
Selection Pressure in CD8+ T-cell Epitopes in the pol Gene of HIV-1 Infected Individuals in Colombia. A Bioinformatic Approach


Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Sources and Sequence Alignments
2.2. Tests of Positive Selection
2.3. Identification of Peptides
2.4. Collection and Preparation of the Three-Dimensional Structure of HLA Molecules
2.5. Structural Prediction of Peptides
2.6. HLA-Peptide Binding Predictions
3. Results
3.1. Z-Test and Maximum Likelihood Analysis of Positive Selection

{kind=link}
| Codons Position (HXB2) | Substitution | dN/dSa | p Value | Location b |
|---|---|---|---|---|
| Protease | ||||
| 12 | T → P/S | 4.4 | 0.0000 * | Epitope |
| 13 | I → V | 5.0 | 0.0002 | Epitope |
| 19 | L → I/Q/V | 1.7 | 0.0015 | Epitope |
| 35 | E → D | 1.9 | 0.0038 | Epitope |
| 37 | S→N/E/D | 5.7 | 0.0000 * | Epitope |
| 41 | R→K | 2.9 | 0.0000 * | Epitope |
| 54 | I → L/V/M/T/A/S | 3.0 | 0.0000 * | Epitope/DRAS |
| 62 | I → V | 2.9 | 0.0002 | DRAS |
| 64 | I → L/M/V | 6.8 | 0.0000 * | DRAS |
| 71 | A → V/I/T/L | 2.3 | 0.0000 * | DRAS |
| 72 | I → V/T/L/R | 2.1 | 0.0115 | Epitope |
| 73 | G → C/S/T/A | 2.1 | 0.0146 | DRAS |
| 74 | T → P | 15.6 | 0.0000 * | DRAS |
| 77 | V → I | 5.3 | 0.0000 * | Epitope/DRAS |
| 82 | V → A/F/S/T | 4.2 | 0.0000 * | Epitope/DRAS |
| 85 | I → V | 36 | 0.0460 | DRAS |
| 90 | L → M | 4.4 | 0.0000 * | DRAS |
| 93 | I → L/M | 8.2 | 0.0000 * | DRAS |
| Reverse transcriptase | ||||
| 39 | T → A/K/S/L | 4.4 | 0.0000 * | Epitope |
| 48 | S → T | 2.6 | 0.0203 | Epitope |
| 69 | T → S/N/D/A/G | 2.2 | 0.0008 | DRAS |
| 74 | L → V/I | 3.0 | 0.0000 * | Epitope/DRAS |
| 75 | V → I | 2.4 | 0.0024 | DRAS |
| 98 | A → S | 2.0 | 0.0106 | Epitope |
| 102 | K → R/Q/E/N/H | 20.1 | 0.0000 * | DRAS |
| 103 | K → N/S | 2.0 | 0.0003 | DRAS |
| 118 | V → I | 1.7 | 0.0179 | Epitope |
| 135 | I → T/V/L/R/M/K | 3.3 | 0.0000 * | Epitope |
| 162 | S → C/A/Y/D/N/H | 2.1 | 0.0003 | Epitope |
| 184 | M → V | 2.5 | 0.0000 * | Epitope/DRAS |
| 188 | Y → L | 4.8 | 0.0002 | DRAS |
| 200 | T → A/I/E | 17.4 | 0.0000 * | Epitope |
| 202 | I → V | 606.1 | 0.0083 | Epitope |
| 211 | R → K/Q/G/T | 1.5 | 0.0051 | Epitope |
| 215 | T → I | 3.4 | 0.0000 * | Epitope |
3.2. Identification of CD8+ T-cell Epitopes with Amino Acid Substitutions
| Mutations | Frequency (%) | Epitope Affected (HLA Alleles) a | Association b |
|---|---|---|---|
| Protease | |||
| I13V | 20.4 | QRPLVTIKI (A*01:01) | NC |
| QRPLVTIKIG (B51) | NC | ||
| VTIKIGGQLK (A*11:01) | SF | ||
| TIKIGGQLK (A3 supertype) | NC | ||
| L19I | 9.0 | VTIKIGGQLK (A*11:01, A*03:01) | SF |
| TIKIGGQLK (A3 supertype) | NC | ||
| E35D | 28.7 | DTVLEEMSL (A*68:02) | NC |
| EEMSLPGRW (B*44:02, B*44:03, B18, B40) | IE | ||
| S37N | 56.0 | DTVLEEMSL (A*68:02) | SF |
| EEMSLPGRW (B*44:02, B*44:03, B18, B40) | SF | ||
