
HPV16 Induces Formation of Virus-p62-PML Hybrid Bodies to Enable Infection

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Pseudoviruses
2.2. Antibodies and Reagents
2.3. Endosomal Preparation and Quantitative Mass Spectrometry
2.4. HPV PsV Infection Assay
2.5. Immunofluorescence Microscopy
2.6. Co-Immunoprecipitation Assay
2.7. Duolink Proximity Ligation Assay (PLA)
2.8. Statistics
3. Results
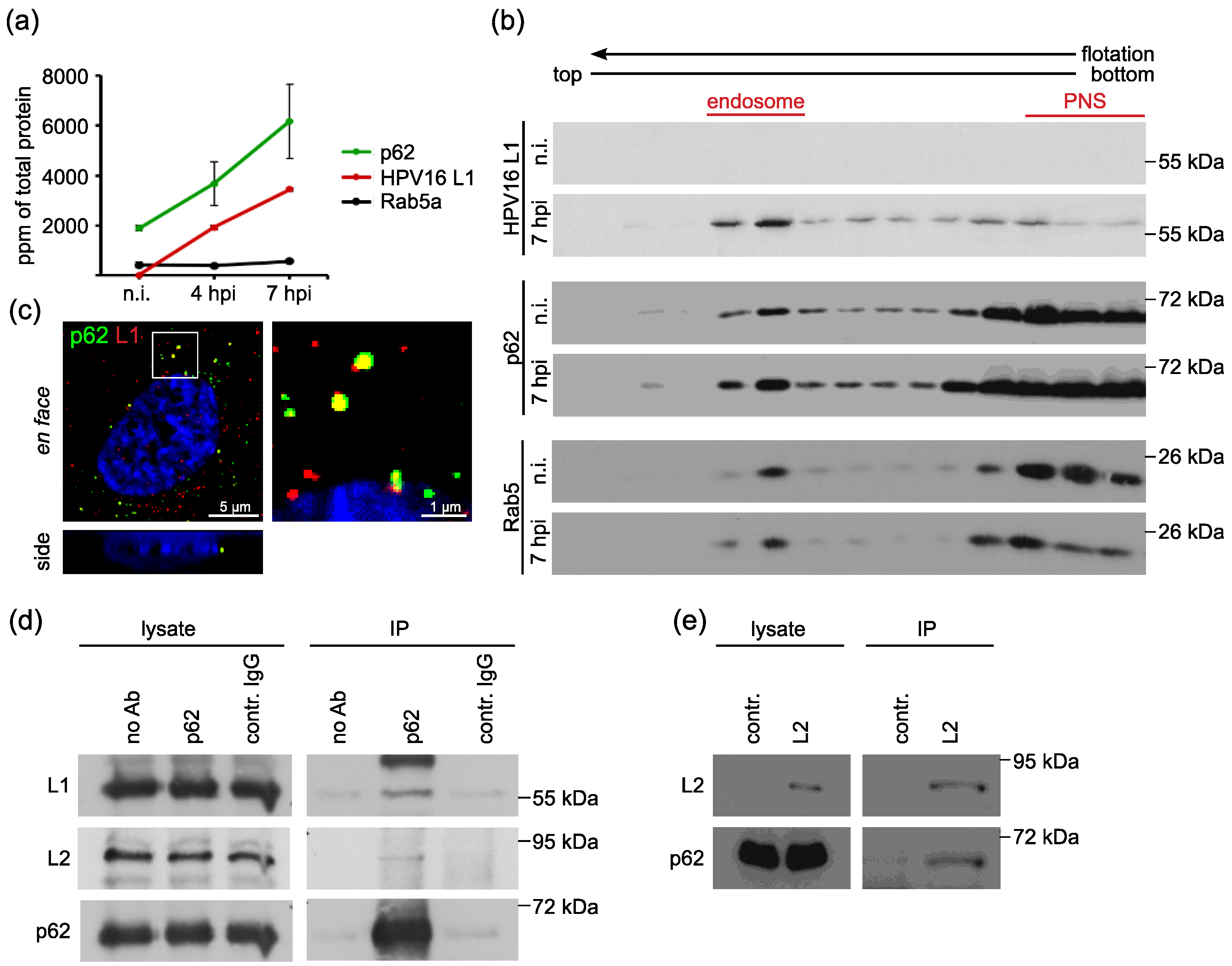
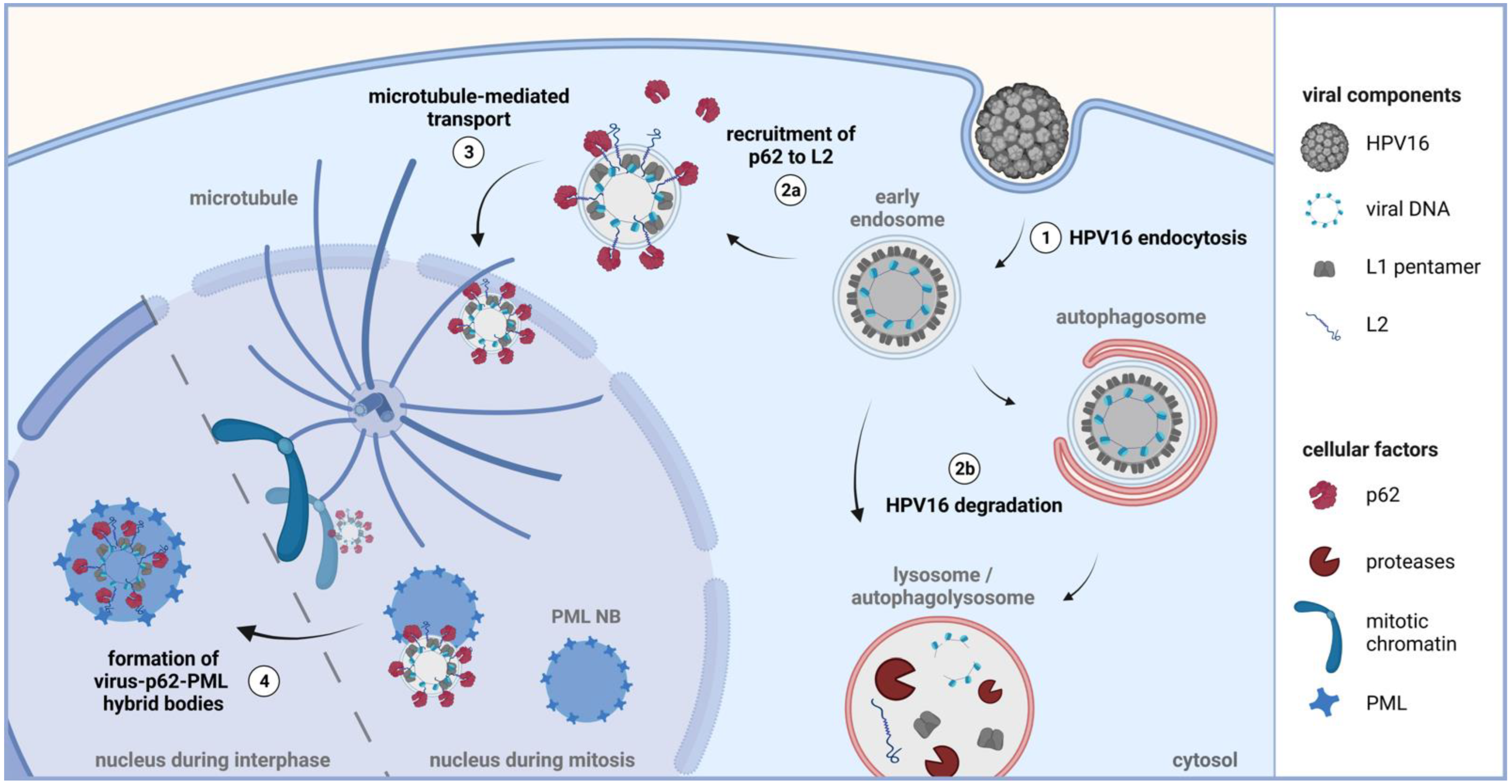
3.1. The Autophagy Adaptor p62 is Recruited to HPV16 Pseudovirus-Filled Endosomes and Interacts with HPV Capsid Proteins
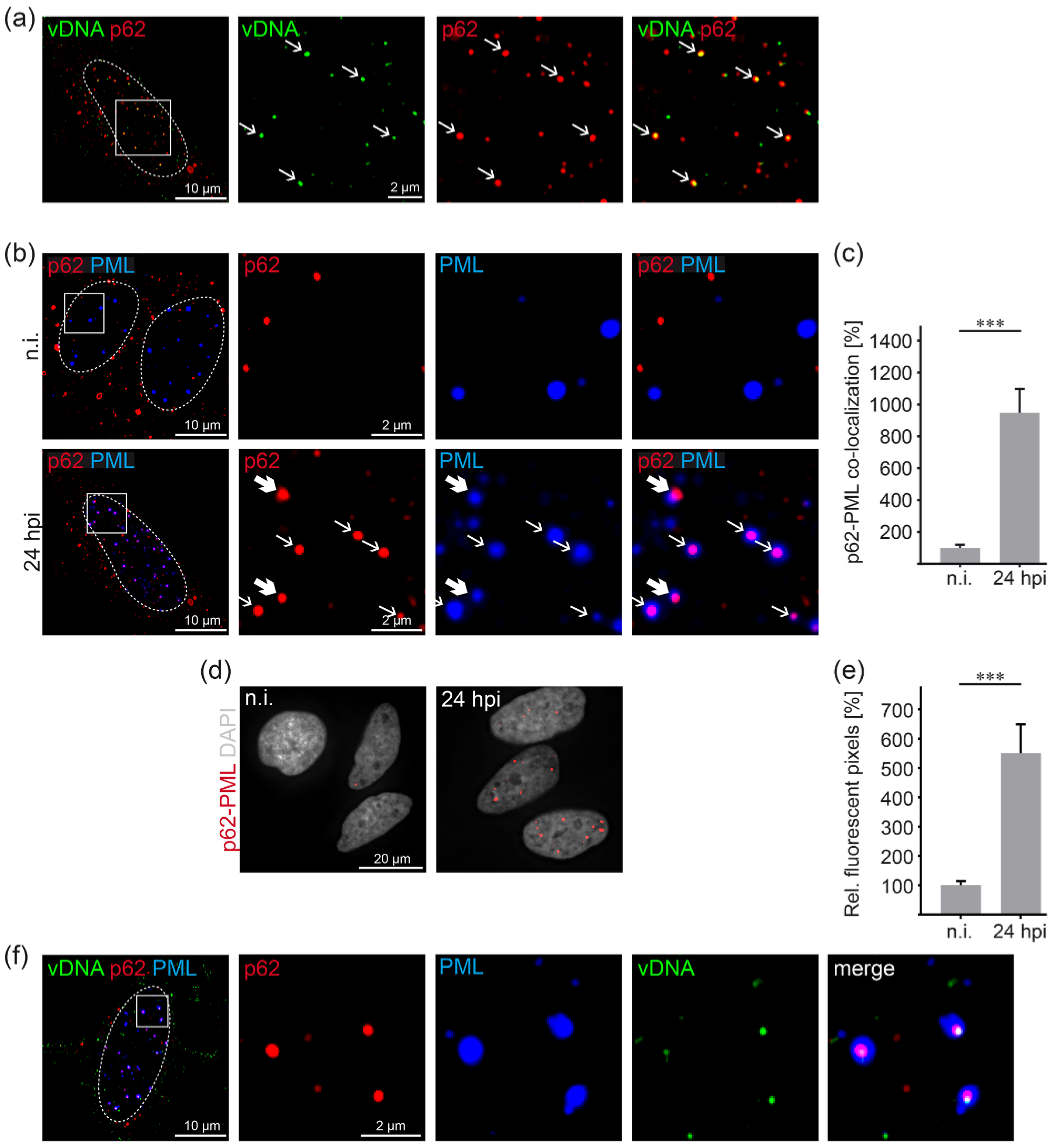
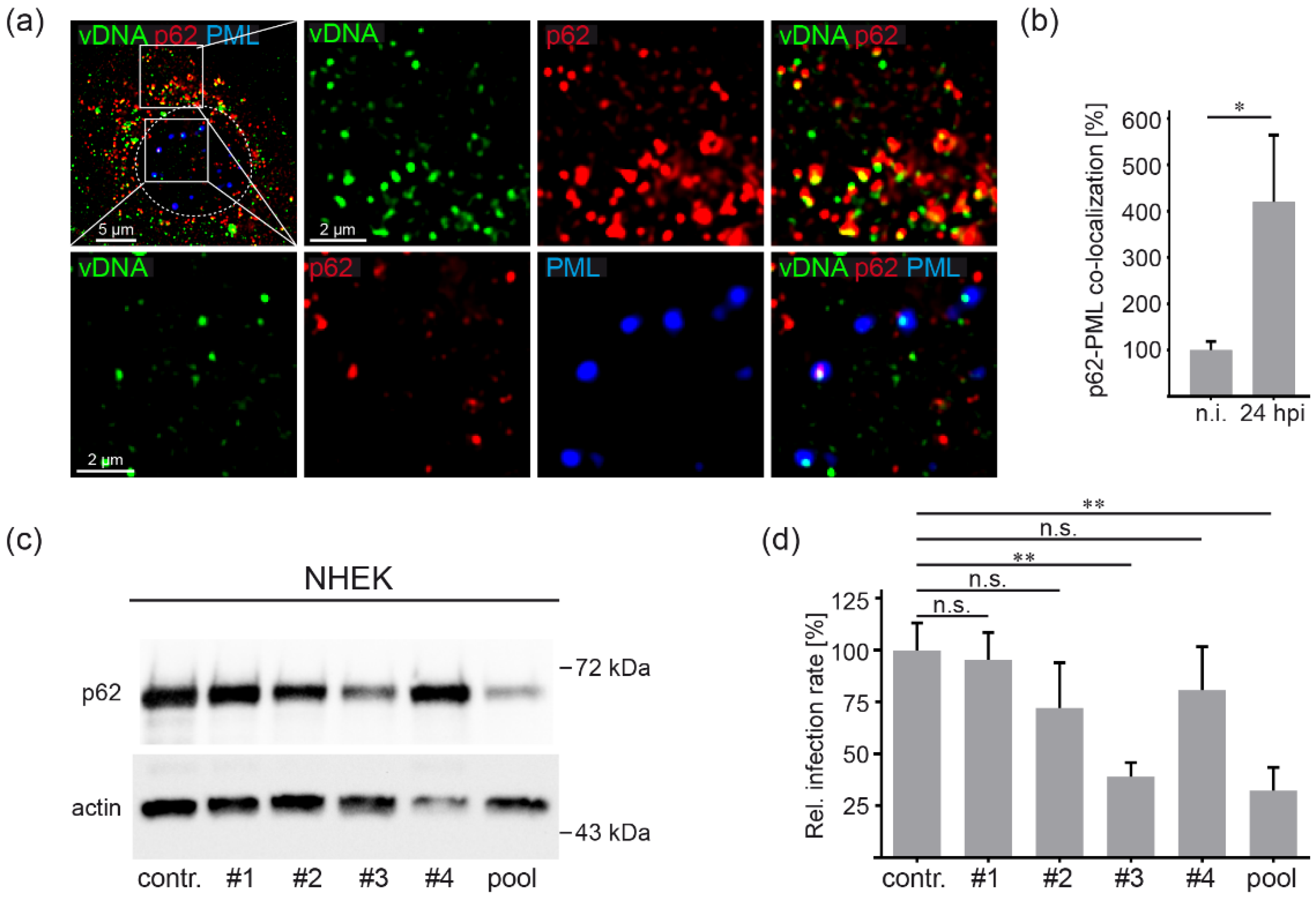
3.2. HPV16 Induces p62-PML Nuclear Body Fusion
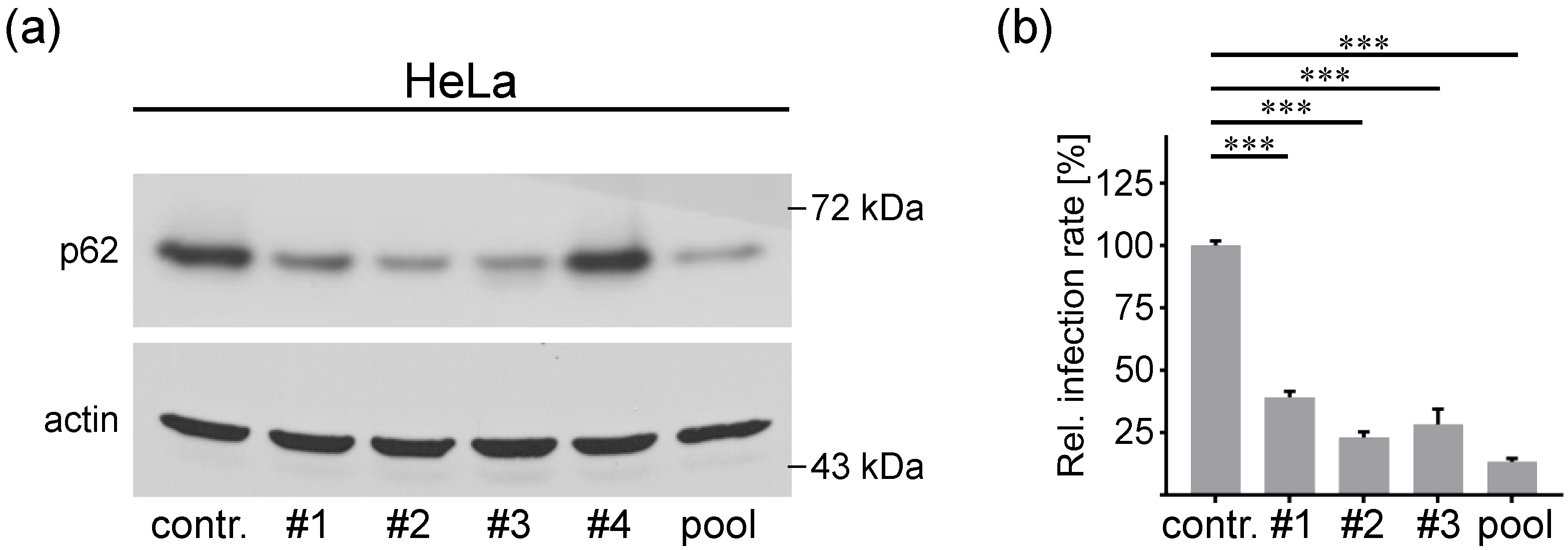
3.3. P62 is a Proviral Factor of HPV16 PsV Infection
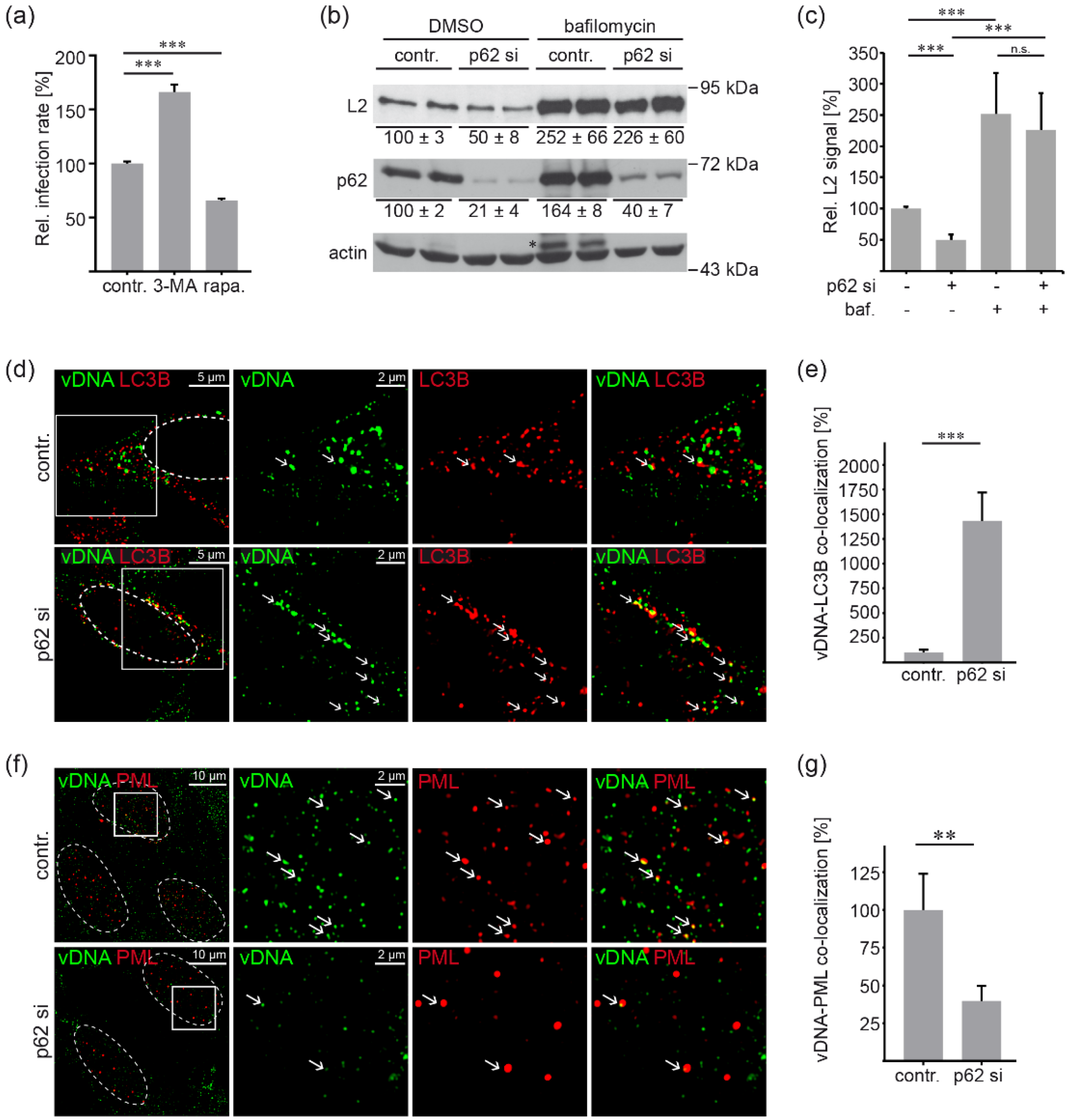
3.4. P62 Protects HPV16 PsV from Autophagic Degradation and Supports Delivery to PML NBs
3.5. HPV16 Induced Formation of Virus Containing p62-PML Hybrid Bodies Is Required for Gene Expression in Primary Keratinocytes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Modis, Y.; Trus, B.L.; Harrison, S.C. Atomic Model of the Papillomavirus Capsid. EMBO J. 2002, 21, 4754–4762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buck, C.B.; Cheng, N.; Thompson, C.D.; Lowy, D.R.; Steven, A.C.; Schiller, J.T.; Trus, B.L. Arrangement of L2 within the Papillomavirus Capsid. J. Virol. 2008, 82, 5190–5197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Villiers, E.-M. Cross-Roads in the Classification of Papillomaviruses. Virology 2013, 445, 2–10. [Google Scholar] [CrossRef] [Green Version]
- McBride, A.A. Human Papillomaviruses: Diversity, Infection and Host Interactions. Nat. Rev. Microbiol. 2021, 20, 95–108. [Google Scholar] [CrossRef]
- zur Hausen, H. Papillomaviruses in the Causation of Human Cancers—A Brief Historical Account. Virology 2009, 384, 260–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kombe, A.J.K.; Li, B.; Zahid, A.; Mengist, H.M.; Bounda, G.-A.; Zhou, Y.; Jin, T. Epidemiology and Burden of Human Papillomavirus and Related Diseases, Molecular Pathogenesis, and Vaccine Evaluation. Front. Public Health 2021, 8, 552028. [Google Scholar] [CrossRef] [PubMed]
