
Exploring Taxifolin Polymorphs: Insights on Hydrate and Anhydrous Forms

 ,
,  , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
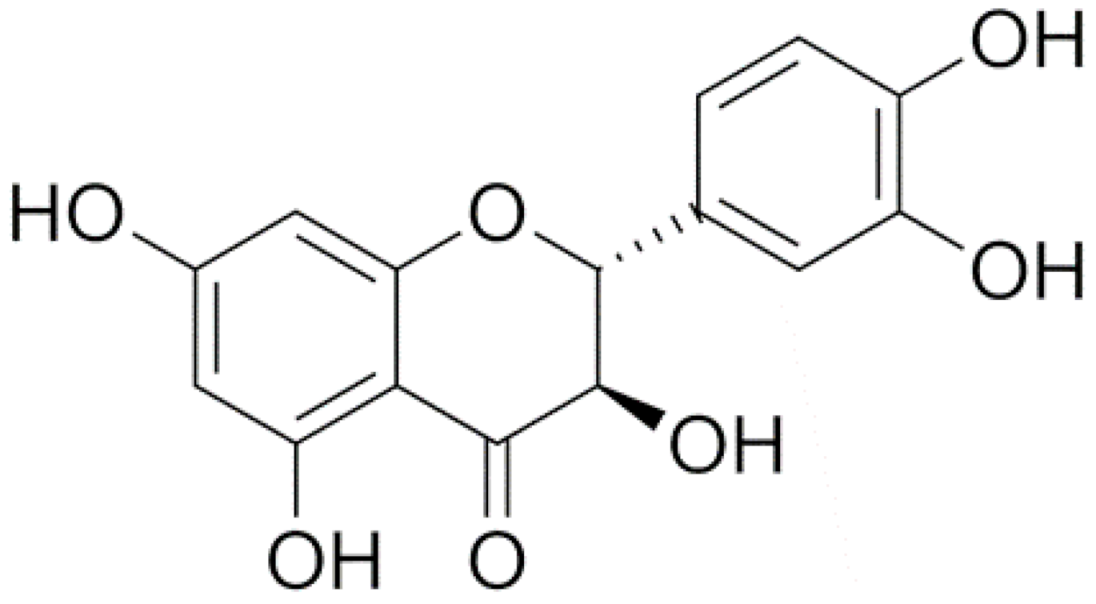
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Crystallization
2.3. X-ray Powder Diffraction
2.4. Scanning Electron Microscopy
2.5. Particle Size Analysis
2.6. Tapped Density Determination
2.7. Thermal Analysis
2.8. Infrared Spectroscopy
2.9. Stability Study
2.10. Solubility Studies
2.10.1. Shake-Flask Method
2.10.2. Intrinsic Dissolution Rate
2.10.3. Tax Dissolution Profile
2.11. Statistical Analysis
3. Results and Discussion
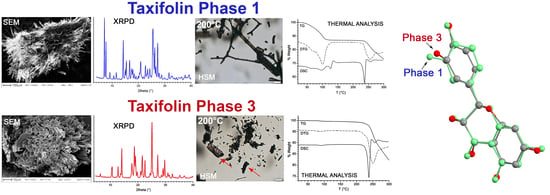
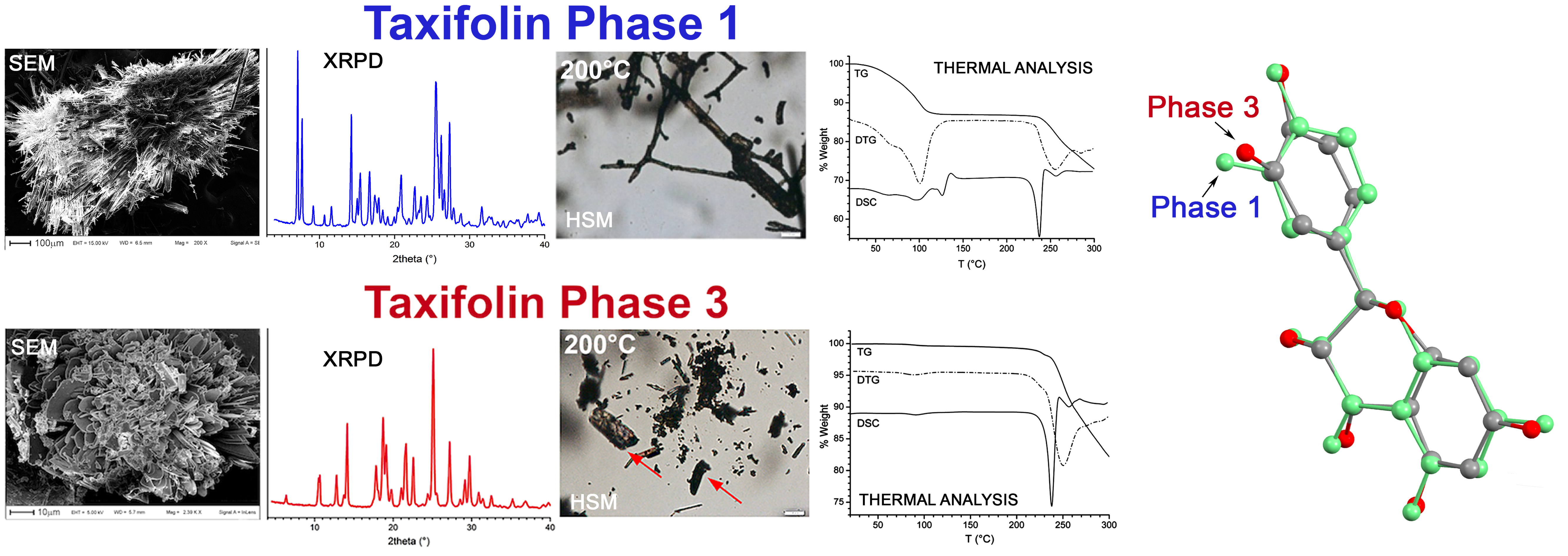
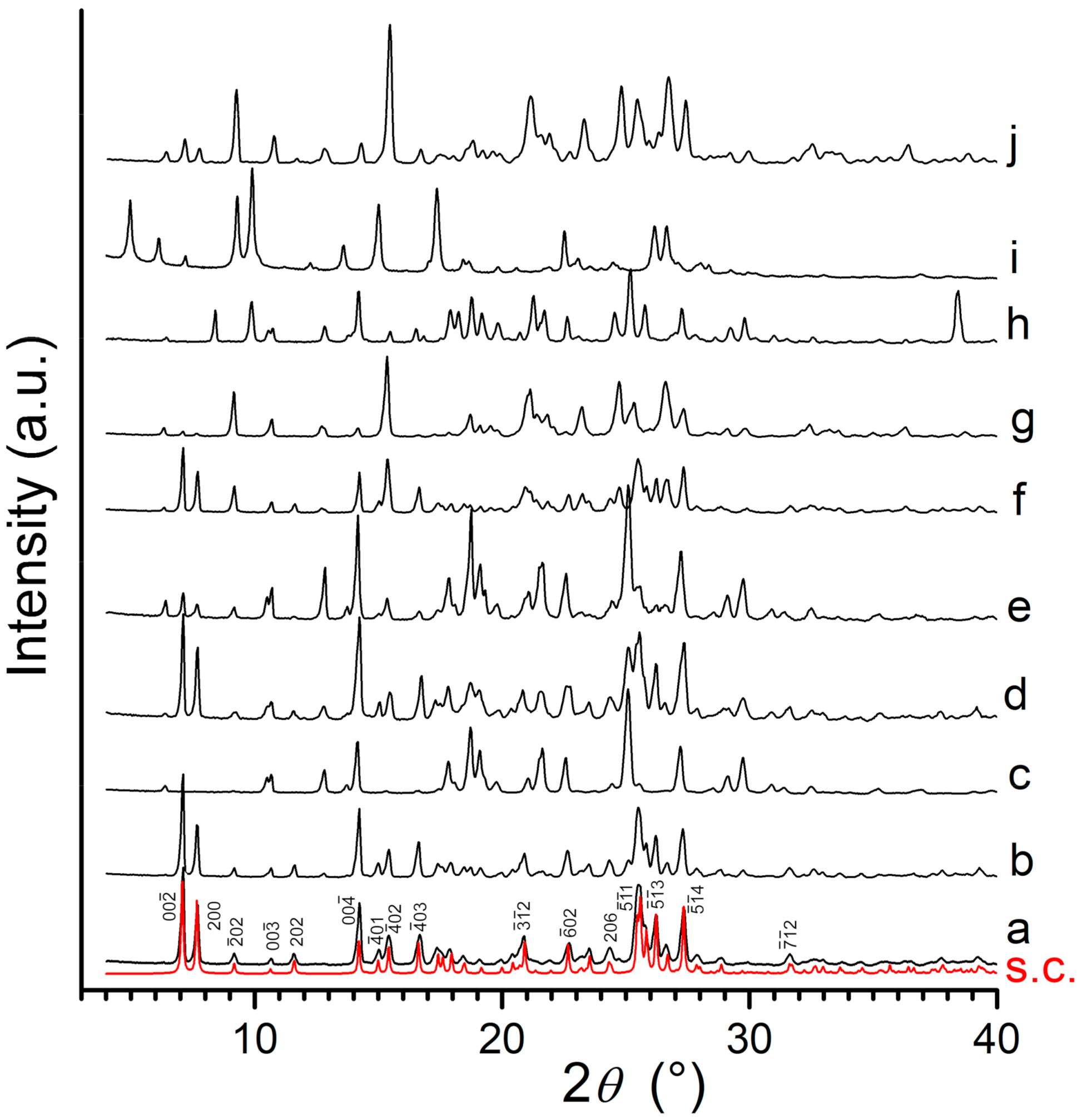
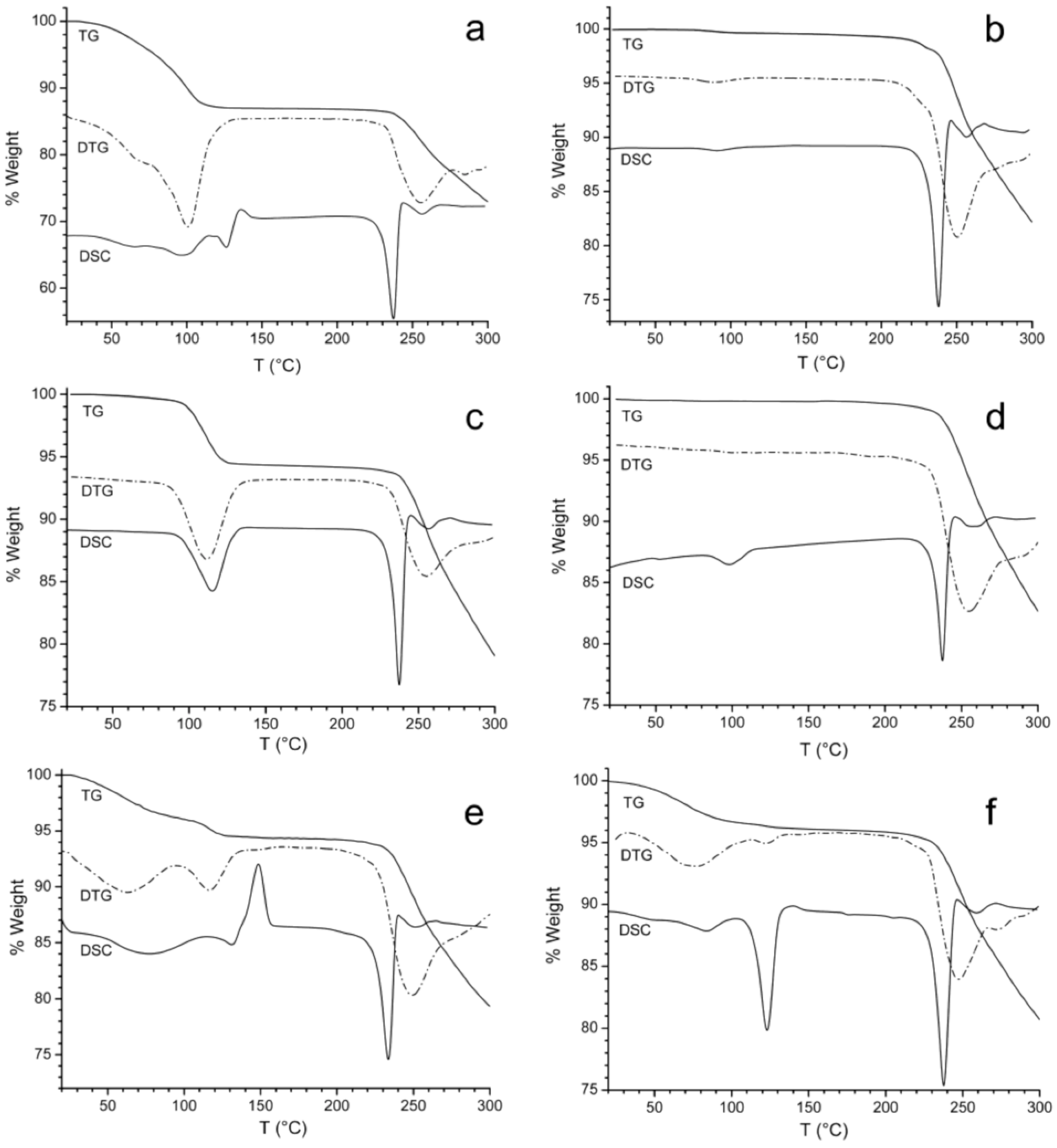
3.1. X-ray Powder Diffraction and Thermal Analysis
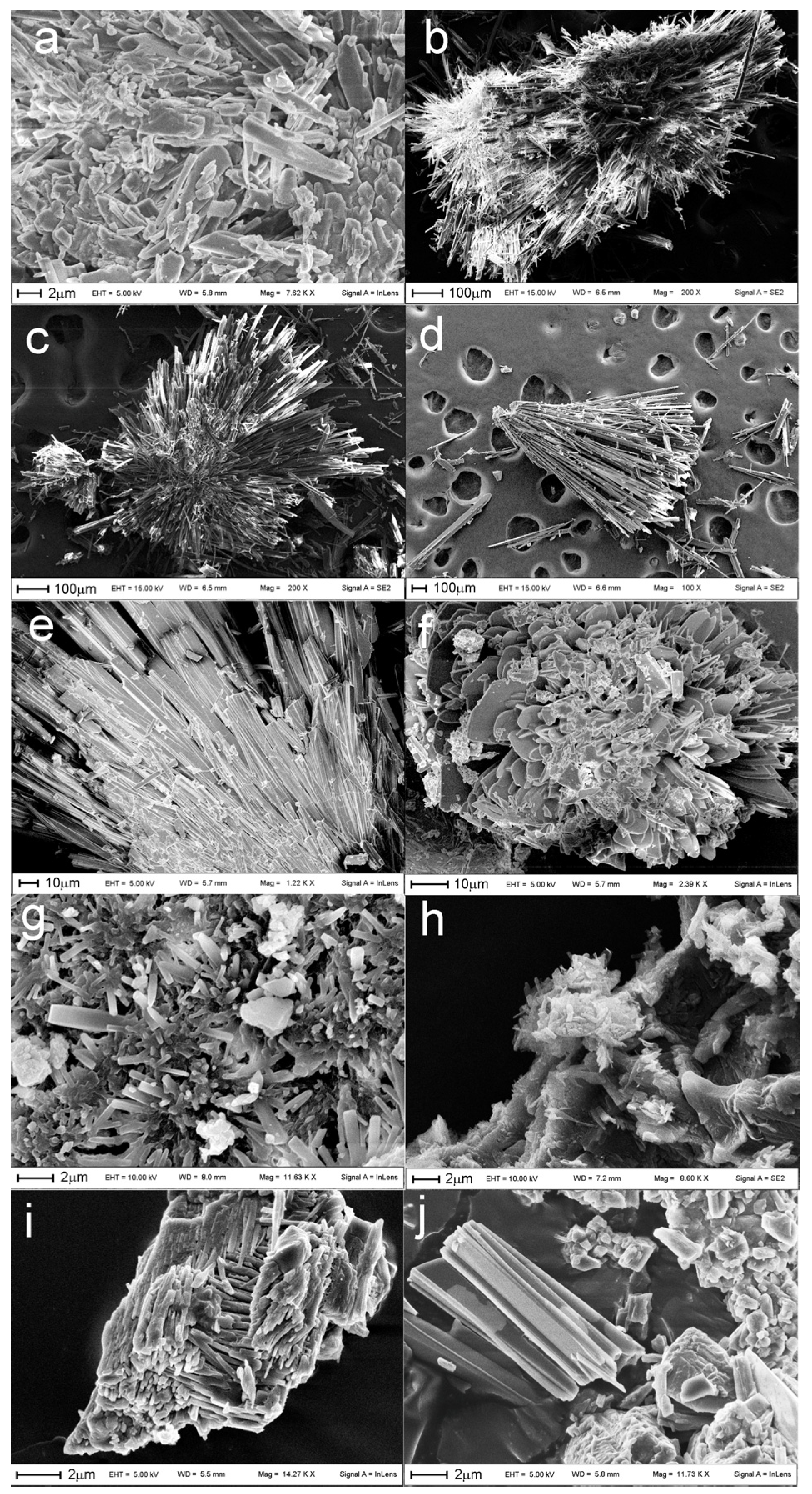
3.2. Scanning Electron Microscopy
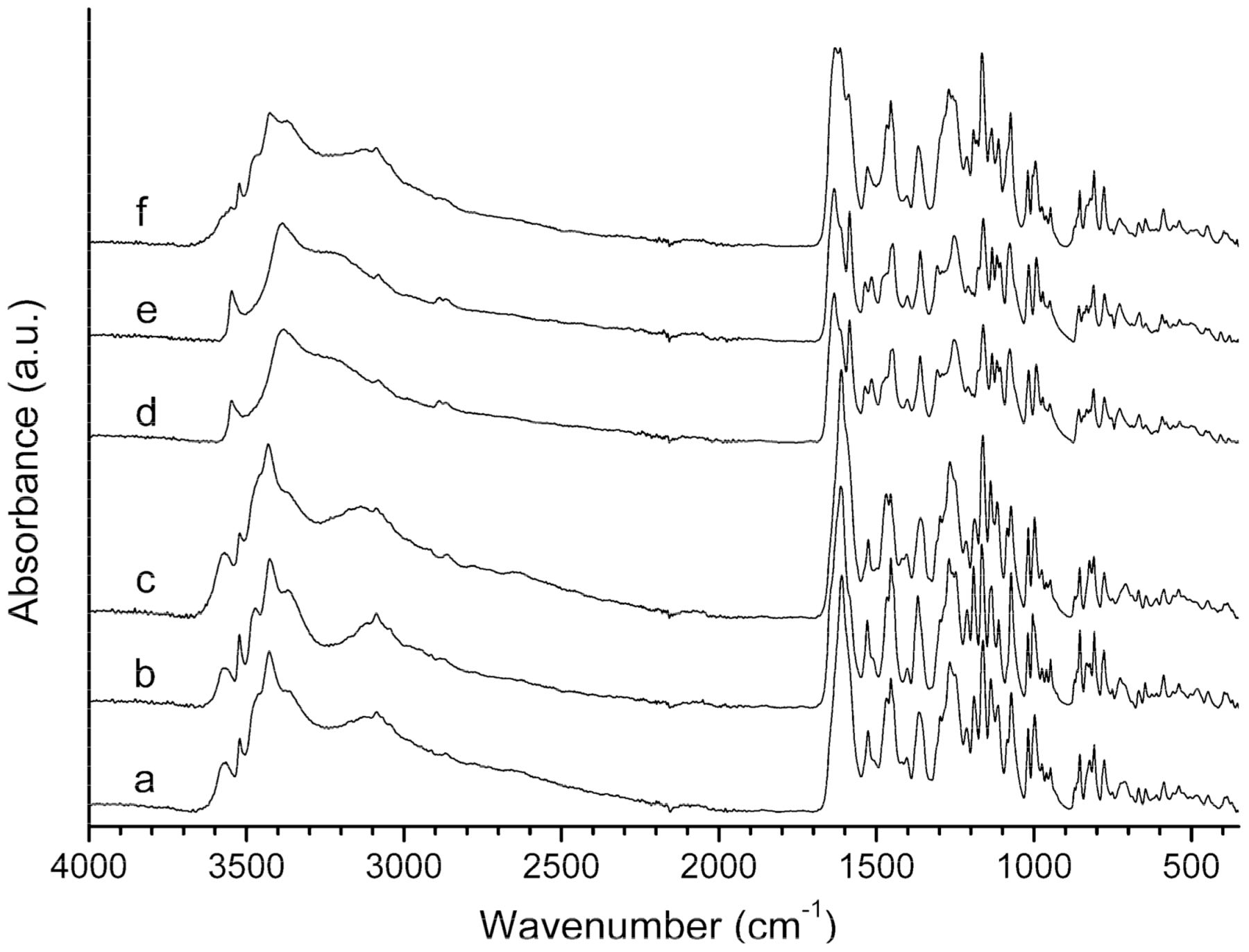
3.3. Infrared Spectroscopy
3.4. Hot Stage Microscopy
3.5. Stability Study
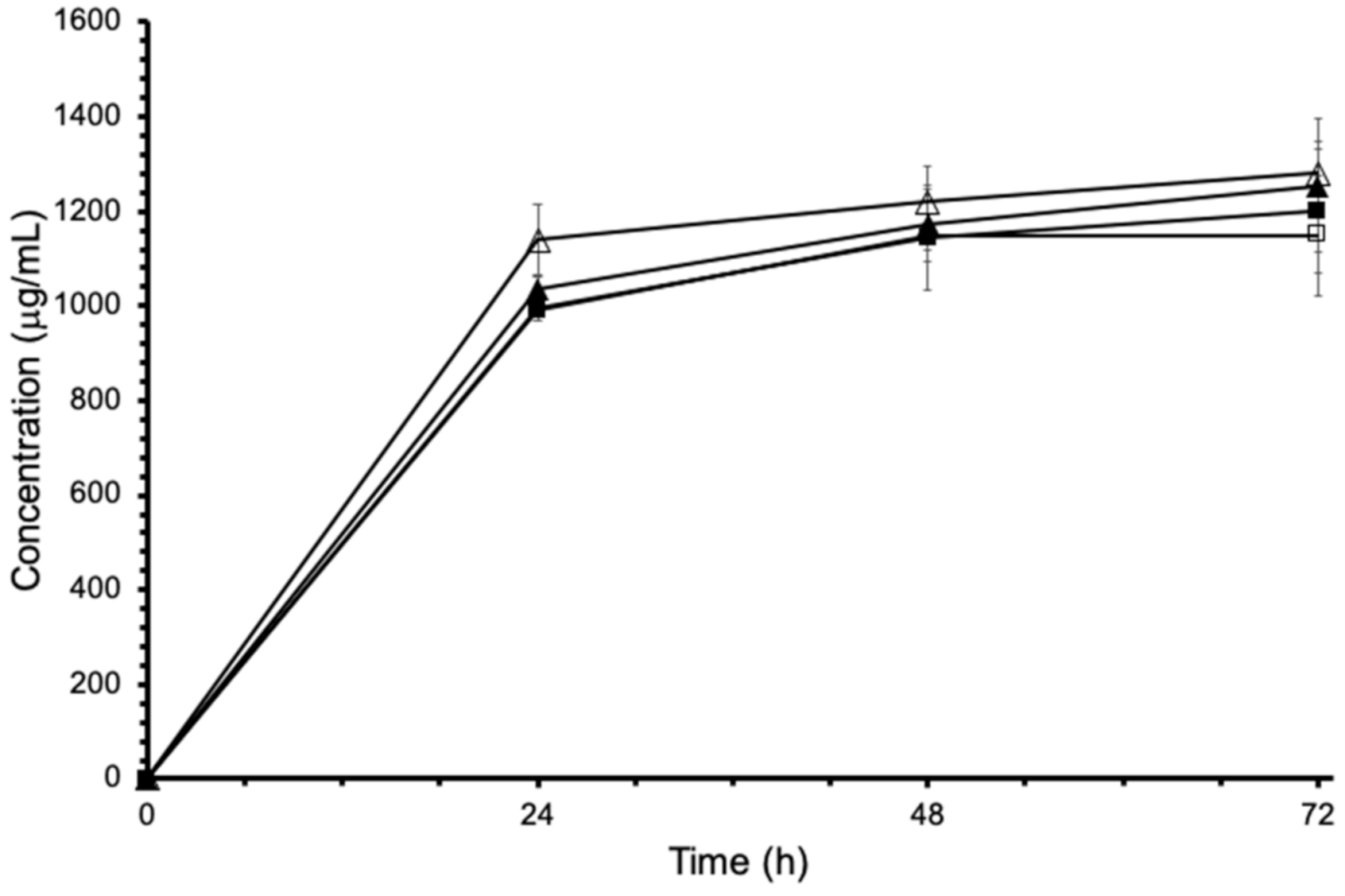
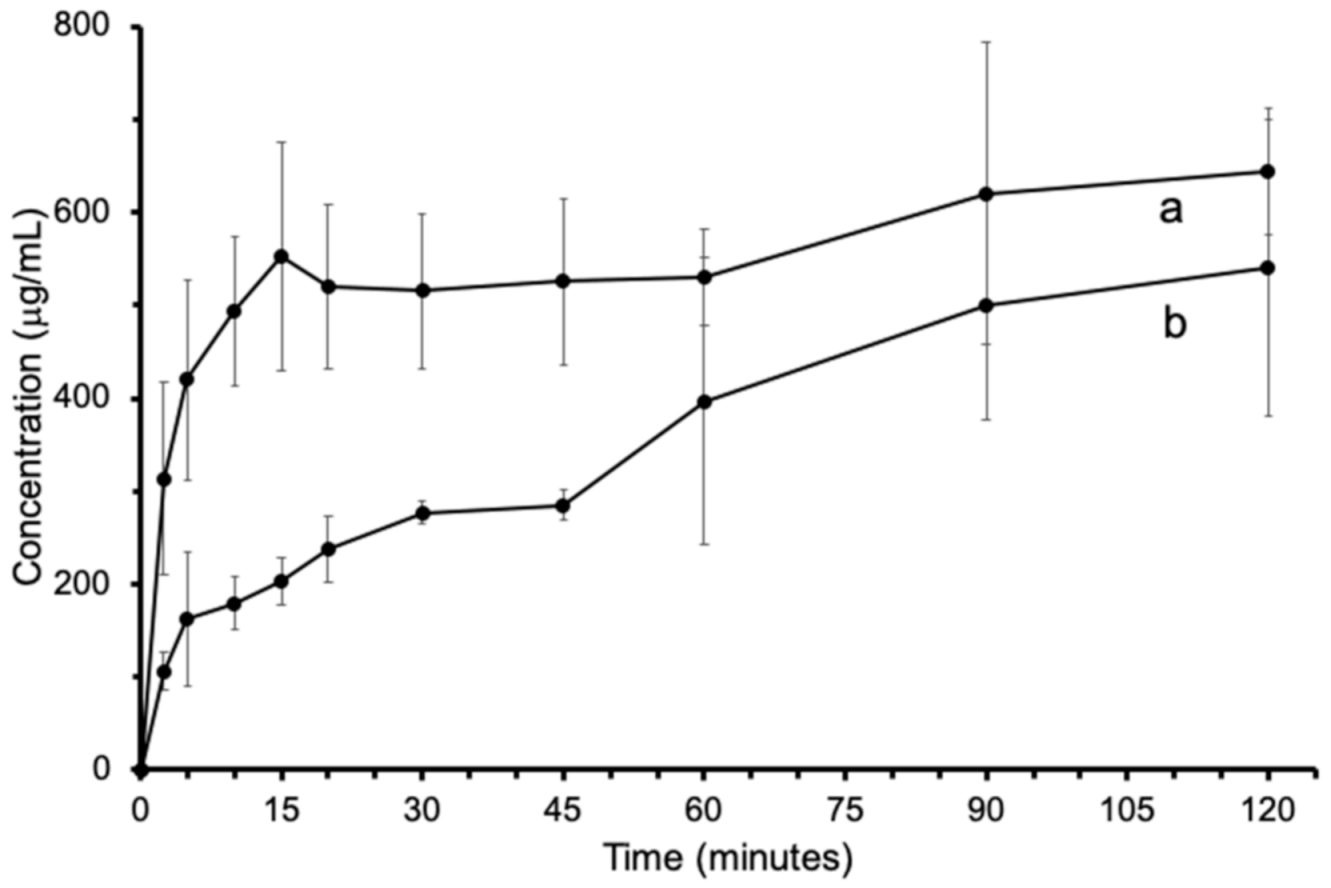
