-L1+/CD8-Low), and Type IV (PD-L1−/CD8-High). The Type I subgroup shows the best survival while the Type III subgroup had the worst survival. EBVaICC is significantly associated with Type I because 90% of EBVaICCs belonged to Type I [210]. Therefore, patients with EBVaICC are more likely to benefit from anti-PD-1/L1 therapy.
4.5. EBV-Associated Smooth Muscle Tumor (EBV-SMT)
EBV-SMT is a rare smooth muscle tumor (<1%) that occurs in patients with any immunocompromised conditions, including transplant patients with long-term immunosuppressive treatment, and patients suffering from human immunodeficiency virus infection or congenital immunodeficiency syndromes [211]. Although the exact mechanism of tumorigenesis is unclear, this tumor is associated with EBV infection and its tumor transformation of smooth muscle cells. EBV-SMT tends to be multifocal and arises in any anatomical location [212]. The prognosis is excellent, with patients dying of opportunistic infections but not the direct effects of the tumor [213].
Histologically, EBV-SMT is recognized as a relatively well-differentiated tumor, presenting with prominent intratumoral T lymphocytes, a low level of mitotic activity, and primitive round cell areas [214]. The tumor is reported to be latency pattern III by expressing EBNA1, EBNA2, LMP1, and LMP2A [215] or a variant of latency type III through the expression of EBNA2 or EBNA3 but no LMP1. This finding determines the mechanism of immune regulation in EBV-SMT because EBNA2 can be highly recognized by cytotoxic T cells [216,217]. Immune reconstitution can help T cells to kill tumor cells, thus contributing to tumor shrinkage [217]. Therefore, although the treatment of EBV-SMT mainly focuses on surgical resection and radiation therapy, immune reconstitution is a potentially promising treatment strategy [218].
5. Immunotherapy by Targeting the TME
Different types of strategies have been developed to target the TME by reactivating antiviral immune response and subverting the immunosuppressive microenvironment.
5.1. Immune Checkpoint Inhibitors
Expression of PD-L1 has been detected in 90% of EBV-positive HL and NPC as well as a wide range of EBV-associated malignancies including extranodal NK/T cell lymphoma, diffuse large B cell lymphoma, and PTLD [219]. Furthermore, PD-1 is also upregulated in EBV-positive tumors. Therefore, these diseases may be beneficial to immune checkpoint inhibitors that target the PD-1/PD-L1 axis [220]. Immune checkpoint inhibitors could impair inhibitory receptors of the immune cells, and thus convert the exhausted state of T cells and restore the cytotoxic EBV-specific T cell activity against malignant cells. PD-1 and PD-L1 inhibitors (e.g., pembrolizumab, nivolumab, atezolizumab, and durvalumab), as well as CTLA-4 inhibitors (e.g., ipilimumab and tremelimumab), are currently under evaluation and some have been proven effective in clinical trials aiming to reverse the immunosuppressive TME [107,108,221].
5.2. T Cell Therapy
EBV-associated malignancies are associated with the latent life cycles of EBV, but the pattern of latency depends on the type of tumor. The viral antigens expressed in various EBV-positive tumors can provide immune-based therapies with target antigens. In immunocompromised patients with EBV-positive tumors of latency III pattern, EBVSTs have shown outstanding success in rapidly restoring EBV-specific immunity [222]. In immunocompetent patients with EBV-positive tumors of latency II pattern, these EBVSTs seem to be less effective because intricate immune evasion strategies have been developed in these patients. However, EBV-positive tumors are frequently associated with exhausted CD8+ T cells and thus some clinical studies have shown that EBVSTs can prolong overall survival in patients with more extensive NPC [155,223]. To ensure clinical efficacy, additional therapies, such as checkpoint inhibitors or EBV vaccines, can be combined with EBVSTs to overcome immune evasion strategies of EBV-associated malignancies [224]. Additionally, gene modifications of EBVSTs may also be useful to reverse the function of inhibitory molecules and provide resistance to growth-promoting genes.
5.3. Therapeutic EBV Vaccine
The immune system plays an important role in controlling tumors. Immune-based tumor-specific therapy is under evaluation in clinical studies because it is highly effective with limited adverse effects [225]. Therapeutic EBV vaccines are designed to stimulate the existing or trigger novel antiviral immune responses in patients with EBV-associated malignancies [153,194]. The targets of EBV therapeutic vaccines are mainly the latent proteins of EBV, such as EBNA1, LMP1, and LMP2. These proteins are expressed in most EBV-associated malignancies and play a key role in the transformation of normal cells into tumors [225,226,227]. Most therapeutic EBV vaccinations concentrate on patients with NPC [194]. DCs, derived from autologous monocytes of NPC patients and pulsed with LMP2 epitopes, can activate and boost the EBV-specific CD8+ T cell responses [228]. Recombinant viral vector vaccines were later developed to provide a wide range of epitopes to the host immune system to improve the vaccines’ efficacy [229].
6. Conclusions
Although it has not been completely understood how EBV initiates infection and develops EBV-associated tumors, it is well recognized that EBV can shape a beneficial and supportive environment by encoding and releasing EBV products, thus intensively interacting with immune cells in the TME. The TME in an already progressing tumor may also facilitate EBV infection in some cases such as EBVaGC, and the EBV-infected cells are capable of forming an immunosuppressive milieu by adopting a variety of immune evasion strategies. Different EBV-associated malignancies have different types of EBV latent patterns, thus having a similar but slightly different interaction with cells in the TME. Different treatment strategies have been under evaluation to reverse the immunosuppressive TME. Immune checkpoint inhibitors have been proven effective in clinical trials in EBV-associated malignancies. However, one single therapeutic strategy might not be enough to control the disease. Therefore, the combination of different therapies targeting the TME might achieve a synergistic effect but needs further clinical evidence.
Author Contributions
Conceptualization, J.Y. and M.C.; Writing—original draft preparation, X.Z., Y.H., K.L. and R.L.; writing—review and editing, J.Y. and M.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Epstein, A. Burkitt lymphoma and the discovery of Epstein-Barr virus. Br. J. Haematol. 2012, 156, 777–779. [Google Scholar] [CrossRef] [PubMed]
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef]
- Wen, K.W.; Wang, L.; Menke, J.R.; Damania, B. Cancers associated with human gammaherpesviruses. FEBS J. 2021; pre-print. [Google Scholar] [CrossRef]
- Crawford, D.H.; Rickinson, A.; Johannessen, I.J.O.U.P. Cancer Virus: The story of Epstein-Barr Virus. Am. J. Epidemiol. 2014, 180, 1213–1215. [Google Scholar]
- Ressing, M.E.; van Gent, M.; Gram, A.M.; Hooykaas, M.J.; Piersma, S.J.; Wiertz, E.J. Immune Evasion by Epstein-Barr Virus. Curr. Top. Microbiol. Immunol. 2015, 391, 355–381. [Google Scholar]
- Machón, C.; Fàbrega-Ferrer, M.; Zhou, D.; Cuervo, A.; Carrascosa, J.L.; Stuart, D.I.; Coll, M. Atomic structure of the Epstein-Barr virus portal. Nat. Commun. 2019, 10, 3891. [Google Scholar] [CrossRef] [Green Version]
- Sample, J.; Young, L.; Martin, B.; Chatman, T.; Kieff, E.; Rickinson, A.; Kieff, E. Epstein-Barr virus types 1 and 2 differ in their EBNA-3A, EBNA-3B, and EBNA-3C genes. J. Virol. 1990, 64, 4084–4092. [Google Scholar] [CrossRef] [Green Version]
- Dambaugh, T.; Hennessy, K.; Chamnankit, L.; Kieff, E. U2 region of Epstein-Barr virus DNA may encode Epstein-Barr nuclear antigen 2. Proc. Natl. Acad. Sci. USA 1984, 81, 7632–7636. [Google Scholar] [CrossRef] [Green Version]
- Zimber, U.; Adldinger, H.K.; Lenoir, G.M.; Vuillaume, M.; Knebel-Doeberitz, M.V.; Laux, G.; Desgranges, C.; Wittmann, P.; Freese, U.K.; Schneider, U. Geographical prevalence of two types of Epstein-Barr virus. Virology 1986, 154, 56–66. [Google Scholar] [CrossRef]
- Luzuriaga, K.; Sullivan, J.L. Infectious mononucleosis. N. Engl. J. Med. 2010, 362, 1993–2000. [Google Scholar] [CrossRef] [Green Version]
- Taylor, G.S.; Long, H.M.; Brooks, J.M.; Rickinson, A.B.; Hislop, A.D. The immunology of Epstein-Barr virus-induced disease. Annu. Rev. Immunol. 2015, 33, 787–821. [Google Scholar] [CrossRef]
- Cesarman, E. Gammaherpesvirus and lymphoproliferative disorders in immunocompromised patients. Cancer Lett. 2011, 305, 163–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesarman, E. Gammaherpesviruses and lymphoproliferative disorders. Annu. Rev. Pathol. 2014, 9, 349–372. [Google Scholar] [CrossRef] [PubMed]
- de Martel, C.; Ferlay, J.; Franceschi, S.; Vignat, J.; Bray, F.; Forman, D.; Plummer, M. Global burden of cancers attributable to infections in 2008: A review and synthetic analysis. Lancet Oncol. 2012, 13, 607–615. [Google Scholar] [CrossRef]
- Khan, G.; Hashim, M.J. Global burden of deaths from Epstein-Barr virus attributable malignancies 1990–2010. Infect. Agents Cancer 2014, 9, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, G.; Fitzmaurice, C.; Naghavi, M.; Ahmed, L.A. Global and regional incidence, mortality and disability-adjusted life-years for Epstein-Barr virus-at tributable malignancies, 1990–2017. BMJ Open 2020, 10, e037505. [Google Scholar] [CrossRef]