| S37D | 14.1 | DTVLEEMSL (A*68:02) | NC |
| EEMSLPGRW (B*44:02, B*44:03, B18, B40) | SF | ||
| R41K | 42.8 | EEMSLPGRW (B*44:02, B*44:03, B18, B40) | NC |
| LPGRWKPKMI (Cw3) | NC | ||
| I54Vc | 19.3 | KMIGGIGGFI (B62) | IE |
| I72V | 9.3 | IEICGHKAIG (B18, B40, B44) | NC |
| GHKAIGTVL (B15) | NC | ||
| I72T | 5.5 | IEICGHKAIG (B18, B40, B44) | NC |
| GHKAIGTVL (B15) | NC | ||
| V77Ic | 27.8 | LVGPTPVNI (A2) | NC |
| V82A c | 13.8 | LVGPTPVNI (A2) | IE |
| Reverse Transcriptase | |||
| T39A | 7.1 | ALVEICTEM (A*02, A*02:01, A2) | NC |
| A98S | 8.7 | GIPHPAGLK (A*03:01) | NC |
| V118I | 19.1 | VLDVGDAYFSV (A*02:01) | NC |
| DAYFSVPL (A24, B*51:01) | NC | ||
| I135T | 30.7 | KYTAFTIPSI (A2) | NC |
| TAFTIPSI (B*51) | IE | ||
| I35V | 7.7 | KYTAFTIPSI (A2) | NC |
| TAFTIPSI (B*51) | IE | ||
| S162C | 10.1 | SPAIFQSSM (B7, B35) | SF |
| AIFQSSMTK (A*03:01) | SF | ||
| T200A | 19.0 | DLEIGQHRTK (A3) | NC |
| I202V | 6.8 | KIEELRQHL (A2) | NC |
| KIEELRQHLL (B58) | NC | ||
| IEELRQHLL (B*40:01, B60, B61) | IE | ||
| R211K | 49.3 | EELRQHLLRW (B44) | NC |
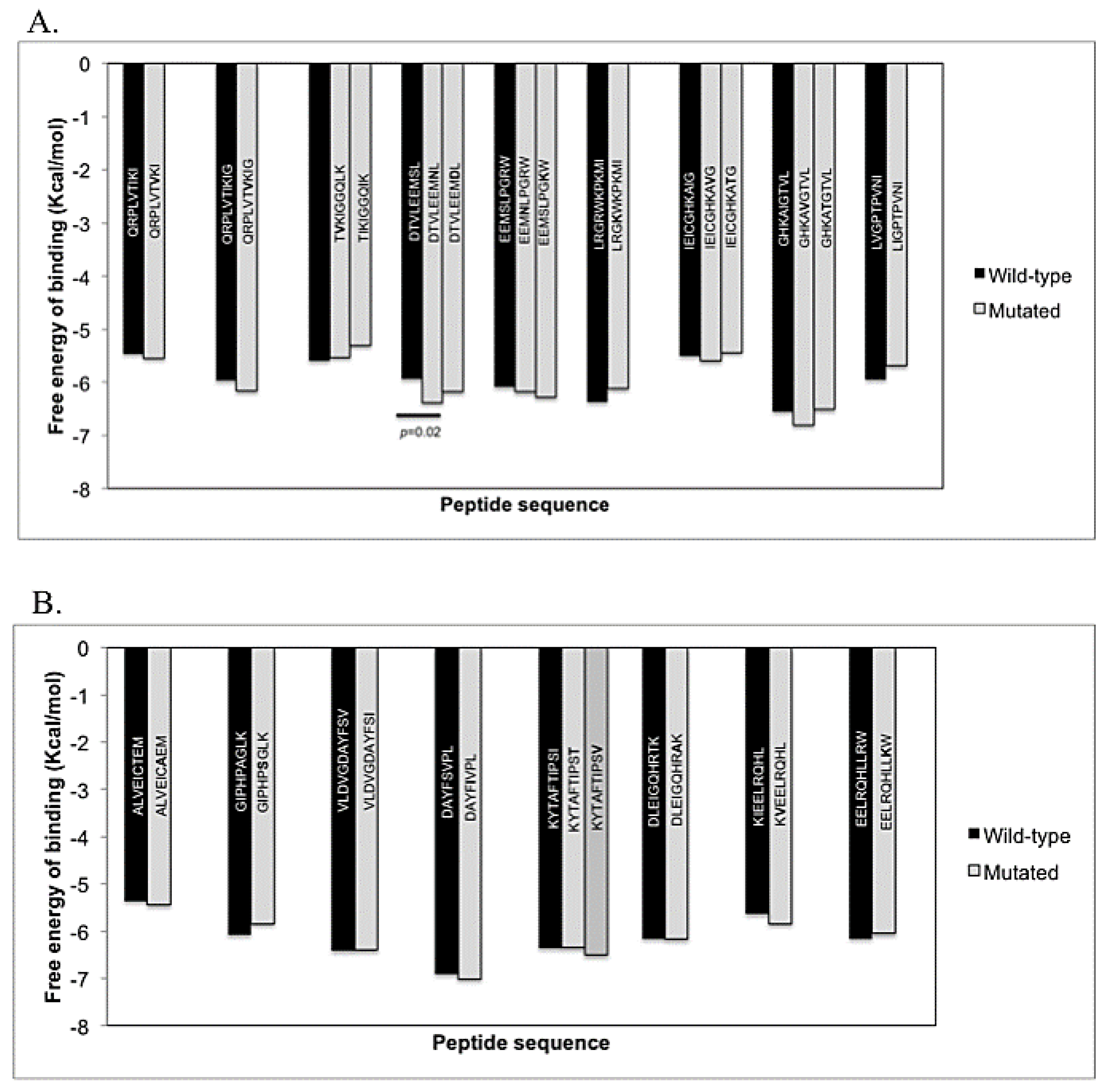
3.3. Docking Simulation and Algorithmic Estimation of the Affinity of Peptides Binding to HLA Molecules

| Amino Acid Sequence | Alleles | SMM | NetMHC | NetMHCpan | ||||
|---|---|---|---|---|---|---|---|---|
| Affinity (nM) | Affinity (nM) | Affinity (nM) | ||||||
| LPPVVAKEI a | B*51 | 172 | 102 | 797 | ||||
| NLVPMVATV a | A*02 | 66 | 29 | 21 | ||||
| Protease | ||||||||
| QRPLVTIKI | A*01:01 | 194334 | 21837 | 36562 | ||||
| QRPLVTVKI | 231499 | 21639 | 37194 | |||||
| QRPLVTIKIG | B51 | 166360 | 30751 | 43536 | ||||
| QRPLVTVKIG | 168286 | 30708 | 43446 | |||||
| TIKIGGQLK | A3 | 432 | 582 | 537 | ||||
| TVKIGGQLK | 456 | 720 | 856 | |||||
| TIKIGGQIK | 1041 b | 2126 b | 1654 b | |||||
| DTVLEEMSL | A*68:02 | 874 | 2686 | 1709 | ||||
| DTVLEEMNL | 1012 | 3795 | 2078 | |||||
| DTVLEEMDL | 2278 b | 12370 b | 6676 b | |||||
| EEMSLPGRW | B*44:02 | 30 | 25 | 14 | ||||
| EEMNLPGRW | 32 | 28 | 21 | |||||
| EEMSLPGKW | 30 | 22 | 14 | |||||
| EDMSLPGRW | 431 b | 565 b | 578 b | |||||
| IEICGHKAIG | B44 | 2093 | 6969 | 6674 | ||||
| IEICGHKAVG | 2122 | 5792 | 5601 | |||||
| IEICGHKATG | 2103 | 5060 | 5162 | |||||
| GHKAIGTVL | B15 | 13091 | 14972 | 15778 | ||||
| GHKAVGTVL | 9840 | 13657 | 14015 | |||||
| GHKATGTVL | 8222 | 12947 | 14673 | |||||
| LVGPTPVNI | A2 | 3027 | 3829 | 4005 | ||||
| LIGPTPVNI | 1945 | 2195 | 1596 | |||||
| LVGPTPANI | 3555 | 3014 | 3912 | |||||
| KMIGGIGGFI | B62 | 514 | 415 | 769 | ||||
| KMIGGIGGFV | 1493 b | 937 b | 1744 b | |||||
| Reverse transcriptase | ||||||||
| ALVEICTEM | A2 | 116 | 70 | 41 | ||||
| ALVEICAEM | 101 | 50 | 32 | |||||
| GIPHPAGLK | A*03:01 | 290 | 108 | 316 | ||||
| GIPHPSGLK | 266 | 99 | 334 | |||||
| VLDVGDAYFSV | A*02:01 | 4314 | 284 | 9 | ||||