3.6. Solubility Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schlickmann, F.; Mota da Silva, L.; Boeing, T.; Bordignon Somensi, L.; de Moura Burci, L.; Santin, J.R.; Filho, V.C.; Faloni de Andrade, S. Gastroprotective Bio-Guiding Study of Fruits from Mimusops Balata. Naunyn Schmiedebergs Arch. Pharmacol. 2015, 388, 1187–1200. [Google Scholar] [CrossRef]
- Kuspradini, H.; Mitsunaga, T.; Ohashi, H. Antimicrobial Activity against Streptococcus Sobrinus and Glucosyltransferase Inhibitory Activity of Taxifolin and Some Flavanonol Rhamnosides from Kempas (Koompassia malaccensis) Extracts. J. Wood Sci. 2009, 55, 308–313. [Google Scholar] [CrossRef]
- Lawrence, R.; Jeyakumar, E.; Gupta, A. Antibacterial Activity of Acacia Arabica (Bark) Extract against Selected Multi Drug Resistant Pathogenic Bacteria. Int. J. Curr. Microbiol. Appl. Sci. 2015, 1, 213–222. [Google Scholar]
- Itaya, S.; Igarashi, K. Effects of Taxifolin on the Serum Cholesterol Level in Rats. Biosci. Biotechnol. Biochem. 1992, 56, 1492–1494. [Google Scholar] [CrossRef]
- Cechinel-Filho, V.; Vaz, Z.R.; Zunino, L.; Calixto, J.B.; Yunes, R.A. Antinociceptive and Anti-Oedematogenic Properties of Astilbin, Taxifolin and Some Related Compounds. Arzneimittelforschung 2000, 50, 281–285. [Google Scholar] [CrossRef]