- Tan, G.W.; Visser, L.; Tan, L.P.; van den Berg, A.; Diepstra, A. The Microenvironment in Epstein-Barr Virus-Associated Malignancies. Pathogens 2018, 7, 40. [Google Scholar] [CrossRef] [Green Version]
- Dolcetti, R. Cross-talk between Epstein-Barr virus and microenvironment in the pathogenesis of lymphomas. Semin. Cancer Biol. 2015, 34, 58–69. [Google Scholar] [CrossRef]
- Shen, Y.; Zhang, S.; Sun, R.; Wu, T.; Qian, J. Understanding the interplay between host immunity and Epstein-Barr virus in NPC patients. Emerg. Microbes Infect. 2015, 4, e20. [Google Scholar] [CrossRef]
- Iizasa, H.; Kim, H.; Kartika, A.V.; Kanehiro, Y.; Yoshiyama, H. Role of Viral and Host microRNAs in Immune Regulation of Epstein-Barr Virus-Associated Diseases. Front. Immunol. 2020, 11, 367. [Google Scholar] [CrossRef] [Green Version]
- Kintner, C.; Sugden, B. Conservation and progressive methylation of Epstein-Barr viral DNA sequences in transformed cells. J. Virol. 1981, 38, 305–316. [Google Scholar] [CrossRef] [Green Version]
- Thorley-Lawson, D.A.; Gross, A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N. Engl. J. Med. 2004, 350, 1328–1337. [Google Scholar] [CrossRef] [PubMed]
- Babcock, G.J.; Hochberg, D.; Thorley-Lawson, A.D. The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiati on stage of the infected B cell. Immunity 2000, 13, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Thorley-Lawson, D.A. Epstein-Barr virus: Exploiting the immune system. Nat. Rev. Immunol. 2001, 1, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Woellmer, A.; Arteaga-Salas, J.M.; Hammerschmidt, W. BZLF1 governs CpG-methylated chromatin of Epstein-Barr Virus reversing epigenetic repression. PLoS Pathog. 2012, 8, e1002902. [Google Scholar] [CrossRef] [PubMed]
- Roughan, J.E.; Thorley-Lawson, D.A. The intersection of Epstein-Barr virus with the germinal center. J. Virol. 2009, 83, 3968–3976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldwell, R.G.; Wilson, J.B.; Anderson, S.J.; Longnecker, R. Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell recep tor signals. Immunity 1998, 9, 405–411. [Google Scholar] [CrossRef] [Green Version]
- Gires, O.; Zimber-Strobl, U.; Gonnella, R.; Ueffing, M.; Marschall, G.; Zeidler, R.; Pich, D.; Hammerschmidt, W. Latent membrane protein 1 of Epstein-Barr virus mimics a constitutively active receptor molecule. EMBO J. 1997, 16, 6131–6140. [Google Scholar] [CrossRef] [Green Version]
- De Leo, A.; Calderon, A.; Lieberman, P.M. Control of Viral Latency by Episome Maintenance Proteins. Trends Microbiol. 2020, 28, 150–162. [Google Scholar] [CrossRef]
- Chiu, Y.F.; Sugden, B. Plasmid Partitioning by Human Tumor Viruses. J. Virol. 2018, 92, e02170-17. [Google Scholar] [CrossRef] [Green Version]
- Laichalk, L.L.; Thorley-Lawson, D.A. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J. Virol. 2005, 79, 1296–1307. [Google Scholar] [CrossRef] [Green Version]
- Amon, W.; Binné, U.K.; Bryant, H.; Jenkins, P.J.; Karstegl, C.E.; Farrell, P.J. Lytic cycle gene regulation of Epstein-Barr virus. J. Virol. 2004, 78, 13460–13469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Countryman, J.; Miller, G. Activation of expression of latent Epstein-Barr herpesvirus after gene transfer with a small cloned s ubfragment of heterogeneous viral DNA. Proc. Natl. Acad. Sci. USA 1985, 82, 4085–4089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, T.Y.; Chu, Y.Y.; Yang, Y.C.; Hsu, S.W.; Liu, S.T.; Chang, L.K. MCAF1 and Rta-activated BZLF1 transcription in Epstein-Barr virus. PLoS ONE 2014, 9, e90698. [Google Scholar] [CrossRef] [PubMed]
- Buschle, A.; Hammerschmidt, W. Epigenetic lifestyle of Epstein-Barr virus. Semin. Immunopathol. 2020, 42, 131–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, T.; Tsurumi, T. Switching of EBV cycles between latent and lytic states. Rev. Med. Virol. 2014, 24, 142–153. [Google Scholar] [CrossRef]
- Dolcetti, R.; Menezes, J. Epstein-Barr virus and undifferentiated nasopharyngeal carcinoma: New immunobiological and molecular insights on a long-standing etiopathogenic association. Adv. Cancer Res. 2003, 87, 127–157. [Google Scholar]
- Rickinson, A.B. Co-infections, inflammation and oncogenesis: Future directions for EBV research. Semin. Cancer Biol. 2014, 26, 99–115. [Google Scholar] [CrossRef]
- Cai, M.B.; Han, H.Q.; Bei, J.X.; Liu, C.C.; Lei, J.J.; Cui, Q.; Feng, Q.S.; Wang, H.Y.; Zhang, J.X.; Liang, Y.; et al. Expression of human leukocyte antigen G is associated with prognosis in nasopharyngeal carcinoma. Int. J. Biol. Sci. 2012, 8, 891–900. [Google Scholar] [CrossRef] [Green Version]
- Ressing, M.E.; van Leeuwen, D.; Verreck, F.A.; Keating, S.; Gomez, R.; Franken, K.L.; Ottenhoff, T.H.; Spriggs, M.; Schumacher, T.N.; Hutt-Fletcher, L.M.; et al. Epstein-Barr virus gp42 is posttranslationally modified to produce soluble gp42 that mediates HLA cla ss II immune evasion. J. Virol. 2005, 79, 841–852. [Google Scholar] [CrossRef] [Green Version]
- Ressing, M.E.; van Leeuwen, D.; Verreck, F.A.; Gomez, R.; Heemskerk, B.; Toebes, M.; Mullen, M.M.; Jardetzky, T.S.; Longnecker, R.; Schilham, M.W.; et al. Interference with T cell receptor-HLA-DR interactions by Epstein-Barr virus gp42 results in reduced T helper cell recognition. Proc. Natl. Acad. Sci. USA 2003, 100, 11583–11588. [Google Scholar] [CrossRef] [Green Version]
- Rowe, M.; Glaunsinger, B.; van Leeuwen, D.; Zuo, J.; Sweetman, D.; Ganem, D.; Middeldorp, J.; Wiertz, E.J.; Ressing, M.E. Host shutoff during productive Epstein-Barr virus infection is mediated by BGLF5 and may contribute to immune evasion. Proc. Natl. Acad. Sci. USA 2007, 104, 3366–3371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, D.G.; Trifilo, M.J.; Edelmann, K.H.; Teyton, L.; McGavern, D.B.; Oldstone, M.B. Interleukin-10 determines viral clearance or persistence in vivo. Nat. Med. 2006, 12, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Marshall, N.A.; Christie, L.E.; Munro, L.R.; Culligan, D.J.; Johnston, P.W.; Barker, R.N.; Vickers, M.A. Immunosuppressive regulatory T cells are abundant in the reactive lymphocytes of Hodgkin lymphoma. Blood 2004, 103, 1755–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baráth, S.; Aleksza, M.; Keresztes, K.; Tóth, J.; Sipka, S.; Szegedi, G.; Illés, A. Immunoregulatory T cells in the peripheral blood of patients with Hodgkin’s lymphoma. Acta Haematol. 2006, 116, 181–185. [Google Scholar] [CrossRef]
- Baumforth, K.R.; Birgersdotter, A.; Reynolds, G.M.; Wei, W.; Kapatai, G.; Flavell, J.R.; Kalk, E.; Piper, K.; Lee, S.; Machado, L.; et al. Expression of the Epstein-Barr virus-encoded Epstein-Barr virus nuclear antigen 1 in Hodgkin’s lympho ma cells mediates Up-regulation of CCL20 and the migration of regulatory T cells. Am. J. Pathol. 2008, 173, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Marshall, N.A.; Culligan, D.J.; Tighe, J.; Johnston, P.W.; Barker, R.N.; Vickers, M.A. The relationships between Epstein-Barr virus latent membrane protein 1 and regulatory T cells in Hodg kin’s lymphoma. Exp. Hematol. 2007, 35, 596–604. [Google Scholar] [CrossRef]
- Morales, O.; Mrizak, D.; François, V.; Mustapha, R.; Miroux, C.; Depil, S.; Decouvelaere, A.V.; Lionne-Huyghe, P.; Auriault, C.; de Launoit, Y.; et al. Epstein-Barr virus infection induces an increase of T regulatory type 1 cells in Hodgkin lymphoma pat ients. Br. J. Haematol. 2014, 166, 875–890. [Google Scholar] [CrossRef]
- Barros, M.H.; Segges, P.; Vera-Lozada, G.; Hassan, R.; Niedobitek, G. Macrophage polarization reflects T cell composition of tumor microenvironment in pediatric classical Hodgkin lymphoma and has impact on survival. PLoS ONE 2015, 10, e0124531. [Google Scholar] [CrossRef]
- Buettner, M.; Meyer, B.; Schreck, S.; Niedobitek, G. Expression of RANTES and MCP-1 in epithelial cells is regulated via LMP1 and CD40. Int. J. Cancer 2007, 121, 2703–2710. [Google Scholar] [CrossRef]
- Huang, H.; Liu, X.; Zhao, F.; Lu, J.; Zhang, B.; Peng, X.; Zhang, M.; Chen, X.; Li, G.; Li, X. M2-polarized tumour-associated macrophages in stroma correlate with poor prognosis and Epstein-Barr v iral infection in nasopharyngeal carcinoma. Acta Otolaryngol. 2017, 137, 888–894. [Google Scholar] [CrossRef]
- Steinman, R.M. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 1991, 9, 271–296. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Chen, X.M.; Huang, H.R.; Zhao, F.P.; Wang, F.; Liu, X.; Li, X.P. Detailed analysis of inflammatory cell infiltration and the prognostic impact on nasopharyngeal carci noma. Head Neck 2018, 40, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Braz-Silva, P.H.; Vitale, S.; Butori, C.; Guevara, N.; Santini, J.; Magalhães, M.; Hofman, P.; Doglio, A. Specific infiltration of langerin-positive dendritic cells in EBV-infected tonsil, Hodgkin lymphoma and nasopharyngeal carcinoma. Int. J. Cancer 2011, 128, 2501–2508. [Google Scholar] [CrossRef]
- Ariza, M.E.; Rivailler, P.; Glaser, R.; Chen, M.; Williams, M.V. Epstein-Barr virus encoded dUTPase containing exosomes modulate innate and adaptive immune responses in human dendritic cells and peripheral blood mononuclear cells. PLoS ONE 2013, 8, e69827. [Google Scholar] [CrossRef] [Green Version]
- Hinata, M.; Kunita, A.; Abe, H.; Morishita, Y.; Sakuma, K.; Yamashita, H.; Seto, Y.; Ushiku, T.; Fukayama, M. Exosomes of Epstein-Barr Virus-Associated Gastric Carcinoma Suppress Dendritic Cell Maturation. Microorganisms 2020, 8, 1776. [Google Scholar] [CrossRef]
- Liu, Y.; He, S.; Wang, X.L.; Peng, W.; Chen, Q.Y.; Chi, D.M.; Chen, J.R.; Han, B.W.; Lin, G.W.; Li, Y.Q.; et al. Tumour heterogeneity and intercellular networks of nasopharyngeal carcinoma at single cell resolution. Nat. Commun. 2021, 12, 741. [Google Scholar] [CrossRef]
