
Frequent Occurrence of Simultaneous Infection with Multiple Rotaviruses in Swiss Pigs

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.1.1. Cross-Sectional Study
2.1.2. Longitudinal Study
2.2. Postmortem Analyses
2.3. RNA Extraction
2.3.1. Viral RNA Mini Kit
2.3.2. Phenol–Chloroform RNA Extraction
2.4. Multiplex Real-Time RT-PCR Assays
2.5. Next-Generation Sequencing (NGS) and Genotyping
2.6. Phylogenetic Analysis
2.7. Statistical Analysis
3. Results
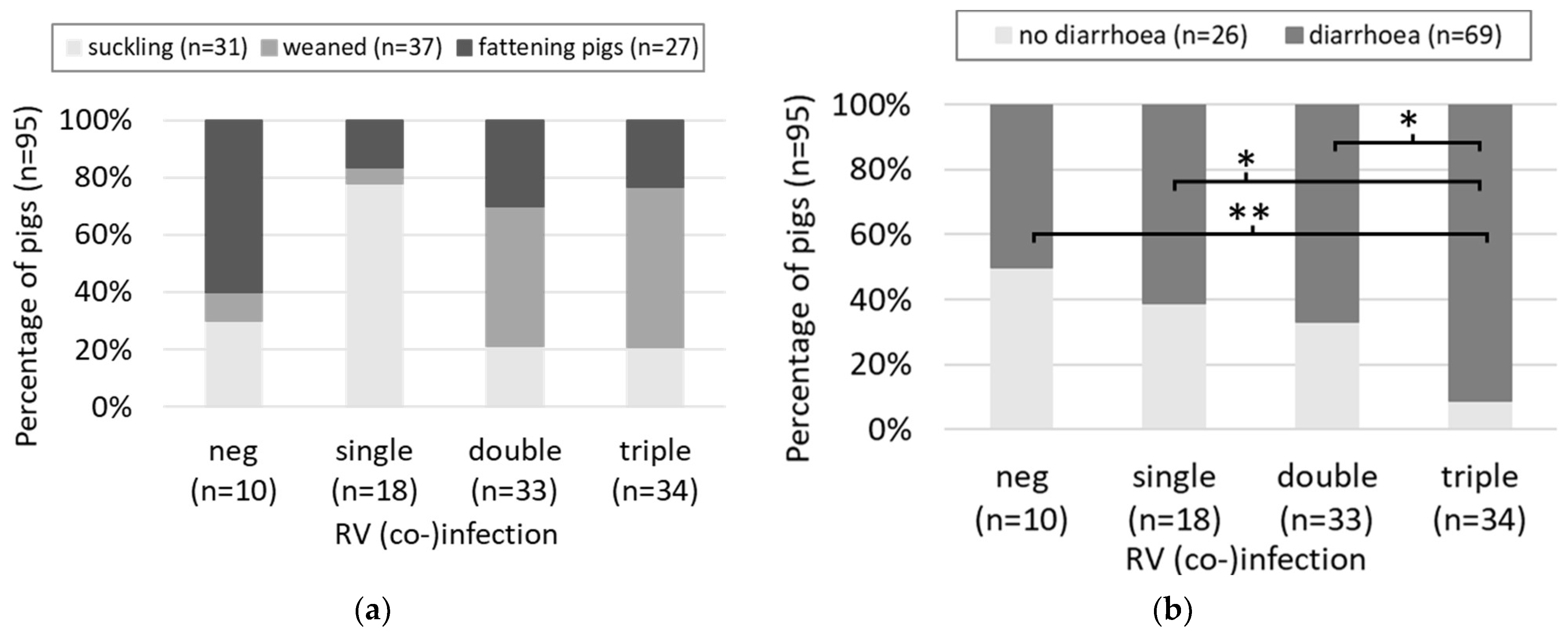
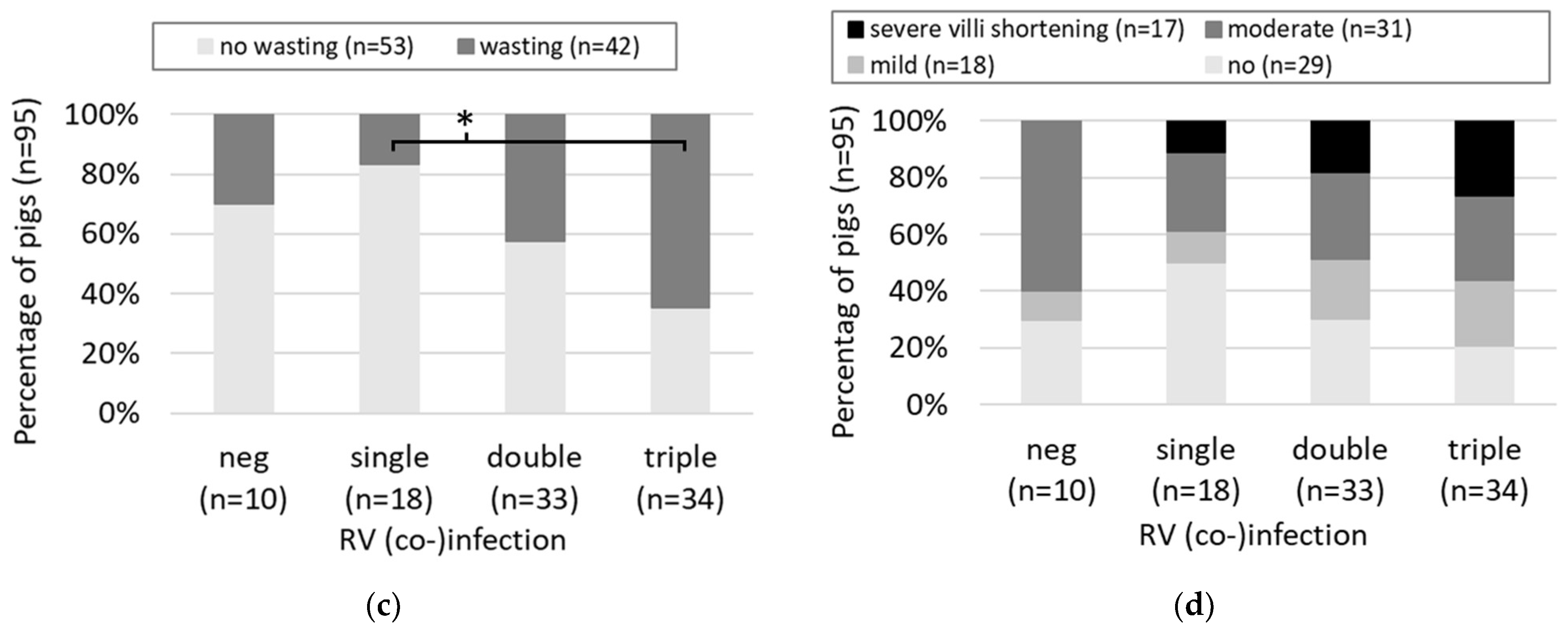
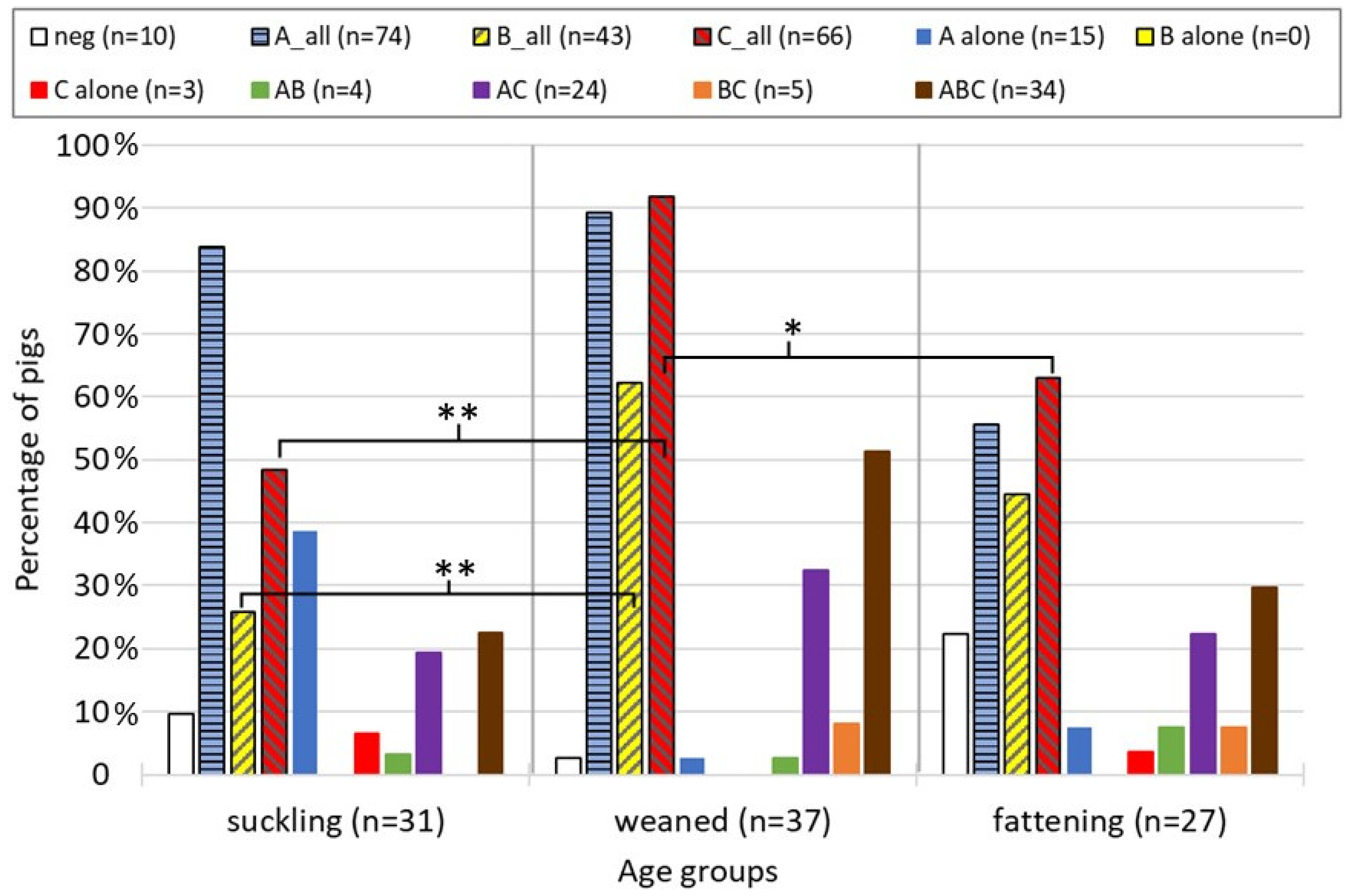
3.1. Cross-Sectional Study
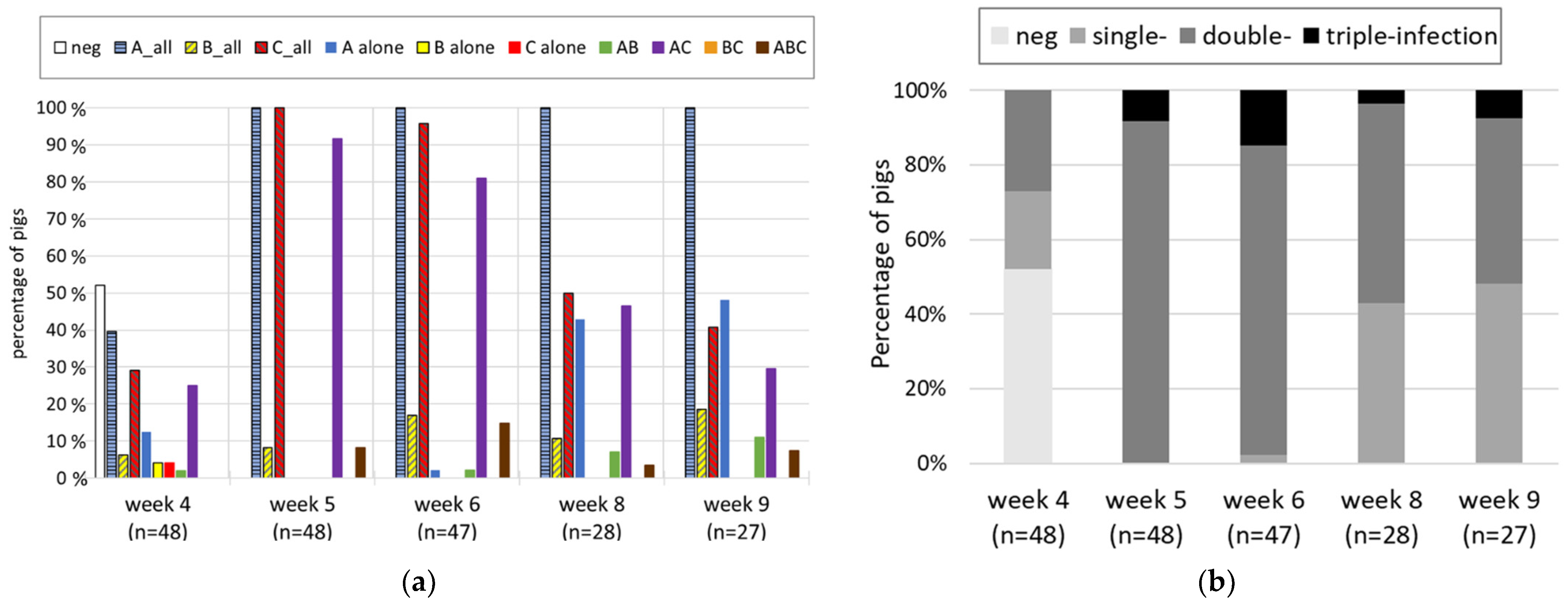
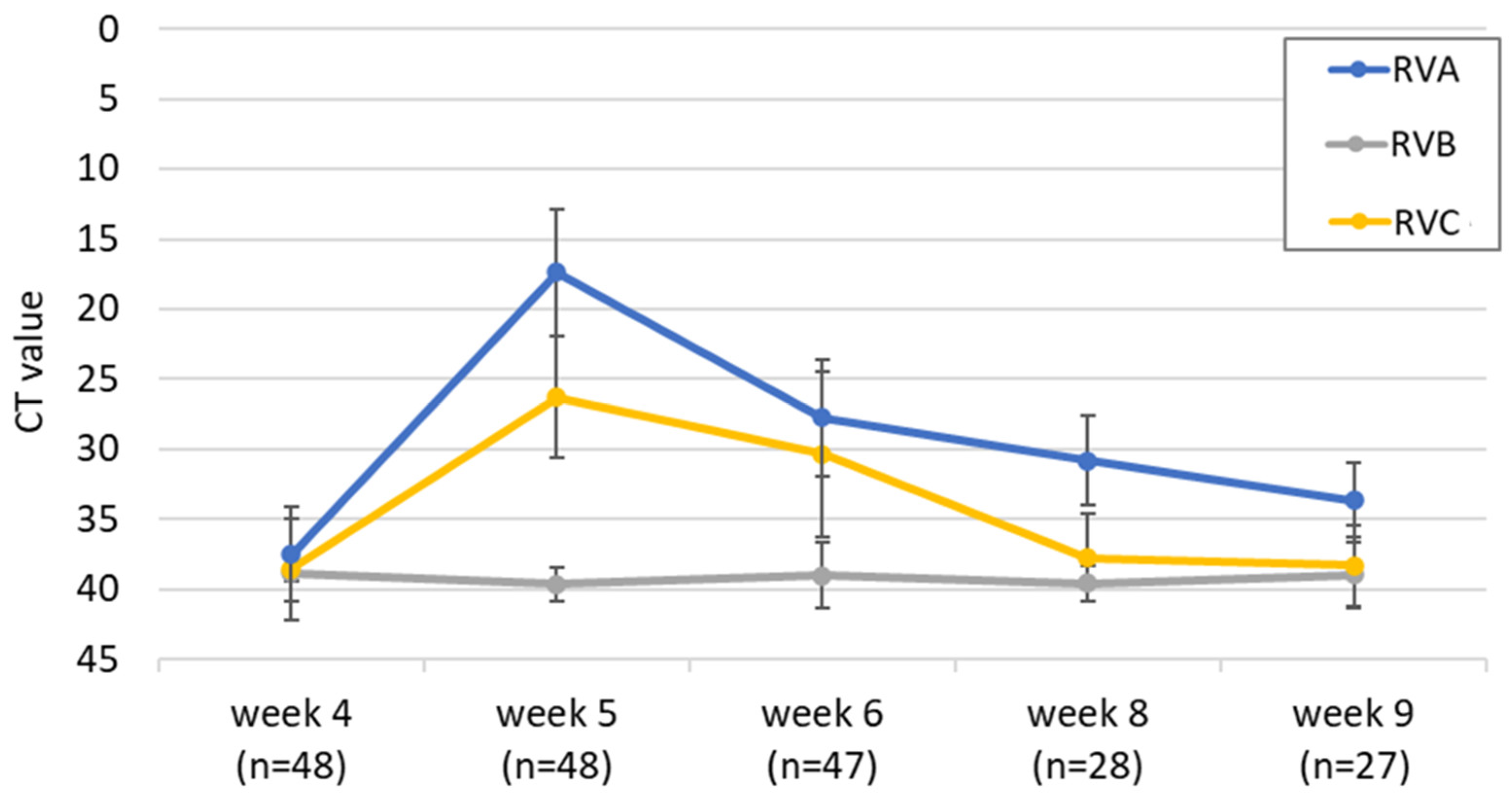
3.2. Longitudinal Study
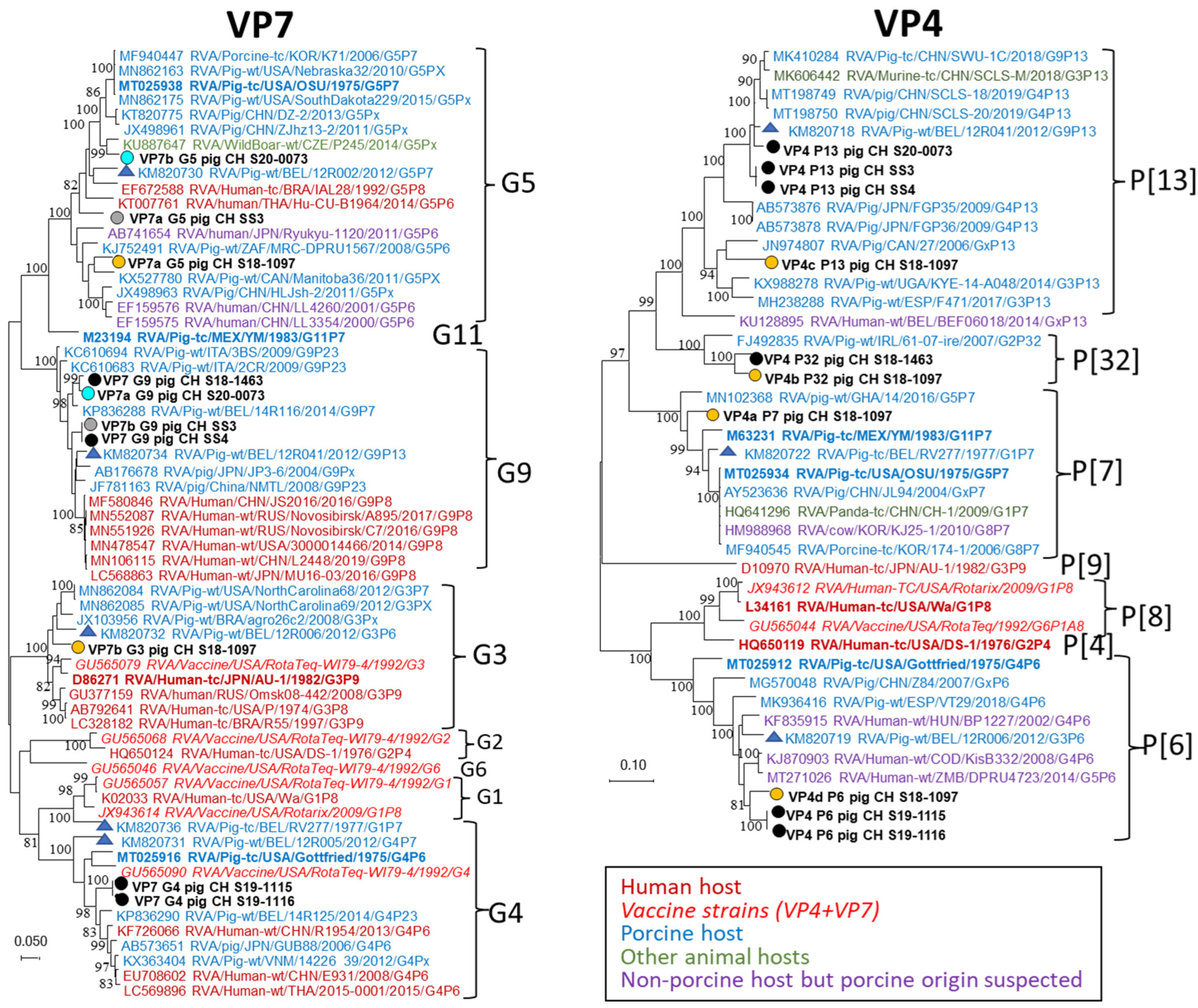
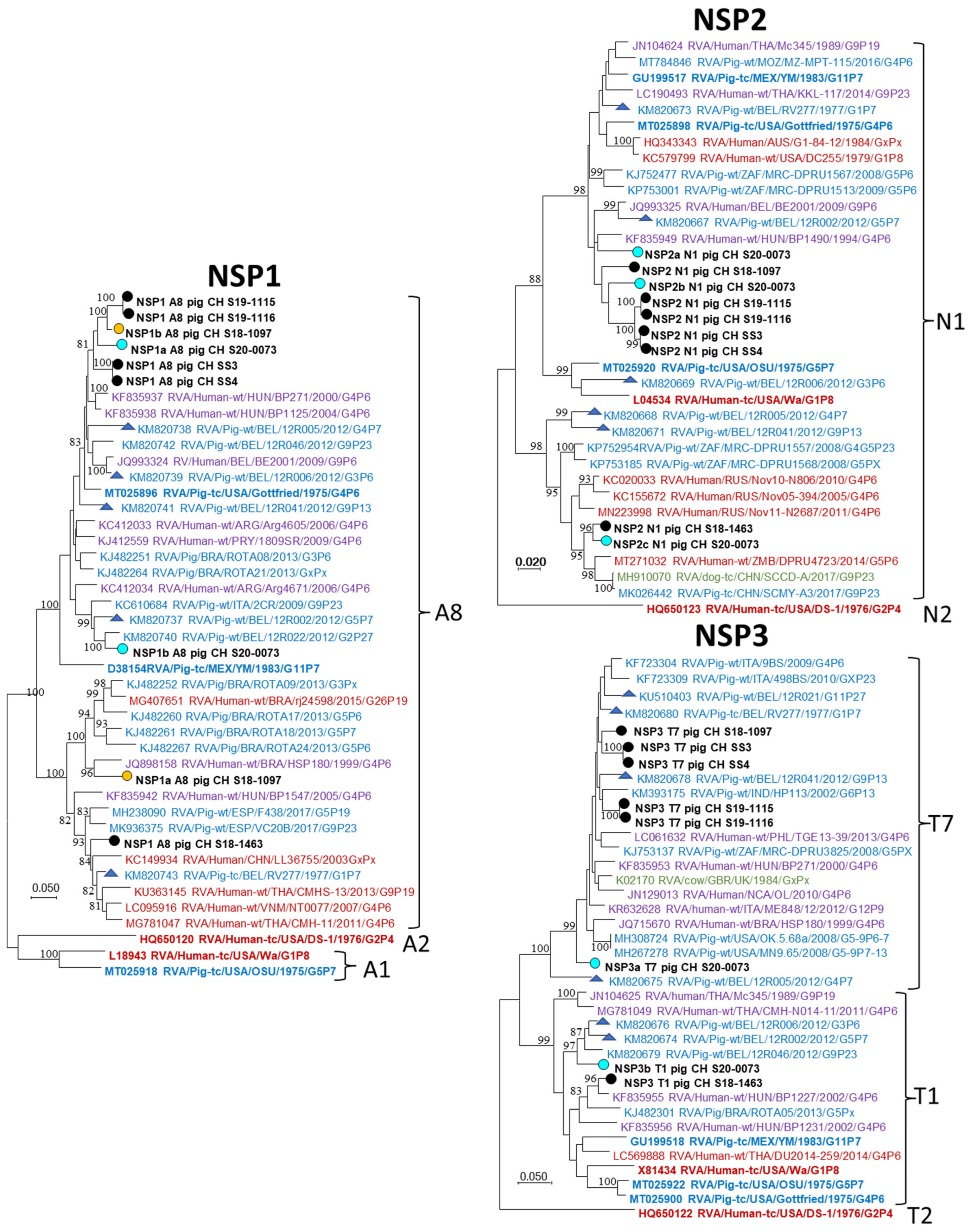
3.3. NGS and Genotyping of RVA Strains
4. Discussion
4.1. Cross-Sectional Study
4.2. Longitudinal Study
4.3. NGS and Genotyping of RVA Strains
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Esona, M.D.; Gautam, R. Rotavirus. Clin. Lab. Med. 2015, 35, 363–391. [Google Scholar] [CrossRef]
- Vlasova, A.; Amimo, J.; Saif, L. Porcine Rotaviruses: Epidemiology, Immune Responses and Control Strategies. Viruses 2017, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Cui, T.; Theuns, S.; Xie, J.; Nauwynck, H.J. Porcine rotavirus mainly infects primary porcine enterocytes at the basolateral surface. Vet. Res. 2019, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crouch, C.F.; Woode, G.N. Serial Studies of Virus Multiplication and Intestinal Damage in Gnotobiotic Piglets Infected with Rotavirus. J. Med. Microbiol. 1978, 11, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Wills, R.W. Diarrhea in Growing-Finishing Swine. Vet. Clin. North Am. Food Animal Pract. 2000, 16, 135–161. [Google Scholar] [CrossRef]