| VLDVGDAYFSI | 8788 b | 534 | 33 b | |||||
| DAYFSVPL | B*51:01 | 7628 | 6527 | 4202 | ||||
| DAYFSIPL | 15150 | 7805 | 4148 | |||||
| KYTAFTIPSI | A2 | 1470 | 5199 | 9216 | ||||
| KYTAFTIPST | 4925 b | 15627 b | 27884 b | |||||
| KYTAFTIPSV | 381 | 1509 | 5743 | |||||
| Reverse transcriptase | ||||||||
| TAFTIPSI | B51 | 996 | 2399 | 1153 | ||||
| TAFTIPST | 1128 | 16898 b | 17009 b | |||||
| TAFTIPSV | 1372 | 4052 | 2887 b | |||||
| DLEIGQHRTK | A3 | 798 | 8188 | 15564 | ||||
| DLEIGQHRAK | 839 | 9011 | 15773 | |||||
| KIEELRQHL | A2 | 5689 | 10072 | 8180 | ||||
| KVEELRQHL | 8853 | 13644 | 14234 | |||||
| KIEELRQHL | A2 | 5689 | 10072 | 8180 | ||||
| KVEELRQHL | 8853 | 13644 | 14234 | |||||
| EELRQHLLRW | B44 | 78 | 104 | 29 | ||||
| EELRQHLLKW | 78 | 84 | 29 | |||||
4. Discussion
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Hemelaar, J.; Gouws, E.; Ghys, P.D.; Osmanov, S. Global and regional distribution of HIV-1 genetic subtypes and recombinants in 2004. Aids 2006, 20, W13–W23. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.L.; Anderson, J.P.; Bradac, J.A.; Carr, J.K.; Foley, B.; Funkhouser, R.K.; Gao, F.; Hahn, B.H.; Kalish, M.L.; Kuiken, C.; et al. Hiv-1 nomenclature proposal. Science 2000, 288, 55–56. [Google Scholar]
- Ayouba, A.; Mauclere, P.; Martin, P.M.; Cunin, P.; Mfoupouendoun, J.; Njinku, B.; Souquieres, S.; Simon, F. HIV-1 group o infection in cameroon, 1986 to 1998. Emerg. Infect. Dis. 2001, 7, 466–467. [Google Scholar] [CrossRef] [PubMed]
- Ayouba, A.; Souquieres, S.; Njinku, B.; Martin, P.M.; Muller-Trutwin, M.C.; Roques, P.; Barre-Sinoussi, F.; Mauclere, P.; Simon, F.; Nerrienet, E. HIV-1 group n among HIV-1-seropositive individuals in cameroon. Aids 2000, 14, 2623–2625. [Google Scholar] [CrossRef] [PubMed]
- Plantier, J.C.; Leoz, M.; Dickerson, J.E.; De Oliveira, F.; Cordonnier, F.; Lemee, V.; Damond, F.; Robertson, D.L.; Simon, F. A new human immunodeficiency virus derived from gorillas. Nat. Med. 2009, 15, 871–872. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.L.; Sharp, P.M.; McCutchan, F.E.; Hahn, B.H. Recombination in hiv-1. Nature 1995, 374, 124–126. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, G.I.; Bautista, C.T.; Eyzaguirre, L.; Carrion, G.; Arias, S.; Sateren, W.B.; Negrete, M.; Montano, S.M.; Sanchez, J.L.; Carr, J.K. Molecular epidemiology of human immunodeficiency virus-infected individuals in Medellin, Colombia. Am. J. Trop. Med. Hyg. 2006, 74, 674–677. [Google Scholar] [PubMed]
- Montano, S.M.; Sanchez, J.L.; Laguna-Torres, A.; Cuchi, P.; Avila, M.M.; Weissenbacher, M.; Serra, M.; Vinoles, J.; Russi, J.C.; Aguayo, N.; et al. Prevalences, genotypes, and risk factors for HIV transmission in south america. J. Acquir. Immune Defic. Syndr. 2005, 40, 57–64. [Google Scholar] [CrossRef]