- Dok-Go, H.; Lee, K.H.; Kim, H.J.; Lee, E.H.; Lee, J.; Song, Y.S.; Lee, Y.-H.; Jin, C.; Lee, Y.S.; Cho, J. Neuroprotective Effects of Antioxidative Flavonoids, Quercetin, (+)-Dihydroquercetin and Quercetin 3-Methyl Ether, Isolated from Opuntia Ficus-Indica Var. Saboten. Brain Res. 2003, 965, 130–136. [Google Scholar] [CrossRef]
- Polyak, S.J.; Morishima, C.; Lohmann, V.; Pal, S.; Lee, D.Y.W.; Liu, Y.; Graf, T.N.; Oberlies, N.H. Identification of Hepatoprotective Flavonolignans from Silymarin. Proc. Natl. Acad. Sci. USA 2010, 107, 5995–5999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidmann, A.E. Dihydroquercetin: More than Just an Impurity? Eur. J. Pharmacol. 2012, 684, 19–26. [Google Scholar] [CrossRef]
- Sun, X.; Chen, R.; Yang, Z.; Sun, G.; Wang, M.; Ma, X.; Yang, L.; Sun, X. Taxifolin Prevents Diabetic Cardiomyopathy in Vivo and in Vitro by Inhibition of Oxidative Stress and Cell Apoptosis. Food Chem. Toxicol. 2014, 63, 221–232. [Google Scholar] [CrossRef]
- Oi, N.; Chen, H.; Ok Kim, M.; Lubet, R.A.; Bode, A.M.; Dong, Z. Taxifolin Suppresses UV-Induced Skin Carcinogenesis by Targeting EGFR and PI3K. Cancer Prev. Res. 2012, 5, 1103–1114. [Google Scholar] [CrossRef] [Green Version]
- Saito, S.; Yamamoto, Y.; Maki, T.; Hattori, Y.; Ito, H.; Mizuno, K.; Harada-Shiba, M.; Kalaria, R.N.; Fukushima, M.; Takahashi, R.; et al. Taxifolin Inhibits Amyloid-β Oligomer Formation and Fully Restores Vascular Integrity and Memory in Cerebral Amyloid Angiopathy. Acta Neuropathol. Commun. 