- Oh, S.A.; Wu, D.C.; Cheung, J.; Navarro, A.; Xiong, H.; Cubas, R.; Totpal, K.; Chiu, H.; Wu, Y.; Comps-Agrar, L.; et al. PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nat. Cancer 2020, 1, 681–691. [Google Scholar] [CrossRef]
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer 2013, 13, 739–752. [Google Scholar] [CrossRef]
- Cai, T.T.; Ye, S.B.; Liu, Y.N.; He, J.; Chen, Q.Y.; Mai, H.Q.; Zhang, C.X.; Cui, J.; Zhang, X.S.; Busson, P.; et al. LMP1-mediated glycolysis induces myeloid-derived suppressor cell expansion in nasopharyngeal carcinoma. PLoS Pathog. 2017, 13, e1006503. [Google Scholar] [CrossRef]
- Hoechst, B.; Gamrekelashvili, J.; Manns, M.P.; Greten, T.F.; Korangy, F. Plasticity of human Th17 cells and iTregs is orchestrated by different subsets of myeloid cells. Blood 2011, 117, 6532–6541. [Google Scholar] [CrossRef] [PubMed]
- Vetsika, E.K.; Koukos, A.; Kotsakis, A. Myeloid-Derived Suppressor Cells: Major Figures that Shape the Immunosuppressive and Angiogenic Network in Cancer. Cells 2019, 8, 1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlecker, E.; Stojanovic, A.; Eisen, C.; Quack, C.; Falk, C.S.; Umansky, V.; Cerwenka, A. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favoring tumor growth. J. Immunol. 2012, 189, 5602–5611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, R.J.; Pachnio, A.; Pedroza-Pacheco, I.; Leese, A.M.; Begum, J.; Long, H.M.; Croom-Carter, D.; Stacey, A.; Moss, P.A.H.; Hislop, A.D.; et al. Asymptomatic Primary Infection with Epstein-Barr Virus: Observations on Young Adult Cases. J. Virol. 2017, 91, e00382-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, H.; McAulay, K.; Macsween, K.F.; Gallacher, N.J.; Higgins, C.D.; Harrison, N.; Swerdlow, A.J.; Crawford, D.H. The immune response to primary EBV infection: A role for natural killer cells. Br. J. Haematol. 2005, 129, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Lo, K.W.; To, K.F.; Huang, D.P. Focus on nasopharyngeal carcinoma. Cancer Cell 2004, 5, 423–428. [Google Scholar] [CrossRef] [Green Version]
- Everett, H.; McFadden, G. Viruses and apoptosis: Meddling with mitochondria. Virology 2001, 288, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Yoshimori, M.; Imadome, K.; Komatsu, H.; Wang, L.; Saitoh, Y.; Yamaoka, S.; Fukuda, T.; Kurata, M.; Koyama, T.; Shimizu, N.; et al. CD137 expression is induced by Epstein-Barr virus infection through LMP1 in T or NK cells and mediate s survival promoting signals. PLoS ONE 2014, 9, e112564. [Google Scholar] [CrossRef]
- Vidard, L.; Dureuil, C.; Baudhuin, J.; Vescovi, L.; Durand, L.; Sierra, V.; Parmantier, E. CD137 (4-1BB) Engagement Fine-Tunes Synergistic IL-15- and IL-21-Driven NK Cell Proliferation. J. Immunol. 2019, 203, 676–685. [Google Scholar] [CrossRef]
- Makowska, A.; Braunschweig, T.; Denecke, B.; Shen, L.; Baloche, V.; Busson, P.; Kontny, U. Interferon β and Anti-PD-1/PD-L1 Checkpoint Blockade Cooperate in NK Cell-Mediated Killing of Nasopharyngeal Carcinoma Cells. Transl. Oncol. 2019, 12, 1237–1256. [Google Scholar] [CrossRef]
- Shah, K.M.; Stewart, S.E.; Wei, W.; Woodman, C.B.; O’Neil, J.D.; Dawson, C.W.; Young, L.S. The EBV-encoded latent membrane proteins, LMP2A and LMP2B, limit the actions of interferon by targeting interferon receptors for degradation. Oncogene 2009, 28, 3903–3914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, T.S.; Chen, S.; Zhang, M.J.; Chan, J.Y.; Gao, W. Epstein-Barr virus-encoded microRNA BART7 downregulates major histocompatibility complex class I chain-related peptide A and reduces the cytotoxicity of natural killer cells to nasopharyngeal carcinoma. Oncol. Lett. 2018, 16, 2887–2892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strowig, T.; Brilot, F.; Arrey, F.; Bougras, G.; Thomas, D.; Muller, W.A.; Münz, C. Tonsilar NK cells restrict B cell transformation by the Epstein-Barr virus via IFN-gamma. PLoS Pathog. 2008, 4, e27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Png, Y.T.; Yang, A.Z.Y.; Lee, M.Y.; Chua, M.J.M.; Lim, C.M. The Role of NK Cells in EBV Infection and EBV-Associated NPC. Viruses 2021, 13, 300. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.S.; Lin, H.; Choy, D.T.; Sham, J.S.; Wei, W.; Chan, K.H.; Ng, M.H. Nasopharyngeal carcinoma and lymphoinfiltration. Oncology 1991, 48, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.Y.; Sakakibara, S.; Yasui, T.; Minamitani, T.; Okuzaki, D.; Kikutani, H. Bystander inhibition of humoral immune responses by Epstein-Barr virus LMP1. Int. Immunol. 2018, 30, 579–590. [Google Scholar] [CrossRef]
- Miao, B.P.; Zhang, R.S.; Li, M.; Fu, Y.T.; Zhao, M.; Liu, Z.G.; Yang, P.C. Nasopharyngeal cancer-derived microRNA-21 promotes immune suppressive B cells. Cell Mol. Immunol. 2015, 12, 750–756. [Google Scholar] [CrossRef]
- Wu, X.; Zhou, Z.; Xu, S.; Liao, C.; Chen, X.; Li, B.; Peng, J.; Li, D.; Yang, L. Extracellular vesicle packaged LMP1-activated fibroblasts promote tumor progression via autophagy and stroma-tumor metabolism coupling. Cancer Lett. 2020, 478, 93–106. [Google Scholar] [CrossRef]
- Davis, A.M.; Rapley, A.; Dawson, C.W.; Young, L.S.; Morris, M.A. The EBV-Encoded Oncoprotein, LMP1, Recruits and Transforms Fibroblasts via an ERK-MAPK-Dependent Mechanism. Pathogens 2021, 10, 982. [Google Scholar] [CrossRef]
- Yamamura, Y.; Asai, N.; Enomoto, A.; Kato, T.; Mii, S.; Kondo, Y.; Ushida, K.; Niimi, K.; Tsunoda, N.; Nagino, M.; et al. Akt-Girdin signaling in cancer-associated fibroblasts contributes to tumor progression. Cancer Res. 2015, 75, 813–823. [Google Scholar] [CrossRef] [Green Version]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Niu, Y.; Yeh, S.; Chang, C. BM-MSCs promote prostate cancer progression via the conversion of normal fibroblasts to cancer-associated fibroblasts. Int. J. Oncol. 2015, 47, 719–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michiels, C. Endothelial cell functions. J. Cell. Physiol. 2003, 196, 430–443. [Google Scholar] [CrossRef] [PubMed]
- Murono, S.; Inoue, H.; Tanabe, T.; Joab, I.; Yoshizaki, T.; Furukawa, M.; Pagano, J.S. Induction of cyclooxygenase-2 by Epstein-Barr virus latent membrane protein 1 is involved in vascular endothelial growth factor production in nasopharyngeal carcinoma cells. Proc. Natl. Acad. Sci. USA 2001, 98, 6905–6910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.; Li, Z.; He, J.; Fu, S.; Duan, Y.; Zhou, Q.; Yan, Y.; Liu, X.; Liu, L.; Feng, C.; et al. Epstein-Barr virus noncoding RNAs from the extracellular vesicles of nasopharyngeal carcinoma (NPC) cells promote angiogenesis via TLR3/RIG-I-mediated VCAM-1 expression. Biochimica et biophysica acta. Mol. Basis Dis. 2019, 1865, 1201–1213. [Google Scholar] [CrossRef]
- Farina, A.; Rosato, E.; York, M.; Gewurz, B.E.; Trojanowska, M.; Farina, G.A. Innate Immune Modulation Induced by EBV Lytic Infection Promotes Endothelial Cell Inflammation and Vascular Injury in Scleroderma. Front. Immunol. 2021, 12, 651013. [Google Scholar] [CrossRef]
- Maggio, E.; van den Berg, A.; Diepstra, A.; Kluiver, J.; Visser, L.; Poppema, S. Chemokines, cytokines and their receptors in Hodgkin’s lymphoma cell lines and tissues. Ann. Oncol. 2002, 13 (Suppl. 1), 52–56. [Google Scholar] [CrossRef]
- Liu, M.; Guo, S.; Stiles, J.K. The emerging role of CXCL10 in cancer (Review). Oncol. Lett. 2011, 2, 583–589. [Google Scholar] [CrossRef] [Green Version]
- Vockerodt, M.; Pinkert, D.; Smola-Hess, S.; Michels, A.; Ransohoff, R.M.; Tesch, H.; Kube, D. The Epstein-Barr virus oncoprotein latent membrane protein 1 induces expression of the chemokine IP-10: Importance of mRNA half-life regulation. Int. J. Cancer 2005, 114, 598–605. [Google Scholar] [CrossRef]
- Gong, L.P.; Chen, J.N.; Xiao, L.; He, Q.; Feng, Z.Y.; Zhang, Z.G.; Liu, J.P.; Wei, H.B.; Shao, C.K. The implication of tumor-infiltrating lymphocytes in Epstein-Barr virus-associated gastric carcinoma. Hum. Pathol. 2019, 85, 82–91. [Google Scholar] [CrossRef]
- Chen, L.C.; Wang, L.J.; Tsang, N.M.; Ojcius, D.M.; Chen, C.C.; Ouyang, C.N.; Hsueh, C.; Liang, Y.; Chang, K.P.; Chen, C.C.; et al. Tumour inflammasome-derived IL-1β recruits neutrophils and improves local recurrence-free survival in EBV-induced nasopharyngeal carcinoma. EMBO Mol. Med. 2012, 4, 1276–1293. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.C.; Lay, J.D.; Chuang, S.E.; Hsieh, W.C.; Chang, Y.; Su, I.J. Epstein-Barr virus (EBV) latent membrane protein-1 down-regulates tumor necrosis factor-alpha (TNF-alpha) receptor-1 and confers resistance to TNF-alpha-induced apoptosis in T cells: Implication for the progression to T-cell lymphoma in EBV-associated hemophagocytic syndrome. Am. J. Pathol. 2007, 170, 1607–1617. [Google Scholar] [PubMed] [Green Version]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; Yang, J.; Li, M.; Huang, N.; Chen, Y.; Wu, X.; Wang, X.; Qiu, S.; Wang, H.; Li, X. Viral IL-10 promotes cell proliferation and cell cycle progression via JAK2/STAT3 signaling pathway in nasopharyngeal carcinoma cells. Biotechnol. Appl. Biochem. 2020, 67, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Roncarolo, M.G.; Bacchetta, R.; Bordignon, C.; Narula, S.; Levings, M.K. Type 1 T regulatory cells. Immunol. Rev. 2001, 182, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Pachnia, D.; Drop, B.; Dworzańska, A.; Kliszczewska, E.; Polz-Dacewicz, M. Transforming Growth Factor-β, Interleukin-10, and Serological Markers in EBV-associated Gastric Carcinoma. Anticancer Res. 2017, 37, 4853–4858. [Google Scholar] [PubMed]