- Papp, H.; László, B.; Jakab, F.; Ganesh, B.; De Grazia, S.; Matthijnssens, J.; Ciarlet, M.; Martella, V.; Bányai, K. Review of group A rotavirus strains reported in swine and cattle. Vet. Microbiol. 2013, 165, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Holland, R.E. Some infectious causes of diarrhea in young farm animals. Clin. Microbiol. Rev. 1990, 3, 345–375. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Otto, P.H.; Ciarlet, M.; Desselberger, U.; Van Ranst, M.; Johne, R. VP6-sequence-based cutoff values as a criterion for rotavirus species demarcation. Arch. Virol. 2012, 157, 1177–1182. [Google Scholar] [CrossRef]
- Johne, R.; Tausch, S.H.; Grützke, J.; Falkenhagen, A.; Patzina-Mehling, C.; Beer, M.; Höper, D.; Ulrich, R.G. Distantly Related Rotaviruses in Common Shrews, Germany, 2004–2014. Emerg. Infect. Dis. 2019, 25, 2310–2314. [Google Scholar] [CrossRef]
- Johne, R.; Schilling-Loeffler, K.; Ulrich, R.G.; Tausch, S.H. Whole Genome Sequence Analysis of a Prototype Strain of the Novel Putative Rotavirus Species, L. Viruses 2022, 14, 462. [Google Scholar] [CrossRef]
- Chasey, D.; Bridger, J.C.; McCrae, M.A. A new type of atypical rotavirus in pigs. Arch. Virol. 1986, 89, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Marthaler, D.; Homwong, N.; Rossow, K.; Culhane, M.; Goyal, S.; Collins, J.; Matthijnssens, J.; Ciarlet, M. Rapid detection and high occurrence of porcine rotavirus A, B, and C by RT-qPCR in diagnostic samples. J. Virol. Methods 2014, 209, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Hull, J.J.A.; Qi, M.; Montmayeur, A.M.; Kumar, D.; Velasquez, D.E.; Moon, S.-S.; Magaña, L.C.; Betrapally, N.; Ng, T.F.F.; Jiang, B.; et al. Metagenomic sequencing generates the whole genomes of porcine rotavirus A, C, and H from the United States. PLoS ONE 2020, 15, e0244498. [Google Scholar] [CrossRef] [PubMed]