2017, 5, 26. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, J.; Dong, X.-D.; Ji, H.-Y. Research on Characteristics, Antioxidant and Antitumor Activities of Dihydroquercetin and Its Complexes. Molecules 2017, 23, 20. [Google Scholar] [CrossRef] [Green Version]
- Schauss, A.G.; Tselyico, S.S.; Kuznetsova, V.A.; Yegorova, I. Toxicological and Genotoxicity Assessment of a Dihydroquercetin-Rich Dahurian Larch Tree (Larix Gmelinii Rupr) Extract (Lavitol). Int. J. Toxicol. 2015, 34, 162–181. [Google Scholar] [CrossRef]
- Borrego-Sánchez, A.; Carazo, E.; Albertini, B.; Passerini, N.; Perissutti, B.; Cerezo, P.; Viseras, C.; Hernández-Laguna, A.; Aguzzi, C.; Sainz-Díaz, C.I. Conformational Polymorphic Changes in the Crystal Structure of the Chiral Antiparasitic Drug Praziquantel and Interactions with Calcium Carbonate. Eur. J. Pharm. Biopharm. 2018, 132, 180–191. [Google Scholar] [CrossRef]
- Rietveld, I.B.; Allouchi, H.; Barrio, M.; Ceolin, R.; Tamarit, J.-L. Polymorphism of Benzylthiouracil, an Active Pharmaceutical Ingredient against Hyperthyroidism. Int. J. Pharm. 2021, 598, 120378. [Google Scholar] [CrossRef]