- Yao, M.; Ohshima, K.; Suzumiya, J.; Kume, T.; Shiroshita, T.; Kikuchi, M. Interleukin-10 expression and cytotoxic-T-cell response in Epstein-Barr-virus-associated nasopharyngeal carcinoma. Int. J. Cancer 1997, 72, 398–402. [Google Scholar] [CrossRef]
- Incrocci, R.; Barse, L.; Stone, A.; Vagvala, S.; Montesano, M.; Subramaniam, V.; Swanson-Mungerson, M. Epstein-Barr Virus Latent Membrane Protein 2A (LMP2A) enhances IL-10 production through the activation of Bruton’s tyrosine kinase and STAT3. Virology 2017, 500, 96–102. [Google Scholar] [CrossRef]
- Sakamoto, T.; Saito, H.; Tatebe, S.; Tsujitani, S.; Ozaki, M.; Ito, H.; Ikeguchi, M. Interleukin-10 expression significantly correlates with minor CD8+ T-cell infiltration and high microvessel density in patients with gastric cancer. Int. J. Cancer 2006, 118, 1909–1914. [Google Scholar] [CrossRef] [Green Version]
- Fior, R.; Vita, N.; Raphael, M.; Minty, A.; Maillot, M.C.; Crevon, M.C.; Caput, D.; Biberfeld, P.; Ferrara, P.; Galanaud, P. Interleukin-13 gene expression by malignant and EBV-transformed human B lymphocytes. Eur. Cytokine Netw. 1994, 5, 593–600. [Google Scholar]
- Kis, L.L.; Gerasimcik, N.; Salamon, D.; Persson, E.K.; Nagy, N.; Klein, G.; Severinson, E.; Klein, E. STAT6 signaling pathway activated by the cytokines IL-4 and IL-13 induces expression of the Epstein-B arr virus-encoded protein LMP-1 in absence of EBNA-2: Implications for the type II EBV latent gene expression in Hodgkin lymphoma. Blood 2011, 117, 165–174. [Google Scholar] [CrossRef]
- Eliopoulos, A.G.; Stack, M.; Dawson, C.W.; Kaye, K.M.; Hodgkin, L.; Sihota, S.; Rowe, M.; Young, L.S. Epstein-Barr virus-encoded LMP1 and CD40 mediate IL-6 production in epithelial cells via an NF-kappaB pathway involving TNF receptor-associated factors. Oncogene 1997, 14, 2899–2916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eliopoulos, A.G.; Gallagher, N.J.; Blake, S.M.; Dawson, C.W.; Young, L.S. Activation of the p38 mitogen-activated protein kinase pathway by Epstein-Barr virus-encoded latent membrane protein 1 coregulates interleukin-6 and interleukin-8 production. J. Biol. Chem. 1999, 274, 16085–16096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, J.; Juszczynski, P.; Rodig, S.J.; Green, M.R.; O’Donnell, E.; Currie, T.; Armant, M.; Takeyama, K.; Monti, S.; Rabinovich, G.A.; et al. Viral induction and targeted inhibition of galectin-1 in EBV+ posttransplant lymphoproliferative disorders. Blood 2011, 117, 4315–4322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhang, W. Expression of PD-L1 in EBV-associated malignancies. Int. Immunopharmacol. 2021, 95, 107553. [Google Scholar] [CrossRef]
- Tsirigotis, P.; Savani, B.N.; Nagler, A. Programmed death-1 immune checkpoint blockade in the treatment of hematological malignancies. Ann. Med. 2016, 48, 428–439. [Google Scholar] [CrossRef]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Green, M.R.; Rodig, S.; Juszczynski, P.; Ouyang, J.; Sinha, P.; O’Donnell, E.; Neuberg, D.; Shipp, M.A. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: Implications for targeted therapy. Clin. Cancer Res. 2012, 18, 1611–1618. [Google Scholar] [CrossRef] [Green Version]
- Gilardini Montani, M.S.; Santarelli, R.; Falcinelli, L.; Gonnella, R.; Granato, M.; Di Renzo, L.; Cuomo, L.; Vitillo, M.; Faggioni, A.; Cirone, M. EBV up-regulates PD-L1 on the surface of primary monocytes by increasing ROS and activating TLR signa ling and STAT3. J. Leukoc. Biol. 2018, 104, 821–832. [Google Scholar] [CrossRef]
- Sasaki, S.; Nishikawa, J.; Sakai, K.; Iizasa, H.; Yoshiyama, H.; Yanagihara, M.; Shuto, T.; Shimokuri, K.; Kanda, T.; Suehiro, Y.; et al. EBV-associated gastric cancer evades T-cell immunity by PD-1/PD-L1 interactions. Gastric Cancer 2019, 22, 486–496. [Google Scholar] [CrossRef] [Green Version]
- Greenough, T.C.; Campellone, S.C.; Brody, R.; Jain, S.; Sanchez-Merino, V.; Somasundaran, M.; Luzuriaga, K. Programmed Death-1 expression on Epstein Barr virus specific CD8+ T cells varies by stage of infection, epitope specificity, and T-cell receptor usage. PLoS ONE 2010, 5, e12926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M.M.; Gebriel, M.G.; Morad, E.A.; Saber, I.M.; Elwan, A.; Salah, M.; Fakhr, A.E.; Shalaby, A.M.; Alabiad, M.A. Expression of Immune Checkpoint Regulators, Cytotoxic T-Lymphocyte Antigen-4, and Programmed Death-Ligand 1 in Epstein-Barr Virus-associated Nasopharyngeal Carcinoma. Appl. Immunohistochem. Mol. Morphol. AIMM 2021, 29, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Zhang, Y.; Jiang, W.; Chen, Y.P.; Xu, S.Y.; Liu, N.; Zhao, Y.; Li, L.; Lei, Y.; Hong, X.H.; et al. Development and validation of an immune checkpoint-based signature to predict prognosis in nasopharyngeal carcinoma using computational pathology analysis. J. Immunother. Cancer 2019, 7, 298. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, M.K.; Lambley, E.; Duraiswamy, J.; Dua, U.; Smith, C.; Elliott, S.; Gill, D.; Marlton, P.; Seymour, J.; Khanna, R. Expression of LAG-3 by tumor-infiltrating lymphocytes is coincident with the suppression of latent membrane antigen-specific CD8+ T-cell function in Hodgkin lymphoma patients. Blood 2006, 108, 2280–2289. [Google Scholar] [CrossRef] [PubMed]
- Meckes, D.G. Exosomal communication goes viral. J. Virol. 2015, 89, 5200–5203. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R. The biology and function of exosomes in cancer. J. Clin. Investig. 2016, 126, 1208–1215. [Google Scholar] [CrossRef]
- Cone, A.S.; York, S.B.; Meckes, D.G. Extracellular Vesicles in Epstein-Barr Virus Pathogenesis. Curr. Clin. Microbiol. Rep. 2019, 6, 121–131. [Google Scholar] [CrossRef]
- Meckes, D.G.; Raab-Traub, N. Microvesicles and viral infection. J. Virol. 2011, 85, 12844–12854. [Google Scholar] [CrossRef] [Green Version]
- Nanbo, A.; Kawanishi, E.; Yoshida, R.; Yoshiyama, H. Exosomes derived from Epstein-Barr virus-infected cells are internalized via caveola-dependent endocytosis and promote phenotypic modulation in target cells. J. Virol. 2013, 87, 10334–10347. [Google Scholar] [CrossRef] [Green Version]
- Gutzeit, C.; Nagy, N.; Gentile, M.; Lyberg, K.; Gumz, J.; Vallhov, H.; Puga, I.; Klein, E.; Gabrielsson, S.; Cerutti, A.; et al. Exosomes derived from Burkitt’s lymphoma cell lines induce proliferation, differentiation, and class- switch recombination in B cells. J. Immunol. 2014, 192, 5852–5862. [Google Scholar] [CrossRef] [Green Version]
- Fang, W.; Zhang, J.; Hong, S.; Zhan, J.; Chen, N.; Qin, T.; Tang, Y.; Zhang, Y.; Kang, S.; Zhou, T.; et al. EBV-driven LMP1 and IFN-γ up-regulate PD-L1 in nasopharyngeal carcinoma: Implications for oncotargeted therapy. Oncotarget 2014, 5, 12189–12202. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, S.; Visco, V.; Raffa, S.; Wakisaka, N.; Pagano, J.S.; Torrisi, M.R. Epstein-Barr virus latent membrane protein 1 promotes concentration in multivesicular bodies of fibroblast growth factor 2 and its release through exosomes. Int. J. Cancer 2007, 121, 1494–1506. [Google Scholar] [CrossRef] [PubMed]
- Meckes, D.G.; Shair, K.H.; Marquitz, A.R.; Kung, C.P.; Edwards, R.H.; Raab-Traub, N. Human tumor virus utilizes exosomes for intercellular communication. Proc. Natl. Acad. Sci. USA 2010, 107, 20370–20375. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, M.; Longnecker, R. Cholesterol is critical for Epstein-Barr virus latent membrane protein 2A trafficking and protein stability. Virology 2007, 360, 461–468. [Google Scholar] [CrossRef]
- Cen, O.; Longnecker, R. Latent Membrane Protein 2 (LMP2). Curr. Top. Microbiol. Immunol. 2015, 391, 151–180. [Google Scholar]
- Ahmed, W.; Tariq, S.; Khan, G. Tracking EBV-encoded RNAs (EBERs) from the nucleus to the excreted exosomes of B-lymphocytes. Sci. Rep. 2018, 8, 15438. [Google Scholar] [CrossRef]
- Iwakiri, D. Epstein-Barr Virus-Encoded RNAs: Key Molecules in Viral Pathogenesis. Cancers 2014, 6, 1615–1630. [Google Scholar] [CrossRef] [Green Version]
- Takada, K. Role of EBER and BARF1 in nasopharyngeal carcinoma (NPC) tumorigenesis. Semin. Cancer Biol. 2012, 22, 162–165. [Google Scholar] [CrossRef]
- Iwakiri, D.; Zhou, L.; Samanta, M.; Matsumoto, M.; Ebihara, T.; Seya, T.; Imai, S.; Fujieda, M.; Kawa, K.; Takada, K. Epstein-Barr virus (EBV)-encoded small RNA is released from EBV-infected cells and activates signaling from Toll-like receptor 3. J. Exp. Med. 2009, 206, 2091–2099. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Meckes, D.G. Methodological Approaches to Study Extracellular Vesicle miRNAs in Epstein–Barr Virus-Associated Cancers. Int. J. Mol. Sci. 2018, 19, 2810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.J.; Huang, T.J.; Yang, C.F.; Peng, L.X.; Liu, R.Y.; Yang, G.D.; Chu, Q.Q.; Huang, J.L.; Liu, N.; Huang, H.B.; et al. Comprehensive profiling of Epstein-Barr virus-encoded miRNA species associated with specific latency types in tumor cells. Virol. J. 2013, 10, 314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tagawa, T.; Albanese, M.; Bouvet, M.; Moosmann, A.; Mautner, J.; Heissmeyer, V.; Zielinski, C.; Lutter, D.; Hoser, J.; Hastreiter, M.; et al. Epstein-Barr viral miRNAs inhibit antiviral CD4+ T cell responses targeting IL-12 and peptide processing. J. Exp. Med. 2016, 213, 2065–2080. [Google Scholar] [CrossRef] [PubMed]
- Toscano, M.A.; Bianco, G.A.; Ilarregui, J.M.; Croci, D.O.; Correale, J.; Hernandez, J.D.; Zwirner, N.W.; Poirier, F.; Riley, E.M.; Baum, L.G.; et al. Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nat. Immunol. 2007, 8, 825–834. [Google Scholar] [CrossRef]