- Martella, V.; Bányai, K.; Lorusso, E.; Bellacicco, A.L.; Decaro, N.; Camero, M.; Bozzo, G.; Moschidou, P.; Arista, S.; Pezzotti, G. Prevalence of group C rotaviruses in weaning and post-weaning pigs with enteritis. Vet. Microbiol. 2007, 123, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Marthaler, D.; Rossow, K.; Gramer, M.; Collins, J.; Goyal, S.; Tsunemitsu, H.; Kuga, K.; Suzuki, T.; Ciarlet, M.; Matthijnssens, J. Detection of substantial porcine group B rotavirus genetic diversity in the United States, resulting in a modified classification proposal for G genotypes. Virology 2012, 433, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Almeida, P.R.; Lorenzetti, E.; Cruz, R.S.; Watanabe, T.T.; Zlotowski, P.; Alfieri, A.A.; Driemeier, D. Diarrhea caused by rotavirus A, B, and C in suckling piglets from southern Brazil: Molecular detection and histologic and immunohistochemical characterization. J. Vet. Diagn. Investig. 2018, 30, 370–376. [Google Scholar] [CrossRef] [Green Version]
- Janke, B.H.; Nelson, J.K.; Benfield, D.A.; Nelson, E.A. Relative Prevalence of Typical and Atypical Strains among Rotaviruses from Diarrheic Pigs in Conventional Swine Herds. J. Vet. Diagn. Investig. 1990, 2, 308–311. [Google Scholar] [CrossRef]
- Kim, Y.; Chang, K.-O.; Straw, B.; Saif, L.J. Characterization of Group C Rotaviruses Associated with Diarrhea Outbreaks in Feeder Pigs. J. Clin. Microbiol. 1999, 37, 1484–1488. [Google Scholar] [CrossRef] [Green Version]
- Nyaga, M.M.; Jere, K.C.; Esona, M.D.; Seheri, M.L.; Stucker, K.M.; Halpin, R.A.; Akopov, A.; Stockwell, T.B.; Peenze, I.; Diop, A.; et al. Whole genome detection of rotavirus mixed infections in human, porcine and bovine samples co-infected with various rotavirus strains collected from sub-Saharan Africa. Infect. Genet. Evol. 2015, 31, 321–334. [Google Scholar] [CrossRef] [Green Version]
- Steyer, A.; Poljšak-Prijatelj, M.; Barlič-Maganja, D.; Marin, J. Human, porcine and bovine rotaviruses in Slovenia: Evidence of interspecies transmission and genome reassortment. J. Gen. Virol. 2008, 89, 1690–1698. [Google Scholar] [CrossRef]