- Wiergowska, G.; Stasiłowicz, A.; Miklaszewski, A.; Lewandowska, K.; Cielecka-Piontek, J. Structural Polymorphism of Sorafenib Tosylate as a Key Factor in Its Solubility Differentiation. Pharmaceutics 2021, 13, 384. [Google Scholar] [CrossRef]
- Tandon, R.; Tandon, N.; Thapar, R.K. Patenting of Polymorphs. Pharm. Pat. Anal. 2018, 7, 59–63. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Olsen, K.W. Unique Mechanism of Facile Polymorphic Conversion of Acetaminophen in Aqueous Medium. Mol. Pharm. 2014, 11, 3056–3067. [Google Scholar] [CrossRef]
- Khoo, J.Y.; Shah, U.V.; Schaepertoens, M.; Williams, D.R.; Heng, J.Y.Y. Process-Induced Phase Transformation of Carbamazepine Dihydrate to Its Polymorphic Anhydrates. Powder Technol. 2013, 236, 114–121. [Google Scholar] [CrossRef]
- Byrn, S.R.; Zografi, G.; Chen, X. (Sean) Accelerating Proof of Concept for Small Molecule Drugs Using Solid-State Chemistry. J. Pharm. Sci. 2010, 99, 3665–3675. [Google Scholar] [CrossRef]
- Palucki, M.; Higgins, J.D.; Kwong, E.; Templeton, A.C. Strategies at the Interface of Drug Discovery and Development: Early Optimization of the Solid State Phase and Preclinical Toxicology Formulation for Potential Drug Candidates. J. Med. Chem. 2010, 53, 5897–5905. [Google Scholar] [CrossRef] [PubMed]