- Ho, D.D.; Neumann, A.U.; Perelson, A.S.; Chen, W.; Leonard, J.M.; Markowitz, M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 1995, 373, 123–126. [Google Scholar] [CrossRef] [PubMed]
- O'Neil, P.K.; Sun, G.; Yu, H.; Ron, Y.; Dougherty, J.P.; Preston, B.D. Mutational analysis of HIV-1 long terminal repeats to explore the relative contribution of reverse transcriptase and RNA polymerase II to viral mutagenesis. J. Biol. Chem. 2002, 277, 38053–38061. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.P.; Loeb, L.A. Fidelity of HIV-1 reverse transcriptase copying RNA in vitro. Biochemistry 1992, 31, 954–958. [Google Scholar] [CrossRef] [PubMed]
- Abram, M.E.; Ferris, A.L.; Shao, W.; Alvord, W.G.; Hughes, S.H. Nature, position, and frequency of mutations made in a single cycle of HIV-1 replication. J. Virol. 2010, 84, 9864–9878. [Google Scholar] [CrossRef] [PubMed]
- Mansky, L.M.; Temin, H.M. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 1995, 69, 5087–5094. [Google Scholar] [PubMed]
- Simon, V.; Ho, D.D. HIV-1 dynamics in vivo: Implications for therapy. Nat. Rev. Microbiol. 2003, 1, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Leslie, A.J.; Pfafferott, K.J.; Chetty, P.; Draenert, R.; Addo, M.M.; Feeney, M.; Tang, Y.; Holmes, E.C.; Allen, T.; Prado, J.G.; et al. HIV evolution: CTL escape mutation and reversion after transmission. Nat. Med. 2004, 10, 282–289. [Google Scholar]
- Marozsan, A.J.; Fraundorf, E.; Abraha, A.; Baird, H.; Moore, D.; Troyer, R.; Nankja, I.; Arts, E.J. Relationships between infectious titer, capsid protein levels, and reverse transcriptase activities of diverse human immunodeficiency virus type 1 isolates. J. Virol. 2004, 78, 11130–11141. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, D.S.; Willey, R.L.; Sato, H.; Chang, L.J.; Blumenthal, R.; Martin, M.A. Quantitation of human immunodeficiency virus type 1 infection kinetics. J. Virol. 1993, 67, 2182–2190. [Google Scholar] [PubMed]
- Hemelaar, J. Implications of HIV diversity for the HIV-1 pandemic. J. Infect. 2013, 66, 391–400. [Google Scholar] [CrossRef] [PubMed]
- De, S.L.E.; Holmes, E.C.; Zanotto, P.M. Distinct patterns of natural selection in the reverse transcriptase gene of HIV-1 in the presence and absence of antiretroviral therapy. Virology 2004, 325, 181–191. [Google Scholar] [CrossRef] [PubMed]
- McMichael, A.J.; Borrow, P.; Tomaras, G.D.; Goonetilleke, N.; Haynes, B.F. The immune response during acute HIV-1 infection: Clues for vaccine development. Nat. Rev. Immunol. 2010, 10, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, Y.; Pfafferott, K.; Frater, J.; Matthews, P.; Payne, R.; Addo, M.; Gatanaga, H.; Fujiwara, M.; Hachiya, A.; Koizumi, H.; et al. Adaptation of HIV-1 to human leukocyte antigen class I. Nature 2009, 458, 641–645. [Google Scholar]