- Roden, R.B.S.; Stern, P.L. Opportunities and Challenges for Human Papillomavirus Vaccination in Cancer. Nat. Rev. Cancer 2018, 18, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Mikuličić, S.; Strunk, J.; Florin, L. HPV16 Entry into Epithelial Cells: Running a Gauntlet. Viruses 2021, 13, 2460. [Google Scholar] [CrossRef] [PubMed]
- Florin, L.; Lang, T. Tetraspanin Assemblies in Virus Infection. Front. Immunol. 2018, 9, 1140. [Google Scholar] [CrossRef]
- Spoden, G.; Freitag, K.; Husmann, M.; Boller, K.; Sapp, M.; Lambert, C.; Florin, L. Clathrin- and Caveolin-Independent Entry of Human Papillomavirus Type 16—Involvement of Tetraspanin-Enriched Microdomains (TEMs). PLoS ONE 2008, 3, e3313. [Google Scholar] [CrossRef] [Green Version]
- Schelhaas, M.; Shah, B.; Holzer, M.; Blattmann, P.; Kühling, L.; Day, P.M.; Schiller, J.T.; Helenius, A. Entry of Human Papillomavirus Type 16 by Actin-Dependent, Clathrin- and Lipid Raft-Independent Endocytosis. PLoS Pathog. 2012, 8, e1002657. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Zhang, P.; Crite, M.; DiMaio, D. Papillomaviruses Go Retro. Pathogens 2020, 9, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guion, L.G.; Sapp, M. The Role of Promyelocytic Leukemia Nuclear Bodies During HPV Infection. Front. Cell. Infect. Microbiol. 2020, 10, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardi, R.; Pandolfi, P.P. Structure, Dynamics and Functions of Promyelocytic Leukaemia Nuclear Bodies. Nat. Rev. Mol. Cell Biol. 2007, 8, 1006–1016. [Google Scholar] [CrossRef]
- Everett, R.D.; Chelbi-Alix, M.K. PML and PML Nuclear Bodies: Implications in Antiviral Defence. Biochimie 2007, 89, 819–830. [Google Scholar] [CrossRef]
- Scherer, M.; Stamminger, T. Emerging Role of PML Nuclear Bodies in Innate Immune Signaling. J. Virol. 2016, 90, 5850–5854. [Google Scholar] [CrossRef] [Green Version]
- DiGiuseppe, S.; Luszczek, W.; Keiffer, T.R.; Bienkowska-Haba, M.; Guion, L.G.M.; Sapp, M.J. Incoming Human Papillomavirus Type 16 Genome Resides in a Vesicular Compartment throughout Mitosis. Proc. Natl. Acad. Sci. USA 2016, 113, 6289–6294. [Google Scholar] [CrossRef] [Green Version]