- Perone, M.J.; Larregina, A.T.; Shufesky, W.J.; Papworth, G.D.; Sullivan, M.L.; Zahorchak, A.F.; Stolz, D.B.; Baum, L.G.; Watkins, S.C.; Thomson, A.W.; et al. Transgenic galectin-1 induces maturation of dendritic cells that elicit contrasting responses in naive and activated T cells. J. Immunol. 2006, 176, 7207–7220. [Google Scholar] [CrossRef] [Green Version]
- Keryer-Bibens, C.; Pioche-Durieu, C.; Villemant, C.; Souquère, S.; Nishi, N.; Hirashima, M.; Middeldorp, J.; Busson, P. Exosomes released by EBV-infected nasopharyngeal carcinoma cells convey the viral latent membrane protein 1 and the immunomodulatory protein galectin 9. BMC Cancer 2006, 6, 283. [Google Scholar] [CrossRef] [Green Version]
- Klibi, J.; Niki, T.; Riedel, A.; Pioche-Durieu, C.; Souquere, S.; Rubinstein, E.; Le Moulec, S.; Moulec, S.L.; Guigay, J.; Hirashima, M.; et al. Blood diffusion and Th1-suppressive effects of galectin-9-containing exosomes released by Epstein-Bar r virus-infected nasopharyngeal carcinoma cells. Blood 2009, 113, 1957–1966. [Google Scholar] [CrossRef] [Green Version]
- Mrizak, D.; Martin, N.; Barjon, C.; Jimenez-Pailhes, A.S.; Mustapha, R.; Niki, T.; Guigay, J.; Pancré, V.; de Launoit, Y.; Busson, P.; et al. Effect of nasopharyngeal carcinoma-derived exosomes on human regulatory T cells. J. Natl. Cancer Inst. 2015, 107, 363. [Google Scholar] [CrossRef] [Green Version]
- Ramayanti, O.; Verkuijlen, S.; Novianti, P.; Scheepbouwer, C.; Misovic, B.; Koppers-Lalic, D.; van Weering, J.; Beckers, L.; Adham, M.; Martorelli, D.; et al. Vesicle-bound EBV-BART13-3p miRNA in circulation distinguishes nasopharyngeal from other head and neck cancer and asymptomatic EBV-infections. Int. J. Cancer 2019, 144, 2555–2566. [Google Scholar] [CrossRef]
- Tsai, C.Y.; Liu, Y.Y.; Liu, K.H.; Hsu, J.T.; Chen, T.C.; Chiu, C.T.; Yeh, T.S. Comprehensive profiling of virus microRNAs of Epstein-Barr virus-associated gastric carcinoma: Highlighting the interactions of ebv-Bart9 and host tumor cells. J. Gastroenterol. Hepatol. 2017, 32, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Xiang, Z.; Liu, Y.; Huang, C.; Pei, Y.; Wang, X.; Zhi, H.; Wong, W.H.; Wei, H.; Ng, I.O.; et al. Exosomes derived from Vδ2-T cells control Epstein-Barr virus-associated tumors and induce T cell antitumor immunity. Sci. Transl. Med. 2020, 12, eaaz3426. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Xie, Y.; Wang, T.; Wang, L. New insights into Epstein-Barr virus-associated tumors: Exosomes (Review). Oncol. Rep. 2022, 47, 13. [Google Scholar] [CrossRef] [PubMed]
- Morscio, J.; Dierickx, D.; Tousseyn, T. Molecular pathogenesis of B-cell posttransplant lymphoproliferative disorder: What do we know so far? Clin. Dev. Immunol. 2013, 2013, 150835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyauchi, K.; Urano, E.; Yoshiyama, H.; Komano, J. Cytokine signatures of transformed B cells with distinct Epstein-Barr virus latencies as a potential diagnostic tool for B cell lymphoma. Cancer Sci. 2011, 102, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Vaysberg, M.; Lambert, S.L.; Krams, S.M.; Martinez, O.M. Activation of the JAK/STAT pathway in Epstein Barr virus+-associated posttransplant lymphoproliferative disease: Role of interferon-gamma. Am. J. Transplant. 2009, 9, 2292–2302. [Google Scholar] [CrossRef] [Green Version]
- Rowe, M.; Peng-Pilon, M.; Huen, D.S.; Hardy, R.; Croom-Carter, D.; Lundgren, E.; Rickinson, A.B. Upregulation of bcl-2 by the Epstein-Barr virus latent membrane protein LMP1: A B-cell-specific response that is delayed relative to NF-kappa B activation and to induction of cell surface markers. J. Virol. 1994, 68, 5602–5612. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Kim, W.S.; Park, C. Epstein-Barr virus latent membrane protein-1 protects B-cell lymphoma from rituximab-induced apoptosis through miR-155-mediated Akt activation and up-regulation of Mcl-1. Leuk. Lymphoma 2012, 53, 1586–1591. [Google Scholar] [CrossRef]
- Incrocci, R.; McCormack, M.; Swanson-Mungerson, M. Epstein-Barr virus LMP2A increases IL-10 production in mitogen-stimulated primary B-cells and B-cell lymphomas. J. Gen. Virol. 2013, 94 (Pt 5), 1127–1133. [Google Scholar] [CrossRef] [Green Version]
- Campion, E.M.; Hakimjavadi, R.; Loughran, S.T.; Phelan, S.; Smith, S.M.; D’Souza, B.N.; Tierney, R.J.; Bell, A.I.; Cahill, P.A.; Walls, D. Repression of the proapoptotic cellular BIK/NBK gene by Epstein-Barr virus antagonizes transforming growth factor β1-induced B-cell apoptosis. J. Virol. 2014, 88, 5001–5013. [Google Scholar] [CrossRef] [Green Version]
- Hong, G.K.; Gulley, M.L.; Feng, W.H.; Delecluse, H.J.; Holley-Guthrie, E.; Kenney, S.C. Epstein-Barr virus lytic infection contributes to lymphoproliferative disease in a SCID mouse model. J. Virol. 2005, 79, 13993–14003. [Google Scholar] [CrossRef] [Green Version]
- Perera, S.M.; Thomas, J.A.; Burke, M.; Crawford, D.H. Analysis of the T-cell micro-environment in Epstein-Barr virus-related post-transplantation B lymphoproliferative disease. J. Pathol. 1998, 184, 177–184. [Google Scholar] [CrossRef]
- Cui, X.; Snapper, C.M. Epstein Barr Virus: Development of Vaccines and Immune Cell Therapy for EBV-Associated Diseases. Front. Immunol. 2021, 12, 734471. [Google Scholar] [CrossRef] [PubMed]
- Rooney, C.M.; Smith, C.A.; Ng, C.Y.; Loftin, S.; Li, C.; Krance, R.A.; Brenner, M.K.; Heslop, H.E. Use of gene-modified virus-specific T lymphocytes to control Epstein-Barr-virus-related lymphoproliferation. Lancet 1995, 345, 9–13. [Google Scholar] [CrossRef]
- Comoli, P.; Labirio, M.; Basso, S.; Baldanti, F.; Grossi, P.; Furione, M.; Viganò, M.; Fiocchi, R.; Rossi, G.; Ginevri, F.; et al. Infusion of autologous Epstein-Barr virus (EBV)-specific cytotoxic T cells for prevention of EBV-related lymphoproliferative disorder in solid organ transplant recipients with evidence of active virus replication. Blood 2002, 99, 2592–2598. [Google Scholar] [CrossRef] [Green Version]
- Aldinucci, D.; Gloghini, A.; Pinto, A.; De Filippi, R.; Carbone, A. The classical Hodgkin’s lymphoma microenvironment and its role in promoting tumour growth and immune escape. J. Pathol. 2010, 221, 248–263. [Google Scholar] [CrossRef]
- Küppers, R. The biology of Hodgkin’s lymphoma. Nat. Rev. Cancer 2009, 9, 15–27. [Google Scholar] [CrossRef]
- Skinnider, B.F.; Mak, T.W. The role of cytokines in classical Hodgkin lymphoma. Blood 2002, 99, 4283–4297. [Google Scholar] [CrossRef] [Green Version]
- Aldinucci, D.; Lorenzon, D.; Cattaruzza, L.; Pinto, A.; Gloghini, A.; Carbone, A.; Colombatti, A. Expression of CCR5 receptors on Reed-Sternberg cells and Hodgkin lymphoma cell lines: Involvement of CCL5/Rantes in tumor cell growth and microenvironmental interactions. Int. J. Cancer 2008, 122, 769–776. [Google Scholar] [CrossRef]
- Shannon-Lowe, C.; Rickinson, A.B.; Bell, A.I. Epstein-Barr virus-associated lymphomas. Philos. Trans. R Soc. Lond B Biol. Sci. 2017, 372, 20160271. [Google Scholar] [CrossRef]
- Roemer, M.G.; Advani, R.H.; Ligon, A.H.; Natkunam, Y.; Redd, R.A.; Homer, H.; Connelly, C.F.; Sun, H.H.; Daadi, S.E.; Freeman, G.J.; et al. PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. J. Clin. Oncol. 2016, 34, 2690–2697. [Google Scholar] [CrossRef] [Green Version]
- Bollard, C.M.; Gottschalk, S.; Torrano, V.; Diouf, O.; Ku, S.; Hazrat, Y.; Carrum, G.; Ramos, C.; Fayad, L.; Shpall, E.J.; et al. Sustained complete responses in patients with lymphoma receiving autologous cytotoxic T lymphocytes targeting Epstein-Barr virus latent membrane proteins. J. Clin. Oncol. 2014, 32, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, D.; Takada, K. Role of EBERs in the pathogenesis of EBV infection. Adv. Cancer Res. 2010, 107, 119–136. [Google Scholar]
- Tsimbouri, P.; Al-Sheikh, Y.; Drotar, M.E.; Cushley, W.; Wilson, J.B. Epstein-Barr virus nuclear antigen-1 renders lymphocytes responsive to IL-2 but not IL-15 for survival. J. Gen. Virol. 2008, 89 (Pt 11), 2821–2832. [Google Scholar] [CrossRef] [PubMed]
- Granai, M.; Mundo, L.; Akarca, A.U.; Siciliano, M.C.; Rizvi, H.; Mancini, V.; Onyango, N.; Nyagol, J.; Abinya, N.O.; Maha, I.; et al. Immune landscape in Burkitt lymphoma reveals M2-macrophage polarization and correlation between PD-L1 expression and non-canonical EBV latency program. Infect. Agents Cancer 2020, 15, 28. [Google Scholar] [CrossRef] [PubMed]
- Crombie, J.; LaCasce, A. The treatment of Burkitt lymphoma in adults. Blood 2021, 137, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Que, Y.; Wang, J.; Zhu, J.; Li, N.; Huang, J.; Lu, S.; Sun, F.; Zhang, L.; Zhen, Z.; Zhang, L.; et al. Combination Therapy With Anti-PD-1 or PD-1 Antibody Alone in Asian Pediatric Patients With Relapsed or Refractory Cancer. Front. Immunol. 2021, 12, 647733. [Google Scholar] [CrossRef]
- Heslop, H.E. Biology and treatment of Epstein-Barr virus-associated non-Hodgkin lymphomas. Hematol. Am. Soc. Hematol. Educ. Program 2005, 260–266. [Google Scholar] [CrossRef] [Green Version]
- Ciavarella, S.; Vegliante, M.C.; Fabbri, M.; De Summa, S.; Melle, F.; Motta, G.; De Iuliis, V.; Opinto, G.; Enjuanes, A.; Rega, S.; et al. Dissection of DLBCL microenvironment provides a gene expression-based predictor of survival applicabl e to formalin-fixed paraffin-embedded tissue. Ann. Oncol. 2018, 29, 2363–2370. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Pittaluga, S.; Jaffe, E.S. The histological classification of diffuse large B-cell lymphomas. Semin. Hematol. 2015, 52, 57–66. [Google Scholar] [CrossRef] [Green Version]
- Kanegane, H.; Nomura, K.; Miyawaki, T.; Tosato, G. Biological aspects of Epstein-Barr virus (EBV)-infected lymphocytes in chronic active EBV infection and associated malignancies. Crit. Rev. Oncol. Hematol. 2002, 44, 239–249. [Google Scholar] [CrossRef]