- Moore, C.B.; John, M.; James, I.R.; Christiansen, F.T.; Witt, C.S.; Mallal, S.A. Evidence of HIV-1 adaptation to HLA-restricted immune responses at a population level. Science 2002, 296, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Matthews, P.C.; Leslie, A.J.; Katzourakis, A.; Crawford, H.; Payne, R.; Prendergast, A.; Power, K.; Kelleher, A.D.; Klenerman, P.; Carlson, J.; et al. HLA footprints on human immunodeficiency virus type 1 are associated with interclade polymorphisms and intraclade phylogenetic clustering. J. Virol. 2009, 83, 4605–4615. [Google Scholar]
- Hemelaar, J. The origin and diversity of the HIV-1 pandemic. Trends Mol. Med. 2012, 18, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Brumme, Z.L.; Brumme, C.J.; Heckerman, D.; Korber, B.T.; Daniels, M.; Carlson, J.; Kadie, C.; Bhattacharya, T.; Chui, C.; Szinger, J.; et al. Evidence of differential HLA class I-mediated viral evolution in functional and accessory/regulatory genes of HIV-1. PLoS Pathog. 2007, 3, e94. [Google Scholar]
- Kelleher, A.D.; Long, C.; Holmes, E.C.; Allen, R.L.; Wilson, J.; Conlon, C.; Workman, C.; Shaunak, S.; Olson, K.; Goulder, P.; et al. Clustered mutations in HIV-1 gag are consistently required for escape from HLA-B27-restricted cytotoxic T lymphocyte responses. J. Exp. Med. 2001, 193, 375–386. [Google Scholar]
- Feeney, M.E.; Tang, Y.; Roosevelt, K.A.; Leslie, A.J.; McIntosh, K.; Karthas, N.; Walker, B.D.; Goulder, P.J. Immune escape precedes breakthrough human immunodeficiency virus type 1 viremia and broadening of the cytotoxic T-lymphocyte response in an HLA-B27-positive long-term-nonprogressing child. J. Virol. 2004, 78, 8927–8930. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, Y.; Kuse, N.; Gatanaga, H.; Naruto, T.; Fujiwara, M.; Dohki, S.; Akahoshi, T.; Maenaka, K.; Goulder, P.; Oka, S.; et al. Long-term control of HIV-1 in hemophiliacs carrying slow-progressing allele HLA-B*5101. J. Virol. 2010, 84, 7151–7160. [Google Scholar]
- Grant, R.M.; Kuritzkes, D.R.; Johnson, V.A.; Mellors, J.W.; Sullivan, J.L.; Swanstrom, R.; D’Aquila, R.T.; van Gorder, M.; Holodniy, M.; Lloyd, R.M., Jr.; et al. Accuracy of the trugene HIV-1 genotyping kit. J. Clin. Microbiol. 2003, 41, 1586–1593. [Google Scholar]
- Gómez, S.; Olaya-García, P.; Díaz, F. Resistencia a los medicamentos antirretrovirales en pacientes que reciben tratamiento para vih-sida en colombia. Infectio 2010, 14, 248–256. [Google Scholar] [CrossRef]
- Pineda-Peña, A.; Rodrigues Faria, N.; Diaz, F.; Olaya-García, P.; Moller Frederiksen, C.; Guangdi, L.; Gomez-Lopez, A.; Lemey, P.; Vandamme, A. The colombian pidemic is dominated by HIV-1subtype b over time: A molecular epidemiolog and phylodynamics study. In Proceedings of the 20th International HIV Dynamics and Evolution, Utrecht, The Netherlands, 8–11 May 2013.