- Day, P.M.; Weisberg, A.S.; Thompson, C.D.; Hughes, M.M.; Pang, Y.Y.; Lowy, D.R.; Schiller, J.T. Human Papillomavirus 16 Capsids Mediate Nuclear Entry during Infection. J. Virol. 2019, 93, e00454-19. [Google Scholar] [CrossRef] [Green Version]
- Calton, C.M.; Bronnimann, M.P.; Manson, A.R.; Li, S.; Chapman, J.A.; Suarez-Berumen, M.; Williamson, T.R.; Molugu, S.K.; Bernal, R.A.; Campos, S.K. Translocation of the Papillomavirus L2/VDNA Complex across the Limiting Membrane Requires the Onset of Mitosis. PLoS Pathog. 2017, 13, e1006200. [Google Scholar] [CrossRef] [Green Version]
- Guion, L.; Bienkowska-Haba, M.; DiGiuseppe, S.; Florin, L.; Sapp, M. PML Nuclear Body-Residing Proteins Sequentially Associate with HPV Genome after Infectious Nuclear Delivery. PLoS Pathog. 2019, 15, e1007590. [Google Scholar] [CrossRef]
- Surviladze, Z.; Sterk, R.T.; Deharo, S.A.; Ozbun, M.A. Cellular Entry of Human Papillomavirus Type 16 Involves Activation of the Phosphatidylinositol 3-Kinase/Akt/MTOR Pathway and Inhibition of Autophagy. J. Virol. 2013, 87, 2508–2517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansen, T.; Lamark, T. Selective Autophagy Mediated by Autophagic Adapter Proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.V.; Mills, J.; Lapierre, L.R. Selective Autophagy Receptor P62/SQSTM1, a Pivotal Player in Stress and Aging. Front. Cell Dev. Biol. 2022, 10, 793328. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. P62/SQSTM1 Binds Directly to Atg8/LC3 to Facilitate Degradation of Ubiquitinated Protein Aggregates by Autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [Green Version]
- Bjørkøy, G.; Lamark, T.; Johansen, T. P62/SQSTM1: A Missing Link between Protein Aggregates and the Autophagy Machinery. Autophagy 2006, 2, 138–139. [Google Scholar] [CrossRef] [Green Version]
- Moscat, J.; Diaz-Meco, M.T. P62 at the Crossroads of Autophagy, Apoptosis, and Cancer. Cell 2009, 137, 1001–1004. [Google Scholar] [CrossRef] [Green Version]