- Wu, F.-T.; Bányai, K.; Jiang, B.; Liu, L.T.-C.; Marton, S.; Huang, Y.-C.; Huang, L.-M.; Liao, M.-H.; Hsiung, C.A. Novel G9 rotavirus strains co-circulate in children and pigs, Taiwan. Sci. Rep. 2017, 7, 40731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthijnssens, J.; Ciarlet, M.; Rahman, M.; Attoui, H.; Bányai, K.; Estes, M.K.; Gentsch, J.R.; Iturriza-Gómara, M.; Kirkwood, C.D.; Martella, V.; et al. Recommendations for the classification of group A rotaviruses using all 11 genomic RNA segments. Arch. Virol. 2008, 153, 1621–1629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthijnssens, J.; Ciarlet, M.; Heiman, E.; Arijs, I.; Delbeke, T.; McDonald, S.M.; Palombo, E.A.; Iturriza-GóMara, M.; Maes, P.; Patton, J.T.; et al. Full Genome-Based Classification of Rotaviruses Reveals a Common Origin between Human Wa-Like and Porcine Rotavirus Strains and Human DS-1-Like and Bovine Rotavirus Strains. J. Virol. 2008, 82, 3204–3219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cates, J.E.; Amin, A.B.; Tate, J.E.; Lopman, B.; Parashar, U. Do Rotavirus Strains Affect Vaccine Effectiveness? A Systematic Review and Meta-analysis. Pediatr. Infect. Dis. J. 2021, 40, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, L.; Galetto-Lacour, A.; Altwegg, M.; Egli, K.; Schmidt, M.; Gervaix, A. Disease burden of rotavirus gastroenteritis in children up to 5 years of age in two Swiss cantons: Paediatrician- and hospital-based surveillance. Eur. J. Pediatr. 2010, 169, 319–325. [Google Scholar] [CrossRef]
- Schediwy, M.; Balmer, S.; Bredtmann, C.; Hadorn, D.; Bless, P.; Rosato, G.; Sydler, T.; Harisberger, M.; Graage, R.; Saura-Martinez, H.; et al. Reviving post-mortem diagnostics as a tool to increase porcine herd health and strengthen early detection of pig diseases—The PathoPig project 2014-2016. Schweiz Arch. Tierheilkd 2018, 160, 375–384. [Google Scholar] [CrossRef]