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. Mega5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Gojobori, T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 1986, 3, 418–426. [Google Scholar] [PubMed]
- Pond, S.L.; Frost, S.D.; Muse, S.V. Hyphy: Hypothesis testing using phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Muse, S.V.; Gaut, B.S. A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with application to the chloroplast genome. Mol. Biol. Evol. 1994, 11, 715–724. [Google Scholar] [PubMed]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press, Inc: New York, NY, USA, 2000; pp. 147–163. [Google Scholar]
- Kosakovsky Pond, S.L.; Frost, S.D. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef]
- Suzuki, Y.; Gojobori, T. A method for detecting positive selection at single amino acid sites. Mol. Biol. Evol. 1999, 16, 1315–1328. [Google Scholar] [CrossRef] [PubMed]
- Delport, W.; Poon, A.F.; Frost, S.D.; Kosakovsky Pond, S.L. Datamonkey 2010: A suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 2010, 26, 2455–2457. [Google Scholar] [CrossRef] [PubMed]
- HIV molecular immunology database. Available online: http://www.hiv.lanl.gov/ (accessed on 12 February 2014).
- Liu, T.F.; Shafer, R.W. Web Resources for HIV type 1 genotypic-resistance Test Interpretation. Clin. Infect. Dis. 2006, 42, 1608–1618. [Google Scholar] [CrossRef] [PubMed]
- HIV drug resistance database – Stanford University. Available online: http://hivdb.stanford.edu/ (accessed on 16 May 2014).
- Johnson, V.A.; Calvez, V.; Gunthard, H.F.; Paredes, R.; Pillay, D.; Shafer, R.W.; Wensing, A.M.; Richman, D.D. Update of the drug resistance mutations in HIV-1: March 2013. Top. Antivir. Med. 2013, 21, 6–14. [Google Scholar] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic. Acids. Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Green, L.G.; Radic, Z.; Taylor, P.; Sharpless, K.B.; Olson, A.J.; Grynszpan, F. Automated docking with protein flexibility in the design of femtomolar “click chemistry” inhibitors of acetylcholinesterase. J. Chem. Inf. Model. 2013, 53, 898–906. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Lambert, C.; Leonard, N.; de Bolle, X.; Depiereux, E. ESyPred3D: Prediction of proteins 3D structures. Bioinformatics 2002, 18, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. Autodock vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [PubMed]
- Hoof, I.; Peters, B.; Sidney, J.; Pedersen, L.E.; Sette, A.; Lund, O.; Buus, S.; Nielsen, M. Netmhcpan, a method for mhc class I binding prediction beyond humans. Immunogenetics 2009, 61, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Lundegaard, C.; Blicher, T.; Lamberth, K.; Harndahl, M.; Justesen, S.; Roder, G.; Peters, B.; Sette, A.; Lund, O.; et al. Netmhcpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS One 2007, 2, e796. [Google Scholar]
- Buus, S.; Lauemoller, S.L.; Worning, P.; Kesmir, C.; Frimurer, T.; Corbet, S.; Fomsgaard, A.; Hilden, J.; Holm, A.; Brunak, S. Sensitive quantitative predictions of peptide-MHC binding by a “query by committee” artificial neural network approach. Tissue Antigens 2003, 62, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Peters, B.; Sette, A. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinform. 2005, 6, 132. [Google Scholar] [CrossRef] [Green Version]
- Immune epitope database and analysis resource. Available online: http://www.iedb.org/ (accessed on 12 January 2015).