- Seibenhener, M.L.; Geetha, T.; Wooten, M.W. Sequestosome 1/P62–More than Just a Scaffold. FEBS Lett. 2007, 581, 175–179. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Li, Y.; Kaminski, P.J.; Andrade, J.; Laimins, L.A. Pathogenesis of Human Papillomaviruses Requires the ATR/P62 Autophagy-Related Pathway. Mbio 2020, 11, e01628-20. [Google Scholar] [CrossRef]
- Buck, C.; Pastrana, D.; Lowy, D.; Schiller, J. Efficient Intracellular Assembly of Papillomaviral Vectors. J. Virol. 2004, 78, 751. [Google Scholar] [CrossRef] [Green Version]
- Leder, C.; Kleinschmidt, J.A.; Wiethe, C.; MÜLLER, M. Enhancement of Capsid Gene Expression: Preparing the Human Papillomavirus Type 16 Major Structural Gene L1 for DNA Vaccination Purposes. J. Virol. 2001, 75, 9201–9209. [Google Scholar] [CrossRef] [Green Version]
- Schneider, M.A.; Spoden, G.A.; Florin, L.; Lambert, C. Identification of the Dynein Light Chains Required for Human Papillomavirus Infection. Cell. Microbiol. 2011, 13, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Finke, J.; Mikuličić, S.; Loster, A.-L.; Gawlitza, A.; Florin, L.; Lang, T. Anatomy of a Viral Entry Platform Differentially Functionalized by Integrins α3 and α6. Sci. Rep. 2020, 10, 5317–5356. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y.; Tanaka, K.; Kondo, K.; Takeuchi, T.; Mori, S.; Kanda, T. Inhibition of Nuclear Entry of HPV16 Pseudovirus-Packaged DNA by an Anti-HPV16 L2 Neutralizing Antibody. Virology 2010, 406, 181–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spoden, G.A.; Besold, K.; Krauter, S.; Plachter, B.; Hanik, N.; Kilbinger, A.F.M.; Lambert, C.; Florin, L. Polyethylenimine Is a Strong Inhibitor of Human Papillomavirus and Cytomegalovirus Infection. Antimicrob. Agents Chemother. 2011, 56, 75–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wüstenhagen, E.; Boukhallouk, F.; Negwer, I.; Rajalingam, K.; Stubenrauch, F.; Florin, L. The Myb-Related Protein MYPOP Is a Novel Intrinsic Host Restriction Factor of Oncogenic Human Papillomaviruses. Oncogene 2018, 37, 6275–6284. [Google Scholar] [CrossRef]
- Rommel, O.; Dillner, J.; Fligge, C.; Bergsdorf, C.; Wang, X.; Selinka, H.-C.; Sapp, M. Heparan Sulfate Proteoglycans Interact Exclusively with Conformationally Intact HPV L1 Assemblies: Basis for a Virus-like Particle ELISA. J. Med. Virol. 2005, 75, 114–121. [Google Scholar] [CrossRef]