- Quan, L.; Chen, X.; Liu, A.; Zhang, Y.; Guo, X.; Yan, S.; Liu, Y. PD-1 Blockade Can Restore Functions of T-Cells in Epstein-Barr Virus-Positive Diffuse Large B-Cell Lymphoma In Vitro. PLoS ONE 2015, 10, e0136476. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.; Vistarop, A.G.; Huaman, F.; Narbaitz, M.; Metrebian, F.; De Matteo, E.; Preciado, M.V.; Chabay, P.A. Cytotoxic response against Epstein Barr virus coexists with diffuse large B-cell lymphoma tolerogenic microenvironment: Clinical features and survival impact. Sci. Rep. 2017, 7, 10813. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas, D.; Vélez, G.; Orfao, A.; Herrera, M.V.; Solano, J.; Olaya, M.; Uribe, A.M.; Saavedra, C.; Duarte, M.; Rodríguez, M.; et al. Epstein-Barr virus-specific CD8(+) T lymphocytes from diffuse large B cell lymphoma patients are functionally impaired. Clin. Exp. Immunol. 2015, 182, 173–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keane, C.; Tobin, J.; Gunawardana, J.; Francis, S.; Gifford, G.; Gabrielli, S.; Gill, A.; Stevenson, W.; Talaulikar, D.; Gould, C.; et al. The tumour microenvironment is immuno-tolerogenic and a principal determinant of patient outcome in E BV-positive diffuse large B-cell lymphoma. Eur. J. Haematol. 2019, 103, 200–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondello, P.; Mian, M. Frontline treatment of diffuse large B-cell lymphoma: Beyond R-CHOP. Hematol. Oncol. 2019, 37, 333–344. [Google Scholar] [CrossRef]
- Chavez, J.C.; Locke, F.L. CAR T cell therapy for B-cell lymphomas. Best Pract. Research. Clin. Haematol. 2018, 31, 135–146. [Google Scholar] [CrossRef]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef] [Green Version]
- Takahara, M.; Kis, L.L.; Nagy, N.; Liu, A.; Harabuchi, Y.; Klein, G.; Klein, E. Concomitant increase of LMP1 and CD25 (IL-2-receptor alpha) expression induced by IL-10 in the EBV-positive NK lines SNK6 and KAI3. Int. J. Cancer 2006, 119, 2775–2783. [Google Scholar] [CrossRef]
- Yoshino, K.; Kishibe, K.; Nagato, T.; Ueda, S.; Komabayashi, Y.; Takahara, M.; Harabuchi, Y. Expression of CD70 in nasal natural killer/T cell lymphoma cell lines and patients; its role for cell proliferation through binding to soluble CD27. Br. J. Haematol. 2013, 160, 331–342. [Google Scholar] [CrossRef]
- Chuang, H.C.; Lay, J.D.; Hsieh, W.C.; Su, I.J. Pathogenesis and mechanism of disease progression from hemophagocytic lymphohistiocytosis to Epstein- Barr virus-associated T-cell lymphoma: Nuclear factor-kappa B pathway as a potential therapeutic target. Cancer Sci. 2007, 98, 1281–1287. [Google Scholar] [CrossRef]
- Bi, X.W.; Wang, H.; Zhang, W.W.; Wang, J.H.; Liu, W.J.; Xia, Z.J.; Huang, H.Q.; Jiang, W.Q.; Zhang, Y.J.; Wang, L. PD-L1 is upregulated by EBV-driven LMP1 through NF-κB pathway and correlates with poor prognosis in natural killer/T-cell lymphoma. J. Hematol. Oncol. 2016, 9, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tse, E.; Kwong, Y.L. The diagnosis and management of NK/T-cell lymphomas. J. Hematol. Oncol. 2017, 10, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwong, Y.L.; Chan, T.S.Y.; Tan, D.; Kim, S.J.; Poon, L.M.; Mow, B.; Khong, P.L.; Loong, F.; Au-Yeung, R.; Iqbal, J.; et al. PD1 blockade with pembrolizumab is highly effective in relapsed or refractory NK/T-cell lymphoma failing l-asparaginase. Blood 2017, 129, 2437–2442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, E.T.; Adami, H.O. The enigmatic epidemiology of nasopharyngeal carcinoma. Cancer Epidemiol. Biomark. Prev. 2006, 15, 1765–1777. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.P.; Chan, A.T.C.; Le, Q.T.; Blanchard, P.; Sun, Y.; Ma, J. Nasopharyngeal carcinoma. Lancet 2019, 394, 64–80. [Google Scholar] [CrossRef]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein-Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef]
- Raab-Traub, N. Nasopharyngeal Carcinoma: An Evolving Role for the Epstein-Barr Virus. Curr. Top. Microbiol. Immunol. 2015, 390 (Pt 1), 339–363. [Google Scholar]
- Lin, D.C.; Meng, X.; Hazawa, M.; Nagata, Y.; Varela, A.M.; Xu, L.; Sato, Y.; Liu, L.Z.; Ding, L.W.; Sharma, A.; et al. The genomic landscape of nasopharyngeal carcinoma. Nat. Genet. 2014, 46, 866–871. [Google Scholar] [CrossRef]
- Lau, K.M.; Cheng, S.H.; Lo, K.W.; Lee, S.A.; Woo, J.K.; van Hasselt, C.A.; Lee, S.P.; Rickinson, A.B.; Ng, M.H. Increase in circulating Foxp3+CD4+CD25(high) regulatory T cells in nasopharyngeal carcinoma patients. Br. J. Cancer 2007, 96, 617–622. [Google Scholar] [CrossRef] [Green Version]
- Mai, H.Q.; Chen, Q.Y.; Chen, D.; Hu, C.; Yang, K.; Wen, J.; Li, J.; Shi, Y.R.; Jin, F.; Xu, R.; et al. Toripalimab or placebo plus chemotherapy as first-line treatment in advanced nasopharyngeal carcinoma: A multicenter randomized phase 3 trial. Nat. Med. 2021, 27, 1536–1543. [Google Scholar] [CrossRef]
- Li, F.; Song, D.; Lu, Y.; Zhu, H.; Chen, Z.; He, X. Delayed-type hypersensitivity (DTH) immune response related with EBV-DNA in nasopharyngeal carcinoma treated with autologous dendritic cell vaccination after radiotherapy. J. Immunother. 2013, 36, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Hui, E.P.; Taylor, G.S.; Jia, H.; Ma, B.B.; Chan, S.L.; Ho, R.; Wong, W.L.; Wilson, S.; Johnson, B.F.; Edwards, C.; et al. Phase I trial of recombinant modified vaccinia ankara encoding Epstein-Barr viral tumor antigens in nasopharyngeal carcinoma patients. Cancer Res. 2013, 73, 1676–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasari, V.; Sinha, D.; Neller, M.A.; Smith, C.; Khanna, R. Prophylactic and therapeutic strategies for Epstein-Barr virus-associated diseases: Emerging strategies for clinical development. Expert Rev. Vaccines 2019, 18, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Cristescu, R.; Lee, J.; Nebozhyn, M.; Kim, K.M.; Ting, J.C.; Wong, S.S.; Liu, J.; Yue, Y.G.; Wang, J.; Yu, K.; et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat. Med. 2015, 21, 449–456. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, S.H.; Han, S.H.; An, J.S.; Lee, E.S.; Kim, Y.S. Clinicopathological and molecular characteristics of Epstein-Barr virus-associated gastric carcinoma: A meta-analysis. J. Gastroenterol. Hepatol. 2009, 24, 354–365. [Google Scholar] [CrossRef]
- Fukayama, M.; Hino, R.; Uozaki, H. Epstein-Barr virus and gastric carcinoma: Virus-host interactions leading to carcinoma. Cancer Sci. 2008, 99, 1726–1733. [Google Scholar] [CrossRef]
- Fukayama, M.; Ushiku, T. Epstein-Barr virus-associated gastric carcinoma. Pathol. Res. Pract. 2011, 207, 529–537. [Google Scholar] [CrossRef]
- Fukayama, M.; Abe, H.; Kunita, A.; Shinozaki-Ushiku, A.; Matsusaka, K.; Ushiku, T.; Kaneda, A. Thirty years of Epstein-Barr virus-associated gastric carcinoma. Virchows Arch. 2020, 476, 353–365. [Google Scholar] [CrossRef]
- Fukayama, M.; Kunita, A.; Kaneda, A. Gastritis-Infection-Cancer Sequence of Epstein-Barr Virus-Associated Gastric Cancer. Adv. Exp. Med. Biol. 2018, 1045, 437–457. [Google Scholar]
- Murphy, G.; Pfeiffer, R.; Camargo, M.C.; Rabkin, C.S. Meta-analysis shows that prevalence of Epstein-Barr virus-positive gastric cancer differs based on sex and anatomic location. Gastroenterology 2009, 137, 824–833. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, J.; Oliveira, C.; Malta, M.; Sousa, H. Epstein-Barr virus gene expression and latency pattern in gastric carcinomas: A systematic review. Future Oncol. 2017, 13, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Park, C.; Kim, H.J.; Park, J.; Hwang, J.; Kim, J.I.; Choi, M.G.; Kim, S.; Kim, K.M.; Kang, M.S. Deregulation of immune response genes in patients with Epstein-Barr virus-associated gastric cancer and outcomes. Gastroenterology 2015, 148, 137–147.e9. [Google Scholar] [CrossRef] [PubMed]
- Song, H.J.; Srivastava, A.; Lee, J.; Kim, Y.S.; Kim, K.M.; Ki Kang, W.; Kim, M.; Kim, S.; Park, C.K.; Kim, S. Host inflammatory response predicts survival of patients with Epstein-Barr virus-associated gastric carcinoma. Gastroenterology 2010, 139, 84–92.e2. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Abe, H.; Kunita, A.; Yamashita, H.; Seto, Y.; Fukayama, M. Overexpression and gene amplification of PD-L1 in cancer cells and PD-L1+ immune cells in Epstein-Barr virus-associated gastric cancer: The prognostic implications. Mod. Pathol. 2017, 30, 427–439. [Google Scholar] [CrossRef]
- Zhang, N.N.; Chen, J.N.; Xiao, L.; Tang, F.; Zhang, Z.G.; Zhang, Y.W.; Feng, Z.Y.; Jiang, Y.; Shao, C.K. Accumulation Mechanisms of CD4(+)CD25(+)FOXP3(+) Regulatory T Cells in EBV-associated Gastric Carcinoma. Sci. Rep. 2015, 5, 18057. [Google Scholar] [CrossRef] [Green Version]
- Ichimura, T.; Abe, H.; Morikawa, T.; Yamashita, H.; Ishikawa, S.; Ushiku, T.; Seto, Y.; Fukayama, M. Low density of CD204-positive M2-type tumor-associated macrophages in Epstein-Barr virus-associated gastric cancer: A clinicopathologic study with digital image analysis. Hum. Pathol. 2016, 56, 74–80. [Google Scholar] [CrossRef]
- Strong, M.J.; Xu, G.; Coco, J.; Baribault, C.; Vinay, D.S.; Lacey, M.R.; Strong, A.L.; Lehman, T.A.; Seddon, M.B.; Lin, Z.; et al. Differences in gastric carcinoma microenvironment stratify according to EBV infection intensity: Implications for possible immune adjuvant therapy. PLoS Pathog. 2013, 9, e1003341. [Google Scholar] [CrossRef]
- Naseem, M.; Barzi, A.; Brezden-Masley, C.; Puccini, A.; Berger, M.D.; Tokunaga, R.; Battaglin, F.; Soni, S.; McSkane, M.; Zhang, W.; et al. Outlooks on Epstein-Barr virus associated gastric cancer. Cancer Treat. Rev. 2018, 66, 15–22. [Google Scholar] [CrossRef]