- Lienhard, J.; Vonlanthen-Specker, I.; Sidler, X.; Bachofen, C. Screening of Swiss Pig Herds for Hepatitis E Virus: A Pilot Study. Animals 2021, 11, 3050. [Google Scholar] [CrossRef]
- Mount, L.E.; Rowell, J.G. Body size, body temperature and age in relation to the metabolic rate of the pig in the first five weeks after birth. J. Physiol. 1960, 154, 408–416. [Google Scholar] [CrossRef] [Green Version]
- Nabuurs, M.J.; Hoogendoorn, A.; van der Molen, E.J.; van Osta, A.L. Villus height and crypt depth in weaned and unweaned pigs, reared under various circumstances in The Netherlands. Res. Vet. Sci 1993, 55, 78–84. [Google Scholar] [CrossRef]
- Kubacki, J.; Fraefel, C.; Bachofen, C. Implementation of next-generation sequencing for virus identification in veterinary diagnostic laboratories. J. Vet. Diagn Invest. 2021, 33, 235–247. [Google Scholar] [CrossRef]
- Kubacki, J.; Flacio, E.; Qi, W.; Guidi, V.; Tonolla, M.; Fraefel, C. Viral Metagenomic Analysis of Aedes albopictus Mosquitos from Southern Switzerland. Viruses 2020, 12, 929. [Google Scholar] [CrossRef] [PubMed]
- Hardmeier, I.; Aeberhard, N.; Qi, W.; Schoenbaechler, K.; Kraettli, H.; Hatt, J.-M.; Fraefel, C.; Kubacki, J. Metagenomic analysis of fecal and tissue samples from 18 endemic bat species in Switzerland revealed a diverse virus composition including potentially zoonotic viruses. PLoS ONE 2021, 16, e0252534. [Google Scholar] [CrossRef] [PubMed]
- Maes, P.; Matthijnssens, J.; Rahman, M.; Van Ranst, M. RotaC: A web-based tool for the complete genome classification of group A rotaviruses. BMC Microbiol. 2009, 9, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Theuns, S.; Heylen, E.; Zeller, M.; Roukaerts, I.D.M.; Desmarets, L.M.B.; Van Ranst, M.; Nauwynck, H.J.; Matthijnssens, J. Complete Genome Characterization of Recent and Ancient Belgian Pig Group A Rotaviruses and Assessment of Their Evolutionary Relationship with Human Rotaviruses. J. Virol. 2015, 89, 1043–1057. [Google Scholar] [CrossRef] [Green Version]