- Volpers, C.; Sapp, M.; Snijders, P.J.; Walboomers, J.M.; Streeck, R.E. Conformational and Linear Epitopes on Virus-like Particles of Human Papillomavirus Type 33 Identified by Monoclonal Antibodies to the Minor Capsid Protein L2. J. Gen. Virol. 1995, 76 Pt 11, 2661–2667. [Google Scholar] [CrossRef]
- Sapp, M.; Kraus, U.; Volpers, C.; Snijders, P.J.; Walboomers, J.M.; Streeck, R.E. Analysis of Type-Restricted and Cross-Reactive Epitopes on Virus-like Particles of Human Papillomavirus Type 33 and in Infected Tissues Using Monoclonal Antibodies to the Major Capsid Protein. J. Gen. Virol. 1994, 75 Pt 12, 3375–3383. [Google Scholar] [CrossRef]
- Gräßel, L.; Fast, L.A.; Scheffer, K.D.; Boukhallouk, F.; Spoden, G.A.; Tenzer, S.; Boller, K.; Bago, R.; Rajesh, S.; Overduin, M.; et al. The CD63-Syntenin-1 Complex Controls Post-Endocytic Trafficking of Oncogenic Human Papillomaviruses. Sci. Rep. 2016, 6, 32337. [Google Scholar] [CrossRef] [Green Version]
- Bund, T.; Spoden, G.A.; Koynov, K.; Hellmann, N.; Boukhallouk, F.; Arnold, P.; Hinderberger, D.; Florin, L. A L2 SUMO Interacting Motif Is Important for PML-Localization and Infection of Human Papillomavirus Type 16. Cell. Microbiol. 2014, 16, 1179–1200. [Google Scholar] [CrossRef]
- Wüstenhagen, E.; Hampe, L.; Boukhallouk, F.; Schneider, M.A.; Spoden, G.A.; Negwer, I.; Koynov, K.; Kast, W.M.; Florin, L. The Cytoskeletal Adaptor Obscurin-Like 1 Interacts with the Human Papillomavirus 16 (HPV16) Capsid Protein L2 and Is Required for HPV16 Endocytosis. J. Virol. 2016, 90, 10629–10641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aniento, F.; Gruenberg, J. Subcellular Fractionation of Tissue Culture Cells. Curr. Protoc. Protein Sci. 2003, 32, 44–47. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, K.; Popa-Wagner, R.; Florin, L. Isolation and Characterization of Pathogen-Bearing Endosomes Enable Analysis of Endosomal Escape and Identification of New Cellular Cofactors of Infection. In Virus-Host Interactions; Methods in Molecular Biology Book Series; Bailer, S., Lieber, D., Eds.; Humana Press: Totowa, NJ, USA, 2013; Volume 1064, pp. 101–113. [Google Scholar] [CrossRef]