- Sette, A.; Vitiello, A.; Reherman, B.; Fowler, P.; Nayersina, R.; Kast, W.M.; Melief, C.J.; Oseroff, C.; Yuan, L.; Ruppert, J.; et al. The relationship between class I binding affinity and immunogenicity of potential cytotoxic T cell epitopes. J. Immunol. 1994, 153, 5586–5592. [Google Scholar]
- Hachiya, A.; Gatanaga, H.; Kodama, E.; Ikeuchi, M.; Matsuoka, M.; Harada, S.; Mitsuya, H.; Kimura, S.; Oka, S. Novel patterns of nevirapine resistance-associated mutations of human immunodeficiency virus type 1 in treatment-naive patients. Virology 2004, 327, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Alteri, C.; Santoro, M.M.; Abbate, I.; Rozera, G.; Bruselles, A.; Bartolini, B.; Gori, C.; Forbici, F.; Orchi, N.; Tozzi, V.; et al. “Sentinel” mutations in standard population sequencing can predict the presence of HIV-1 reverse transcriptase major mutations detectable only by ultra-deep pyrosequencing. J. Antimicrob. Chemother. 2011, 66, 2615–2623. [Google Scholar]
- Sidney, J.; Southwood, S.; Sette, A. HLA Binding Peptides and Their Uses. U.S. Patent 20,060,079,453, 13 April 2006. [Google Scholar]
- Manosuthi, W.; Thongyen, S.; Nilkamhang, S.; Manosuthi, S.; Sungkanuparph, S. HIV-1 drug resistance-associated mutations among antiretroviral-naive thai patients with chronic HIV-1 infection. J. Med. Virol. 2013, 85, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Brockman, M.A.; Schneidewind, A.; Lahaie, M.; Schmidt, A.; Miura, T.; Desouza, I.; Ryvkin, F.; Derdeyn, C.A.; Allen, S.; Hunter, E.; et al. Escape and compensation from early HLA-B57-mediated cytotoxic T-lymphocyte pressure on human immunodeficiency virus type 1 gag alter capsid interactions with cyclophilin a. J. Virol. 2007, 81, 12608–12618. [Google Scholar]
- Smith, J.M.; Haigh, J. The hitch-hiking effect of a favourable gene. Genet. Res. 2007, 89, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Pennings, P.S.; Kryazhimskiy, S.; Wakeley, J. Loss and recovery of genetic diversity in adapting populations of HIV. PLoS Genet. 2014, 10, e1004000. [Google Scholar] [CrossRef] [PubMed]
- Neher, R.A.; Leitner, T. Recombination rate and selection strength in HIV intra-patient evolution. PLoS Comput. Biol. 2010, 6, e1000660. [Google Scholar] [CrossRef] [PubMed]
- Madden, D.R. The three-dimensional structure of peptide-MHC complexes. Annu. Rev. Immunol. 1995, 13, 587–622. [Google Scholar] [CrossRef] [PubMed]
- Ladell, K.; Hashimoto, M.; Iglesias, M.C.; Wilmann, P.G.; McLaren, J.E.; Gras, S.; Chikata, T.; Kuse, N.; Fastenackels, S.; Gostick, E.; et al. A molecular basis for the control of preimmune escape variants by HIV-specific CD8+ T cells. Immunity 2013, 38, 425–436. [Google Scholar]
- Reid, S.W.; McAdam, S.; Smith, K.J.; Klenerman, P.; O'Callaghan, C.A.; Harlos, K.; Jakobsen, B.K.; McMichael, A.J.; Bell, J.I.; Stuart, D.I.; et al. Antagonist HIV-1 Gag peptides induce structural changes in HLA B8. J. Exp. Med. 1996, 184, 2279–2286. [Google Scholar]
- Davenport, M.P. Antagonists or altruists: Do viral mutants modulate T-cell responses? Immunol. Today 1995, 16, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; O’Connor, D.H.; Jing, P.; Dzuris, J.L.; Mothe, B.R.; Vogel, T.U.; Dunphy, E.; Liebl, M.E.; Emerson, C.; Wilson, N.; et al. Tat-specific cytotoxic T lymphocytes select for siv escape variants during resolution of primary viraemia. Nature 2000, 407, 386–390. [Google Scholar]
- Koup, R.A.; Safrit, J.T.; Cao, Y.; Andrews, C.A.; McLeod, G.; Borkowsky, W.; Farthing, C.; Ho, D.D. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J. Virol. 1994, 68, 4650–4655. [Google Scholar] [PubMed]