- Collins, P.J.; Martella, V.; Buonavoglia, C.; O’Shea, H. Identification of a G2-like porcine rotavirus bearing a novel VP4 type, P[32]. Vet. Res. 2010, 41, 73. [Google Scholar] [CrossRef] [Green Version]
- Wenske, O.; Rückner, A.; Piehler, D.; Schwarz, B.-A.; Vahlenkamp, T.W. Epidemiological analysis of porcine rotavirus A genotypes in Germany. Vet. Microbiol. 2018, 214, 93–98. [Google Scholar] [CrossRef]
- Chandler-Bostock, R.; Hancox, L.R.; Nawaz, S.; Watts, O.; Iturriza-Gomara, M.; Mellits, K.M. Genetic diversity of porcine group A rotavirus strains in the UK. Vet. Microbiol. 2014, 173, 27–37. [Google Scholar] [CrossRef]
- Midgley, S.E.; Bányai, K.; Buesa, J.; Halaihel, N.; Hjulsager, C.K.; Jakab, F.; Kaplon, J.; Larsen, L.E.; Monini, M.; Poljšak-Prijatelj, M.; et al. Diversity and zoonotic potential of rotaviruses in swine and cattle across Europe. Vet. Microbiol. 2012, 156, 238–245. [Google Scholar] [CrossRef]
- Basso, W.; Marti, H.; Hilbe, M.; Sydler, T.; Stahel, A.; Bürgi, E.; Sidler, X. Clinical cystoisosporosis associated to porcine cytomegalovirus (PCMV, Suid herpesvirus 2) infection in fattening pigs. Parasitol. Int. 2017, 66, 806–809. [Google Scholar] [CrossRef]
- Chepngeno, J.; Diaz, A.; Paim, F.C.; Saif, L.J.; Vlasova, A.N. Rotavirus C: Prevalence in suckling piglets and development of virus-like particles to assess the influence of maternal immunity on the disease development. Vet. Res. 2019, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsuda, K.; Kohmoto, M.; Kawashima, K.; Tsunemitsu, H. Frequency of Enteropathogen Detection in Suckling and Weaned Pigs with Diarrhea in Japan. J. Vet. Diagn. Investig. 2006, 18, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Vonlanthen-Specker, I.; Stephan, R.; Sidler, X.; Moor, D.; Fraefel, C.; Bachofen, C. Genetic Diversity of Hepatitis E Virus Type 3 in Switzerland—From Stable to Table. Animals 2021, 11, 3177. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, C.; Stalder, H.; Sidler, X.; Renzullo, S.; Gurtner, C.; Grahofer, A.; Schweizer, M. Long-Term Circulation of Atypical Porcine Pestivirus (APPV) within Switzerland. Viruses 2019, 11, 653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phan, M.V.T.; Anh, P.H.; Cuong, N.V.; Munnink, B.B.O.; Van Der Hoek, L.; My, P.T.; Tri, T.N.; Bryant, J.E.; Baker, S.; Thwaites, G.; et al. Unbiased whole-genome deep sequencing of human and porcine stool samples reveals circulation of multiple groups of rotaviruses and a putative zoonotic infection. Virus Evol. 2016, 2, vew027. [Google Scholar] [CrossRef] [Green Version]
- Le Pendu, J.; Ruvoën-Clouet, N. Fondness for sugars of enteric viruses confronts them with human glycans genetic diversity. Human Genet. 2020, 139, 903–910. [Google Scholar] [CrossRef]
- Guo, Y.; Candelero-Rueda, R.A.; Saif, L.J.; Vlasova, A.N. Infection of porcine small intestinal enteroids with human and pig rotavirus A strains reveals contrasting roles for histo-blood group antigens and terminal sialic acids. PLOS Pathog. 2021, 17, e1009237. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain, Host | VP7 | VP4 | VP6 | VP1 | VP2 | VP3 | NSP1 | NSP2 | NSP3 | NSP4 | NSP5 | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SS3, pig * | G9 | P[13] | I5 | R1 | C1 | M1 | A8 | N1 | T7 | E1 | H1 | |
| G5 | ||||||||||||
| SS4, pig * | G9 | P[13] | I5 | R1 | C1 | M1 | A8 | N1 | T7 | E1 | H1 | |
| S19-1115, pig * | G4 | P[6] | I5 | R1 | C1 | M1 | A8 | N1 | T7 | E1 | H1 | |
| S19-1116, pig * | G4 | P[6] | I5 | R1 | C1 | M1 | A8 | N1 | T7 | E1 | H1 | |
| S18-1463, pig * | G9 | P[32] | I5 | R1 | C1 | M1 | A8 | N1 | T1 | E9 | H1 | |
| S18-1097, pig * | G5 | P[13] | 2 × I5 | R1 | C1 | M1 | 2 × A8 | N1 | T7 | E1 | H1 | |
| P[6] | ||||||||||||
| G3 | P[7] | |||||||||||
| P[32] | ||||||||||||