- DiGiuseppe, S.; Keiffer, T.R.; Bienkowska-Haba, M.; Luszczek, W.; Guion, L.G.M.; Müller, M.; Sapp, M. Topography of the Human Papillomavirus Minor Capsid Protein L2 during Vesicular Trafficking of Infectious Entry. J. Virol. 2015, 89, 10442–10452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagchi, S.; Fredriksson, R.; Wallén-Mackenzie, Å. In Situ Proximity Ligation Assay (PLA). Methods Mol. Biol. 2015, 1318, 149–159. [Google Scholar] [CrossRef]
- Mendez, R.; Banerjee, S. Proximal Ligation Assay (PLA) on Lung Tissue and Cultured Macrophages to Demonstrate Protein-Protein Interaction. Bio-Protocol 2017, 7, e2602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, L.M.; Cicchini, L.; Pyeon, D. Human Papillomavirus Infection Is Inhibited by Host Autophagy in Primary Human Keratinocytes. Virology 2013, 437, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Cerqueira, C.; Ventayol, P.S.; Vogeley, C.; Schelhaas, M. Kallikrein-8 Proteolytically Processes Human Papillomaviruses in the Extracellular Space to Facilitate Entry into Host Cells. J. Virol. 2015, 89, 7038–7052. [Google Scholar] [CrossRef] [Green Version]
- Popa, A.; Zhang, W.; Harrison, M.S.; Goodner, K.; Kazakov, T.; Goodwin, E.C.; Lipovsky, A.; Burd, C.G.; Dimaio, D. Direct Binding of Retromer to Human Papillomavirus Type 16 Minor Capsid Protein L2 Mediates Endosome Exit during Viral Infection. PLoS Pathog. 2015, 11, e1004699. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-C.; Chathuranga, K.; Lee, J.-S. Intracellular Sensing of Viral Genomes and Viral Evasion. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Chan, Y.K.; Gack, M.U. Viral Evasion of Intracellular DNA and RNA Sensing. Nat. Rev. Microbiol. 2016, 14, 360–373. [Google Scholar] [CrossRef]
- Xie, J.; Zhang, P.; Crite, M.; Lindsay, C.V.; DiMaio, D. Retromer Stabilizes Transient Membrane Insertion of L2 Capsid Protein during Retrograde Entry of Human Papillomavirus. Sci. Adv. 2021, 7, eabh4276. [Google Scholar] [CrossRef] [PubMed]