- Stenger, F.C.; Couto, A.G.; Bresolin, T.M.B.; Cechinel Filho, V. Taxifolin: Extraction Optimization and Development of Analytical Method by Hplc to Assess the Purity. In Proceedings of the VIII Simpósio Ibero-Americano de Plantas Medicinais. III Simpósio Ibero-Americano em Câncer, Itajaí, Santa Catarina, Brazil, 24–27 October 2016; Volume 1, p. 463. [Google Scholar]
- Stenger Moura, F.C.; dos Santos Machado, C.L.; Reisdorfer Paula, F.; Garcia Couto, A.; Ricci, M.; Cechinel-Filho, V.; Bonomini, T.J.; Sandjo, L.P.; Bellé Bresolin, T.M. Taxifolin Stability: In Silico Prediction and in Vitro Degradation with HPLC-UV/UPLC–ESI-MS Monitoring. J. Pharm. Anal. 2021, 11, 232–240. [Google Scholar] [CrossRef]
- Werner, P.-E.; Eriksson, L.; Westdahl, M. TREOR, a Semi-Exhaustive Trial-and-Error Powder Indexing Program for All Symmetries. J. Appl. Cryst. 1985, 18, 367–370. [Google Scholar] [CrossRef]
- Altomare, A.; Cuocci, C.; Giacovazzo, C.; Moliterni, A.; Rizzi, R.; Corriero, N.; Falcicchio, A. EXPO2013: A Kit of Tools for Phasing Crystal Structures from Powder Data. J. Appl. Cryst. 2013, 46, 1231–1235. [Google Scholar] [CrossRef]
- Larson, A.C.; von Dreele, R.B. GSAS General Structure Analysis System; LANL Report LAUR 86-748; Los Alamos National Laboratory: Los Alamos, NM, USA, 2001.