- Huang, Y.H.; Zhang, C.Z.; Huang, Q.S.; Yeong, J.; Wang, F.; Yang, X.; He, Y.F.; Zhang, X.L.; Zhang, H.; Chen, S.L.; et al. Clinicopathologic features, tumor immune microenvironment and genomic landscape of Epstein-Barr virus -associated intrahepatic cholangiocarcinoma. J. Hepatol. 2021, 74, 838–849. [Google Scholar] [CrossRef]
- Hussein, K.; Maecker-Kolhoff, B.; Donnerstag, F.; Laenger, F.; Kreipe, H.; Jonigk, D. Epstein-Barr virus-associated smooth muscle tumours after transplantation, infection with human immunodeficiency virus and congenital immunodeficiency syndromes. Pathobiology 2013, 80, 297–301. [Google Scholar] [CrossRef]
- Lee, E.S.; Locker, J.; Nalesnik, M.; Reyes, J.; Jaffe, R.; Alashari, M.; Nour, B.; Tzakis, A.; Dickman, P.S. The association of Epstein-Barr virus with smooth-muscle tumors occurring after organ transplantation. N. Engl. J. Med. 1995, 332, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Calafiore, R.; Mouchtouris, N.; Flomenberg, N.; Harrop, J.S. Epstein-Barr Virus-Associated Smooth Muscle Tumor of the Spine After Bone Marrow Transplant: Case Report and Review of Literature. World Neurosurg. 2020, 135, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Hussein, K.; Rath, B.; Ludewig, B.; Kreipe, H.; Jonigk, D. Clinico-pathological characteristics of different types of immunodeficiency-associated smooth muscle tumours. Eur. J. Cancer 2014, 50, 2417–2424. [Google Scholar] [CrossRef] [PubMed]
- Rogatsch, H.; Bonatti, H.; Menet, A.; Larcher, C.; Feichtinger, H.; Dirnhofer, S. Epstein-Barr virus-associated multicentric leiomyosarcoma in an adult patient after heart transplantation: Case report and review of the literature. Am. J. Surg. Pathol. 2000, 24, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Deyrup, A.T.; Lee, V.K.; Hill, C.E.; Cheuk, W.; Toh, H.C.; Kesavan, S.; Chan, E.W.; Weiss, S.W. Epstein-Barr virus-associated smooth muscle tumors are distinctive mesenchymal tumors reflecting multiple infection events: A clinicopathologic and molecular analysis of 29 tumors from 19 patients. Am. J. Surg. Pathol. 2006, 30, 75–82. [Google Scholar] [CrossRef]
- Creager, A.J.; Maia, D.M.; Funkhouser, W.K. Epstein-Barr virus-associated renal smooth muscle neoplasm: Report of a case with review of the literature. Arch. Pathol. Lab. Med. 1998, 122, 277–281. [Google Scholar]
- Chong, Y.B.; Lu, P.-L.; Ma, Y.-C.; Yin, H.-L.; Chang, C.-H. Epstein-Barr Virus-Associated Smooth Muscle Tumor and Its Correlation With CD4 Levels in a Patient With HIV Infection. Front. Cell. Infect. Microbiol. 2022, 12, 725342. [Google Scholar] [CrossRef]
- Chen, B.J.; Chapuy, B.; Ouyang, J.; Sun, H.H.; Roemer, M.G.; Xu, M.L.; Yu, H.; Fletcher, C.D.; Freeman, G.J.; Shipp, M.A.; et al. PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated malignancies. Clin. Cancer Res. 2013, 19, 3462–3473. [Google Scholar] [CrossRef] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Masterson, L.; Howard, J.; Gonzalez-Cruz, J.; Jackson, C.; Barnett, C.; Overton, L.; Liu, H.; Ladwa, R.; Simpson, F.; McGrath, M.; et al. Immune checkpoint inhibitors in advanced nasopharyngeal carcinoma: Beyond an era of chemoradiation? Int. J. Cancer 2020, 146, 2305–2314. [Google Scholar] [CrossRef]
- Burns, D.M.; Crawford, D.H. Epstein-Barr virus-specific cytotoxic T-lymphocytes for adoptive immunotherapy of post-transplant lymphoproliferative disease. Blood Rev. 2004, 18, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Burton, R.; Reddy, V.; Lucas, K.G. Safety of allogeneic Epstein-Barr virus (EBV)-specific cytotoxic T lymphocytes for patients with refractory EBV-related lymphoma. Br. J. Haematol. 2002, 118, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Poppema, S. Immunobiology and pathophysiology of Hodgkin lymphomas. Hematol. Am. Soc. Hematol. Educ. Program 2005, 231–238. [Google Scholar] [CrossRef] [Green Version]
- Duraiswamy, J.; Sherritt, M.; Thomson, S.; Tellam, J.; Cooper, L.; Connolly, G.; Bharadwaj, M.; Khanna, R. Therapeutic LMP1 polyepitope vaccine for EBV-associated Hodgkin disease and nasopharyngeal carcinoma. Blood 2003, 101, 3150–3156. [Google Scholar] [CrossRef] [Green Version]
- Münz, C.; Bickham, K.L.; Subklewe, M.; Tsang, M.L.; Chahroudi, A.; Kurilla, M.G.; Zhang, D.; O’Donnell, M.; Steinman, R.M. Human CD4(+) T lymphocytes consistently respond to the latent Epstein-Barr virus nuclear antigen EBNA 1. J. Exp. Med. 2000, 191, 1649–1660. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.P.; Tierney, R.J.; Thomas, W.A.; Brooks, J.M.; Rickinson, A.B. Conserved CTL epitopes within EBV latent membrane protein 2: A potential target for CTL-based tumor therapy. J. Immunol. 1997, 158, 3325–3334. [Google Scholar]
- Lin, C.L.; Lo, W.F.; Lee, T.H.; Ren, Y.; Hwang, S.L.; Cheng, Y.F.; Chen, C.L.; Chang, Y.S.; Lee, S.P.; Rickinson, A.B.; et al. Immunization with Epstein-Barr Virus (EBV) peptide-pulsed dendritic cells induces functional CD8+ T-c ell immunity and may lead to tumor regression in patients with EBV-positive nasopharyngeal carcinoma. Cancer Res. 2002, 62, 6952–6958. [Google Scholar]
- Si, Y.; Deng, Z.; Lan, G.; Du, H.; Wang, Y.; Si, J.; Wei, J.; Weng, J.; Qin, Y.; Huang, B.; et al. The Safety and Immunological Effects of rAd5-EBV-LMP2 Vaccine in Nasopharyngeal Carcinoma Patients: A Phase I Clinical Trial and Two-Year Follow-Up. Chem. Pharm. Bull. 2016, 64, 1118–1123. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Progress and distinguished gene expression profiles of B cells infected by EBV. The germinal center model is the widely held model to show how the virus enters memory B cells. Orally acquired virus particles may initially infect oropharyngeal B cells, resulting in EBV-transformed B cell growth through a growth program by expressing all the viral proteins (latency III), including EBNA1, EBNA2, EBNA3A-3C, EBNA-LP, LMP1, LPM2A, and LMP2B. Being highly immunogenic, these cells can be recognized and rapidly eliminated by host immunity. Then, the virus-infected B cells go through a physiologic germinal center reaction, by limiting protein expression of latency II (EBNA1, LMP1, LMP2A, and LMP2B). After that, B cells leave the germinal center and enter the memory B cell pool by shutting down all the viral genes (latency 0). When the viral genome replicates along with the cell, EBV-infected memory B cells need to express EBNA1 (latency I). The latently infected memory B cells occasionally reactivate and differentiate into plasma cells, which is associated with the initiation of the lytic program and production virions.
Figure 1.
Progress and distinguished gene expression profiles of B cells infected by EBV. The germinal center model is the widely held model to show how the virus enters memory B cells. Orally acquired virus particles may initially infect oropharyngeal B cells, resulting in EBV-transformed B cell growth through a growth program by expressing all the viral proteins (latency III), including EBNA1, EBNA2, EBNA3A-3C, EBNA-LP, LMP1, LPM2A, and LMP2B. Being highly immunogenic, these cells can be recognized and rapidly eliminated by host immunity. Then, the virus-infected B cells go through a physiologic germinal center reaction, by limiting protein expression of latency II (EBNA1, LMP1, LMP2A, and LMP2B). After that, B cells leave the germinal center and enter the memory B cell pool by shutting down all the viral genes (latency 0). When the viral genome replicates along with the cell, EBV-infected memory B cells need to express EBNA1 (latency I). The latently infected memory B cells occasionally reactivate and differentiate into plasma cells, which is associated with the initiation of the lytic program and production virions.

Figure 2.
Components of tumor microenvironment in EBV-associated malignancies. EBV-associated malignancies are characterized by the distinct components of the tumor microenvironment. To suppress the host’s immune system and evade immune surveillance, the EBV-infected malignant cells shape and establish an immunosuppressive tumor microenvironment by communicating the cellular components, altering the molecular factors, and releasing extracellular vesicles. TAMs, tumor-associated macrophages; CAFs, cancer-associated fibroblasts; MDSCs, myeloid-derived suppressor cells; DCs, dendritic cells.
Figure 2.
Components of tumor microenvironment in EBV-associated malignancies. EBV-associated malignancies are characterized by the distinct components of the tumor microenvironment. To suppress the host’s immune system and evade immune surveillance, the EBV-infected malignant cells shape and establish an immunosuppressive tumor microenvironment by communicating the cellular components, altering the molecular factors, and releasing extracellular vesicles. TAMs, tumor-associated macrophages; CAFs, cancer-associated fibroblasts; MDSCs, myeloid-derived suppressor cells; DCs, dendritic cells.

Figure 3.
Extracellular vesicles released by EBV-infected cells. The EBV-infected cells release exosomes (exos), containing different components, such as LMP1, LMP2, EBERs, miRNAs, galectin-1, galectin-9, and dUTPase. These extracellular vesicles interact with different immune cells to form a supportive and immunosuppressive tumor microenvironment.
Figure 3.
Extracellular vesicles released by EBV-infected cells. The EBV-infected cells release exosomes (exos), containing different components, such as LMP1, LMP2, EBERs, miRNAs, galectin-1, galectin-9, and dUTPase. These extracellular vesicles interact with different immune cells to form a supportive and immunosuppressive tumor microenvironment.