- Broniarczyk, J.; Bergant, M.; Goździcka-Józefiak, A.; Banks, L. Human Papillomavirus Infection Requires the TSG101 Component of the ESCRT Machinery. Virology 2014, 460, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. P62/SQSTM1 Functions as a Signaling Hub and an Autophagy Adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souquere, S.; Weil, D.; Pierron, G. Comparative Ultrastructure of CRM1-Nucleolar Bodies (CNoBs), Intranucleolar Bodies (INBs) and Hybrid PML/P62 Bodies Uncovers New Facets of Nuclear Body Dynamic and Diversity. Nucleus 2015, 6, 326–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Wu, R.; Zheng, J.; Li, P.; Yu, L. Polyubiquitin Chain-Induced P62 Phase Separation Drives Autophagic Cargo Segregation. Cell Res. 2018, 28, 405–415. [Google Scholar] [CrossRef] [Green Version]
- Fu, A.; Cohen-Kaplan, V.; Avni, N.; Livneh, I.; Ciechanover, A. P62-Containing, Proteolytically Active Nuclear Condensates, Increase the Efficiency of the Ubiquitin–Proteasome System. Proc. Natl. Acad. Sci. USA 2021, 118, e2107321118. [Google Scholar] [CrossRef]
- Ishii, Y. Electron Microscopic Visualization of Autophagosomes Induced by Infection of Human Papillomavirus Pseudovirions. Biochem. Biophys. Res. Commun. 2013, 433, 385–389. [Google Scholar] [CrossRef]
- Besemer, A.S.; Maus, J.; Ax, M.D.A.; Stein, A.; Vo, S.; Freese, C.; Nalbach, K.; von Hilchen, C.; Pfalzgraf, I.F.; Koziollek-Drechsler, I.; et al. Receptor-Mediated Endocytosis 8 (RME-8)/DNAJC13 Is a Novel Positive Modulator of Autophagy and Stabilizes Cellular Protein Homeostasis. Cell. Mol. Life Sci. 2020, 21, 1406–1416. [Google Scholar] [CrossRef] [Green Version]
- Korolchuk, V.I.; Mansilla, A.; Menzies, F.M.; Rubinsztein, D.C. Autophagy Inhibition Compromises Degradation of Ubiquitin-Proteasome Pathway Substrates. Mol. Cell 2009, 33, 517–527. [Google Scholar] [CrossRef]
- Krause, M.; Yakimovich, A.; Kriston-Vizi, J.; Huttunen, M.; Mercer, J. Vaccinia Virus Subverts Xenophagy through Phosphorylation and Nuclear Targeting of P62. Biorxiv 2021. [Google Scholar] [CrossRef]