- Toby, B.H. EXPGUI, a Graphical User Interface for GSAS. J. Appl. Cryst. 2001, 34, 210–213. [Google Scholar] [CrossRef] [Green Version]
- Nicoli, D.F.; Wu, J.S.; Chang, Y.J.; McKenzie, D.C.; Hasapidis, K. Automatic, High Resolution Particle Size Analysis by Single-Particle Optical Sensing. Am. Lab. 1992, 24, 39–44. [Google Scholar]
- WHO. Protocol to conduct equilibrium solubility experiments for the purpose of Biopharmaceutics ClassificationSystem-based classification of active pharmaceuticalingredients for biowaiver. In WHO Expert Committee on Specifications for Pharmaceutical Preparations; WHO: Geneva, Switzerland, 2019; pp. 203–218. [Google Scholar]
- Avdeef, A.; Fuguet, E.; Llinàs, A.; Ràfols, C.; Bosch, E.; Völgyi, G.; Verbić, T.; Boldyreva, E.; Takács-Novák, K. Equilibrium Solubility Measurement of Ionizable Drugs—Consensus Recommendations for Improving Data Quality. ADMET DMPK 2016, 4, 62. [Google Scholar] [CrossRef] [Green Version]
- Council of Europe. European Pharmacopoeia Commission Intrinsic Dissolution. In European Pharmacopoeia, 10th ed.; Strasbourg, FR Council of Europe: Strasbourg, France, 2019; pp. 372–373. ISBN 978-92-871-6054-6. [Google Scholar]
- Lee, E.H. A Practical Guide to Pharmaceutical Polymorph Screening & Selection. Asian J. Pharm. Sci. 2014, 9, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Newman, A. Specialized Solid Form Screening Techniques. Org. Process. Res. Dev. 2013, 17, 457–471. [Google Scholar] [CrossRef]
- Selivanova, I.A.; Tyukavkina, N.A.; Kolesnik, Y.A.; Nesterov, V.N.; Kuleshova, L.N.; Khutoryanskii, V.A.; Bazhenov, B.N.; Saibotalov, M.Y. Study of the Crystalline Structure of Dihydroquercetin. Pharm. Chem. J. 1999, 33, 222–224. [Google Scholar] [CrossRef]
- Van Eerdenbrugh, B.; Baird, J.A.; Taylor, L.S. Crystallization Tendency of Active Pharmaceutical Ingredients Following Rapid Solvent Evaporation—Classification and Comparison with Crystallization Tendency from Undercooled Melts. J. Pharm. Sci. 2010, 99, 3826–3838. [Google Scholar] [CrossRef]
- Kiehlmann, E.; Li, E.P.M. Isomerization of Dihydroquercetin. J. Nat. Prod. 1995, 58, 450–455. [Google Scholar] [CrossRef]
- Zu, S.; Yang, L.; Huang, J.; Ma, C.; Wang, W.; Zhao, C.; Zu, Y. Micronization of Taxifolin by Supercritical Antisolvent Process and Evaluation of Radical Scavenging Activity. Int. J. Mol. Sci 2012, 13, 8869–8881. [Google Scholar] [CrossRef] [Green Version]
- Florence, A.T.; Attwood, D. Properties of the Solid State. In Physicochemical Principles of Pharmacy; Florence, A.T., Attwood, D., Eds.; Macmillan Education UK: London, UK, 1998; pp. 5–35. ISBN 978-1-349-14416-7. [Google Scholar]
- Baranović, G.; Šegota, S. Infrared Spectroscopy of Flavones and Flavonols. Reexamination of the Hydroxyl and Carbonyl Vibrations in Relation to the Interactions of Flavonoids with Membrane Lipids. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2018, 192, 473–486. [Google Scholar] [CrossRef]
- Bakkialakshmi, S.; Roy, J. Infrared Spectrum Analysis of Some Flavonoids with Hemoglobin. Int. J. Appl. Adv. Sci. Res. 2017, 2, 107–110. [Google Scholar]
- Heneczkowski, M.; Kopacz, M.; Nowak, D.; Kuźniar, A. Infrared Spectrum Analysis of Some Flavonoids. Acta Pol. Pharm. 2001, 58, 415–420. [Google Scholar] [PubMed]
- Stieger, N.; Aucamp, M.; Zhang, S.-W.; de Villiers, M.M. Hot-Stage Optical Microscopy as an Analytical Tool to Understand Solid-State Changes in Pharmaceutical Materials. Available online: http://www.americanpharmaceuticalreview.com/Featured-Articles/39283-Hot-stage-Optical-Microscopy-as-an-Analytical-Tool-to-Understand-Solid-state-Changes-in-Pharmaceutical-Materials/ (accessed on 3 September 2019).