- Borrow, P.; Lewicki, H.; Wei, X.; Horwitz, M.S.; Peffer, N.; Meyers, H.; Nelson, J.A.; Gairin, J.E.; Hahn, B.H.; Oldstone, M.B.; et al. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat. Med. 1997, 3, 205–211. [Google Scholar]
- Brumme, Z.L.; John, M.; Carlson, J.M.; Brumme, C.J.; Chan, D.; Brockman, M.A.; Swenson, L.C.; Tao, I.; Szeto, S.; Rosato, P.; et al. HLA-associated immune escape pathways in HIV-1 subtype B Gag, Pol and Nef proteins. PLoS One 2009, 4, e6687. [Google Scholar]
- Prado, J.G.; Honeyborne, I.; Brierley, I.; Puertas, M.C.; Martinez-Picado, J.; Goulder, P.J. Functional consequences of human immunodeficiency virus escape from an HLA-B*13-restricted CD8+ T-cell epitope in p1 Gag protein. J. Virol. 2009, 83, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.D.; Korber, B.T. Immune control of HIV: The obstacles of HLA and viral diversity. Nat. Immunol. 2001, 2, 473–475. [Google Scholar] [CrossRef]
- Carlson, J.M.; Brumme, Z.L. HIV evolution in response to HLA-restricted CTL selection pressures: A population-based perspective. Microbes Infect./Inst. Pasteur 2008, 10, 455–461. [Google Scholar] [CrossRef]
- Goulder, P.J.; Brander, C.; Tang, Y.; Tremblay, C.; Colbert, R.A.; Addo, M.M.; Rosenberg, E.S.; Nguyen, T.; Allen, R.; Trocha, A.; et al. Evolution and transmission of stable CTL escape mutations in HIV infection. Nature 2001, 412, 334–338. [Google Scholar]
- Rodriguez, L.M.; Giraldo, M.C.; Garcia, N.; Velasquez, L.; Paris, S.C.; Alvarez, C.M.; Garcia, L.F. Human leucocyte antigen gene (HLA-A, HLA-B, HLA-DRB1) frequencies in deceased organ donors. Biomedica 2007, 27, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Ponomarenko, J.; Zhu, Z.; Tamang, D.; Wang, P.; Greenbaum, J.; Lundegaard, C.; Sette, A.; Lund, O.; Bourne, P.E.; et al. Immune epitope database analysis resource. Nucleic Acids Res. 2012, 40, W525–W530. [Google Scholar]
- Peters, B.; Bui, H.H.; Frankild, S.; Nielson, M.; Lundegaard, C.; Kostem, E.; Basch, D.; Lamberth, K.; Harndahl, M.; Fleri, W.; et al. A community resource benchmarking predictions of peptide binding to MHC-I molecules. PLoS Comput. Biol. 2006, 2, e65. [Google Scholar]
- Dhanik, A.; McMurray, J.S.; Kavraki, L.E. Dinc: A new autodock-based protocol for docking large ligands. BMC Struct. Biol. 2013, 13, S11. [Google Scholar] [CrossRef] [PubMed]
- Konya, J.; Stuber, G.; Bjorndal, A.; Fenyo, E.M.; Dillner, J. Primary induction of human cytotoxic lymphocytes against a synthetic peptide of the human immunodeficiency virus type 1 protease. J. Gen. Virol. 1997, 78, 2217–2224. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acevedo-Sáenz, L.; Ochoa, R.; Rugeles, M.T.; Olaya-García, P.; Velilla-Hernández, P.A.; Diaz, F.J. Selection Pressure in CD8+ T-cell Epitopes in the pol Gene of HIV-1 Infected Individuals in Colombia. A Bioinformatic Approach. Viruses 2015, 7, 1313-1331. https://0-doi-org.brum.beds.ac.uk/10.3390/v7031313
Acevedo-Sáenz L, Ochoa R, Rugeles MT, Olaya-García P, Velilla-Hernández PA, Diaz FJ. Selection Pressure in CD8+ T-cell Epitopes in the pol Gene of HIV-1 Infected Individuals in Colombia. A Bioinformatic Approach. Viruses. 2015; 7(3):1313-1331. https://0-doi-org.brum.beds.ac.uk/10.3390/v7031313
Chicago/Turabian StyleAcevedo-Sáenz, Liliana, Rodrigo Ochoa, Maria Teresa Rugeles, Patricia Olaya-García, Paula Andrea Velilla-Hernández, and Francisco J. Diaz. 2015. "Selection Pressure in CD8+ T-cell Epitopes in the pol Gene of HIV-1 Infected Individuals in Colombia. A Bioinformatic Approach" Viruses 7, no. 3: 1313-1331. https://0-doi-org.brum.beds.ac.uk/10.3390/v7031313