- Pankiv, S.; Lamark, T.; Bruun, J.-A.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. Nucleocytoplasmic Shuttling of P62/SQSTM1 and Its Role in Recruitment of Nuclear Polyubiquitinated Proteins to Promyelocytic Leukemia Bodies. J. Biol. Chem. 2010, 285, 5941–5953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, C.; Krämer, N.; Krauter, S.; Strand, D.; Sehn, E.; Wolfrum, U.; Freiwald, A.; Butter, F.; Plachter, B. Autophagy Interferes with Human Cytomegalovirus Genome Replication, Morphogenesis, and Progeny Release. Autophagy 2020, 17, 779–795. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Luo, Z.; Yan, C.-Y.; Wang, X.-H.; He, Z.-J.; Ouyang, S.-H.; Liu, L.-F.; Zhou, Q.-Q.; Mu, H.-L.; Gong, H.-B.; et al. Autophagic Degradation of PML Promotes Susceptibility to HSV-1 by Stress-Induced Corticosterone. Theranostics 2020, 10, 9032–9049. [Google Scholar] [CrossRef] [PubMed]
- Stepp, W.H.; Meyers, J.M.; McBride, A.A. Sp100 Provides Intrinsic Immunity against Human Papillomavirus Infection. Mbio 2013, 4, e00845-13. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Cao, L.; Kang, R.; Yang, M.; Liu, L.; Zhao, Y.; Yu, Y.; Xie, M.; Yin, X.; Livesey, K.M.; et al. Autophagy Regulates Myeloid Cell Differentiation by P62/SQSTM1-Mediated Degradation of PML-RARα Oncoprotein. Autophagy 2011, 7, 401–411. [Google Scholar] [CrossRef] [Green Version]
- Skilling, J.; Denny, R.; Richardson, K.; Young, P.; McKenna, T.; Campuzano, I.; Ritchie, M. ProbSeq—A Fragmentation Model for Interpretation of Electrospray Tandem Mass Spectrometry Data. Comp. Funct. Genom. 2004, 5, 61–68. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schweiger, L.; Lelieveld-Fast, L.A.; Mikuličić, S.; Strunk, J.; Freitag, K.; Tenzer, S.; Clement, A.M.; Florin, L. HPV16 Induces Formation of Virus-p62-PML Hybrid Bodies to Enable Infection. Viruses 2022, 14, 1478. https://0-doi-org.brum.beds.ac.uk/10.3390/v14071478
Schweiger L, Lelieveld-Fast LA, Mikuličić S, Strunk J, Freitag K, Tenzer S, Clement AM, Florin L. HPV16 Induces Formation of Virus-p62-PML Hybrid Bodies to Enable Infection. Viruses. 2022; 14(7):1478. https://0-doi-org.brum.beds.ac.uk/10.3390/v14071478
Chicago/Turabian StyleSchweiger, Linda, Laura A. Lelieveld-Fast, Snježana Mikuličić, Johannes Strunk, Kirsten Freitag, Stefan Tenzer, Albrecht M. Clement, and Luise Florin. 2022. "HPV16 Induces Formation of Virus-p62-PML Hybrid Bodies to Enable Infection" Viruses 14, no. 7: 1478. https://0-doi-org.brum.beds.ac.uk/10.3390/v14071478