- Bartolomei, M. Solid-State Studies on the Hemihydrate and the Anhydrous Forms of Flunisolide. J. Pharm. Biomed. Anal. 2000, 24, 81–93. [Google Scholar] [CrossRef]
- Poole, J.W.; Bahal, C.K. Dissolution Behavior and Solubility of Anhydrous and Dihydrate Forms of Wy-4508, an Aminoalicyclic Penicillin. J. Pharm. Sci. 1970, 59, 1265–1267. [Google Scholar] [CrossRef]
- van Tonder, E.C.; Maleka, T.S.P.; Liebenberg, W.; Song, M.; Wurster, D.E.; de Villiers, M.M. Preparation and Physicochemical Properties of Niclosamide Anhydrate and Two Monohydrates. Int. J. Pharm. 2004, 269, 417–432. [Google Scholar] [CrossRef]
- Tieger, E.; Kiss, V.; Pokol, G.; Finta, Z.; Rohlíček, J.; Skořepová, E.; Dušek, M. Rationalization of the Formation and Stability of Bosutinib Solvated Forms. CrystEngComm 2016, 18, 9260–9274. [Google Scholar] [CrossRef] [Green Version]
- Baka, E.; Comer, J.E.A.; Takács-Novák, K. Study of Equilibrium Solubility Measurement by Saturation Shake-Flask Method Using Hydrochlorothiazide as Model Compound. J. Pharm. Biomed. Anal. 2008, 46, 335–341. [Google Scholar] [CrossRef]
- Lagas, M.; Lerk, C.F. The Polymorphism of Sulphathiazole. Int. J. Pharm. 1981, 8, 11–24. [Google Scholar] [CrossRef]
- Gu, C.H.; Young, V.; Grant, D.J. Polymorph Screening: Influence of Solvents on the Rate of Solvent-Mediated Polymorphic Transformation. J. Pharm. Sci. 2001, 90, 1878–1890. [Google Scholar] [CrossRef]
- Byrn, S.R.; Zografi, G.; Chen, X. Solid-State Properties of Pharmaceutical Materials; John Wiley & Sons: Hoboken, NJ, USA, 2017; ISBN 978-1-118-14530-2. [Google Scholar]
- Mullin, J.W. Crystallization; Elsevier: Amsterdam, The Netherlands, 2001; ISBN 978-0-7506-4833-2. [Google Scholar]
- Bartolomei, M.; Bertocchi, P.; Antoniella, E.; Rodomonte, A. Physico-Chemical Characterisation and Intrinsic Dissolution Studies of a New Hydrate Form of Diclofenac Sodium: Comparison with Anhydrous Form. J. Pharm. Biomed. Anal. 2006, 40, 1105–1113. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.X.; Carlin, A.S.; Amidon, G.L.; Hussain, A.S. Feasibility Studies of Utilizing Disk Intrinsic Dissolution Rate to Classify Drugs. Int. J. Pharm. 2004, 270, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Andersson, S.B.E.; Alvebratt, C.; Bergström, C.A.S. Controlled Suspensions Enable Rapid Determinations of Intrinsic Dissolution Rate and Apparent Solubility of Poorly Water-Soluble Compounds. Pharm. Res. 2017, 34, 1805–1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagerberg, J.H.; Tsinman, O.; Sun, N.; Tsinman, K.; Avdeef, A.; Bergström, C.A.S. Dissolution Rate and Apparent Solubility of Poorly Soluble Drugs in Biorelevant Dissolution Media. Mol. Pharm. 2010, 7, 1419–1430. [Google Scholar] [CrossRef]
- Zu, Y.; Wu, W.; Zhao, X.; Li, Y.; Wang, W.; Zhong, C.; Zhang, Y.; Zhao, X. Enhancement of Solubility, Antioxidant Ability and Bioavailability of Taxifolin Nanoparticles by Liquid Antisolvent Precipitation Technique. Int. J. Pharm. 2014, 471, 366–376. [Google Scholar] [CrossRef]
- Shikov, A.N.; Pozharitskaya, O.N.; Miroshnyk, I.; Mirza, S.; Urakova, I.N.; Hirsjärvi, S.; Makarov, V.G.; Heinämäki, J.; Yliruusi, J.; Hiltunen, R. Nanodispersions of Taxifolin: Impact of Solid-State Properties on Dissolution Behavior. Int. J. Pharm. 2009, 377, 148–152. [Google Scholar] [CrossRef]
- Yang, L.-J.; Chen, W.; Ma, S.-X.; Gao, Y.-T.; Huang, R.; Yan, S.-J.; Lin, J. Host–Guest System of Taxifolin and Native Cyclodextrin or Its Derivative: Preparation, Characterization, Inclusion Mode, and Solubilization. Carbohydr. Polym. 2011, 85, 629–637. [Google Scholar] [CrossRef]
- Baranov, I.A.; Dzhons, D.Y.; Budruev, A.V.; Mochalova, A.E.; Smirnova, L.A.; Koryagin, A.S. Long-Acting Bioactive Composition Based on Chitosan and Taxifolin. Inorg. Mater. Appl. Res. 2015, 6, 479–484. [Google Scholar] [CrossRef]
- Stenger Moura, F.C.; Perioli, L.; Pagano, C.; Vivani, R.; Ambrogi, V.; Bresolin, T.M.; Ricci, M.; Schoubben, A. Chitosan Composite Microparticles: A Promising Gastroadhesive System for Taxifolin. Carbohydr. Polym. 2019, 218, 343–354. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stenger Moura, F.C.; Pinna, N.; Vivani, R.; Nunes, G.E.; Schoubben, A.; Bellé Bresolin, T.M.; Bechold, I.H.; Ricci, M. Exploring Taxifolin Polymorphs: Insights on Hydrate and Anhydrous Forms. Pharmaceutics 2021, 13, 1328. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13091328
Stenger Moura FC, Pinna N, Vivani R, Nunes GE, Schoubben A, Bellé Bresolin TM, Bechold IH, Ricci M. Exploring Taxifolin Polymorphs: Insights on Hydrate and Anhydrous Forms. Pharmaceutics. 2021; 13(9):1328. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13091328
Chicago/Turabian StyleStenger Moura, Fernanda Cristina, Nicola Pinna, Riccardo Vivani, Gisele Elias Nunes, Aurélie Schoubben, Tania Mari Bellé Bresolin, Ivan Helmuth Bechold, and Maurizio Ricci. 2021. "Exploring Taxifolin Polymorphs: Insights on Hydrate and Anhydrous Forms" Pharmaceutics 13, no. 9: 1328. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13091328