| S20-0073, pig * | G5 | P[13] | 2 × I5 | 2 × R1 | C1 | M1 | 2 × A8 | 3 × N1 | T1 | E9 | H1 | |
| G9 | T7 | |||||||||||
| S18-1093, pig | G5 | P[13] | 3 × I5 | 2 × R1 | 2 × C1 | M1 | 3 × A8 | N1 | 4 × T7 | 2 × E1 | H1 | |
| G11 | P[6] | |||||||||||
| G9 | P[7] | |||||||||||
| 12R002, pig | G5 | P[7] | I5 | R1 | C1 | M1 | A8 | N1 | T1 | E1 | H1 | [35] |
| 12R005, pig | G4 | P[7] | I5 | R1 | C1 | M1 | A8 | N1 | T7 | E1 | H1 | |
| 12R006, pig | G3 | P[6] | I5 | R1 | C1 | M1 | A8 | N1 | T1 | E1 | H1 | |
| 12R041, pig | G9 | P[13] | I5 | R1 | C1 | M1 | A8 | N1 | T7 | E1 | H1 | |
| RV277, pig | G1 | P[7] | I1 | R1 | C1 | M1 | A8 | N1 | T7 | E1 | H1 | |
| Gottfried, pig | G4 | P[6] | I1 | R1 | C1 | M1 | A8 | N1 | T1 | E1 | H1 | [22] |
| OSU, pig | G5 | P[7] | I5 | R1 | C1 | M1 | A1 | N1 | T1 | E1 | H1 | |
| YM, pig | G11 | P[7] | I5 | R1 | C1 | M1 | A8 | N1 | T1 | E1 | H1 | |
| Wa, human | G1 | P[1] | I1 | R1 | C1 | M1 | A1 | N1 | T1 | E1 | H1 | |
| DS-1, human | G2 | P[2] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 | |
| Au-1, human | G3 | P[3] | I3 | R3 | C3 | M3 | A3 | N3 | T3 | E3 | H3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baumann, S.; Sydler, T.; Rosato, G.; Hilbe, M.; Kümmerlen, D.; Sidler, X.; Bachofen, C. Frequent Occurrence of Simultaneous Infection with Multiple Rotaviruses in Swiss Pigs. Viruses 2022, 14, 1117. https://0-doi-org.brum.beds.ac.uk/10.3390/v14051117
Baumann S, Sydler T, Rosato G, Hilbe M, Kümmerlen D, Sidler X, Bachofen C. Frequent Occurrence of Simultaneous Infection with Multiple Rotaviruses in Swiss Pigs. Viruses. 2022; 14(5):1117. https://0-doi-org.brum.beds.ac.uk/10.3390/v14051117
Chicago/Turabian StyleBaumann, Sibylle, Titus Sydler, Giuliana Rosato, Monika Hilbe, Dolf Kümmerlen, Xaver Sidler, and Claudia Bachofen. 2022. "Frequent Occurrence of Simultaneous Infection with Multiple Rotaviruses in Swiss Pigs" Viruses 14, no. 5: 1117. https://0-doi-org.brum.beds.ac.uk/10.3390/v14051117