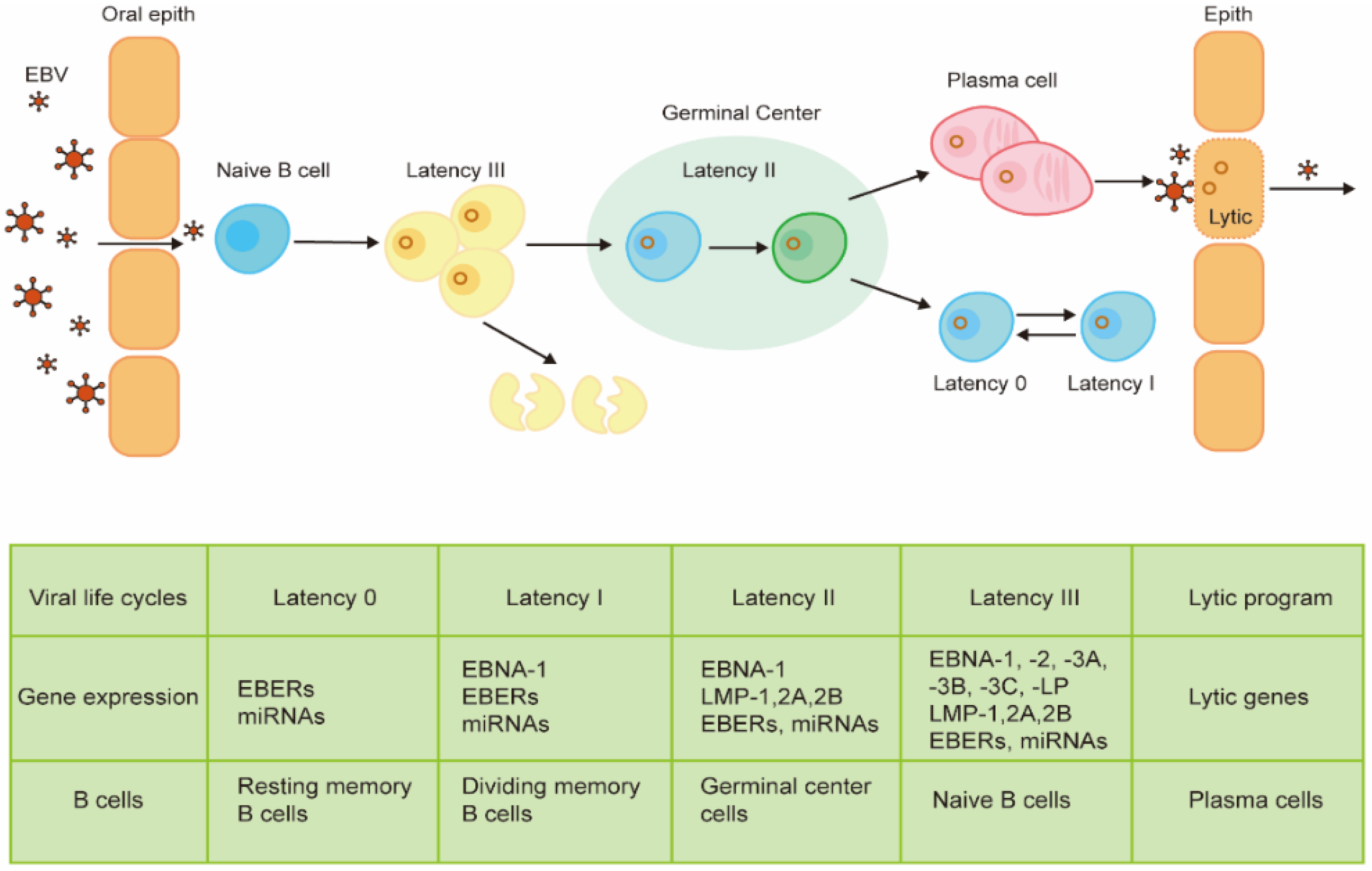{kind=link}
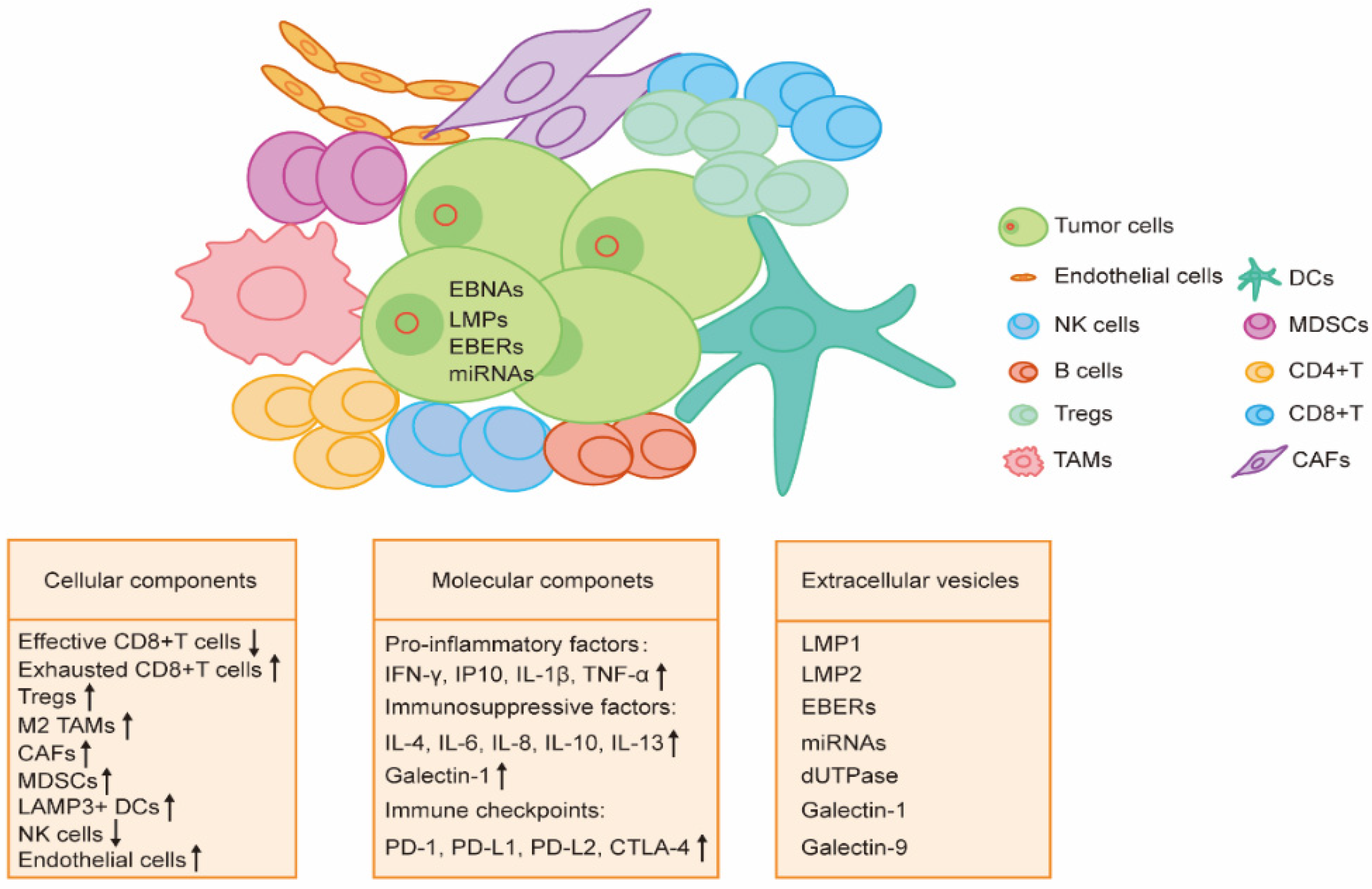{kind=link}
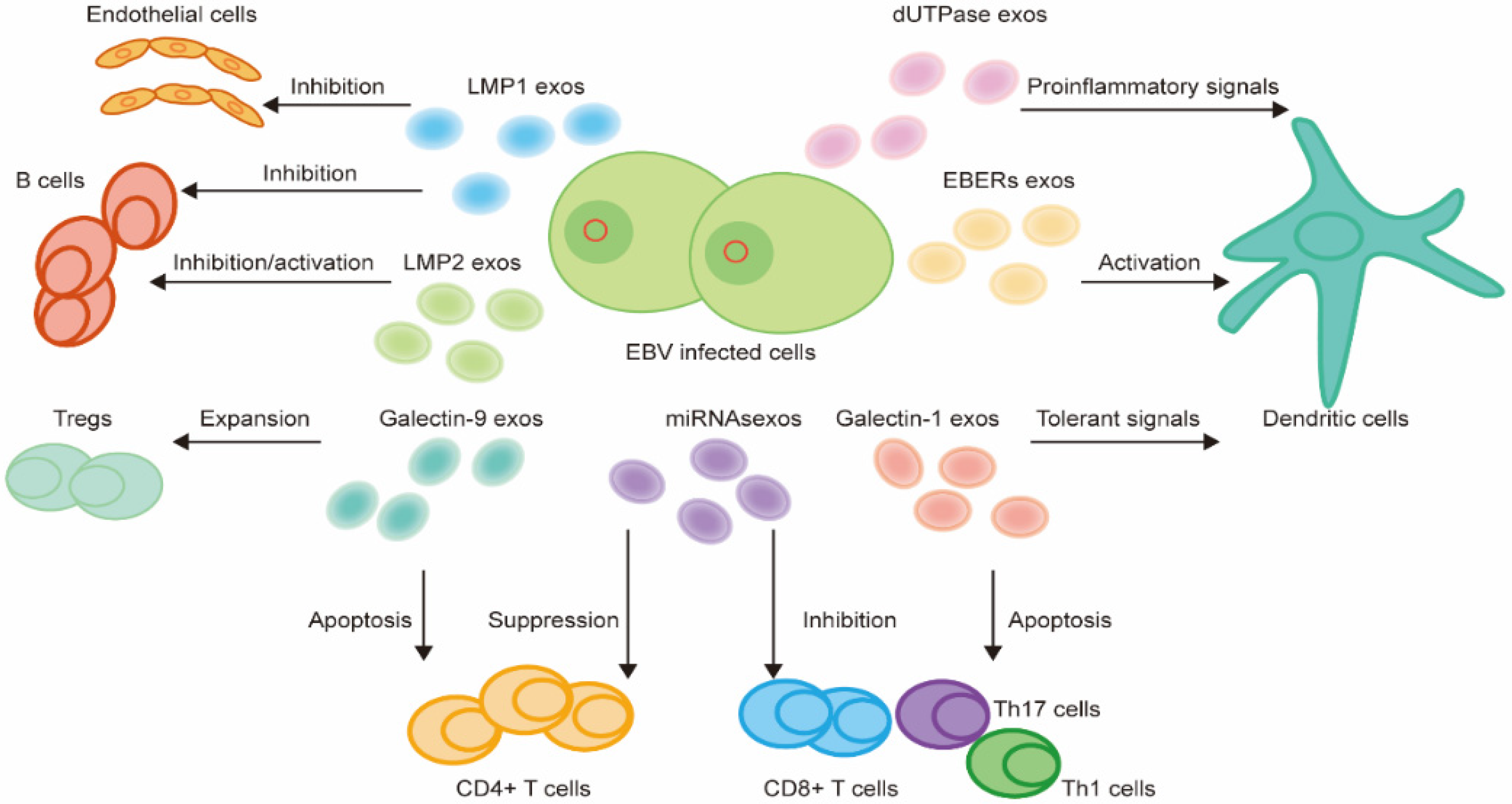{kind=link}
Table 1.
Features of EBV-associated malignancies.
| EBV-Associated Malignancies | EBV Positive Rate | Latency Pattern | Immune Markers | Immunotherapy | |
|---|---|---|---|---|---|
| Immune Cells | Immune Molecules | ||||
| PTLD | 100% | Latency III | Memory/helper T cells Decreased cytotoxic T cells | IFN-γ, IL-6, IL-10, IL-13 | Adoptive T cell therapy |
| Classical HL | 50% | Latency II | Tregs Exhausted CD8+ T cells | PD-L1, TNFR, Th2 cytokines and chemokines, IL-10, galectin-1, TGF-β | PD-1 inhibitors T cell therapy |
| Epidemic BL | 100% | Latency I | M2 TAMs | IL-2, IL-6, IL-10 | NA |
| DLBCL | 10% | Latency II or III | M2 TAMs Exhausted CD8+ T cells | IL-10, PD-1, PD-L1, PD-L2, LAG3, TIM3 | Chimeric antigen receptor T cell therapy |
| Extranodal NK/T cell lymphomas | 100% | Latency II | Activated T cells and macrophages | IL-2, IL-10, CD27, TNF-α, PD-L1 | PD-1 inhibitors |
| Undifferentiated NPC | 100% | Latency I/II | Exhausted CD8+ T cells LAMP3+ DCs | PD-L1, PD-L2, CTLA-4, IDO1, HLA-G | PD-1 inhibitors Therapeutic EBV vaccines |
| GC | 10% | Latency I or I/II | CD8+ T cells Tregs TAMs | PD-L1, IDO1 | PD-1 inhibitors |
| ICC | 6.6% | Latency I | CD8+ T cells M1 TAMs CD20+ B cells | PD-L1 | NA |
| SMT | <1% | Latency III | T cells | NA | NA |
Note: PTLD, Post-transplant lymphoproliferative disease; HL, Hodgkin lymphoma; BL, Burkitt lymphoma; DLBCL, diffuse large B cell lymphoma; GC, gastric carcinoma; NPC, nasopharyngeal carcinoma; ICC, intrahepatic cholangiocarcinoma; SMT, smooth muscle tumor; NA, not applicable.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zheng, X.; Huang, Y.; Li, K.; Luo, R.; Cai, M.; Yun, J. Immunosuppressive Tumor Microenvironment and Immunotherapy of Epstein–Barr Virus-Associated Malignancies. Viruses 2022, 14, 1017. https://0-doi-org.brum.beds.ac.uk/10.3390/v14051017
AMA Style
Zheng X, Huang Y, Li K, Luo R, Cai M, Yun J. Immunosuppressive Tumor Microenvironment and Immunotherapy of Epstein–Barr Virus-Associated Malignancies. Viruses. 2022; 14(5):1017. https://0-doi-org.brum.beds.ac.uk/10.3390/v14051017
Chicago/Turabian StyleZheng, Xueyi, Yuhua Huang, Kai Li, Rongzhen Luo, Muyan Cai, and Jingping Yun. 2022. "Immunosuppressive Tumor Microenvironment and Immunotherapy of Epstein–Barr Virus-Associated Malignancies" Viruses 14, no. 5: 1017. https://0-doi-org.brum.beds.ac.uk/10.3390/v14051017
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.