
T22-PE24-H6 Nanotoxin Selectively Kills CXCR4-High Expressing AML Patient Cells In Vitro and Potently Blocks Dissemination In Vivo

 , , , , , and
, , , , , and
Abstract
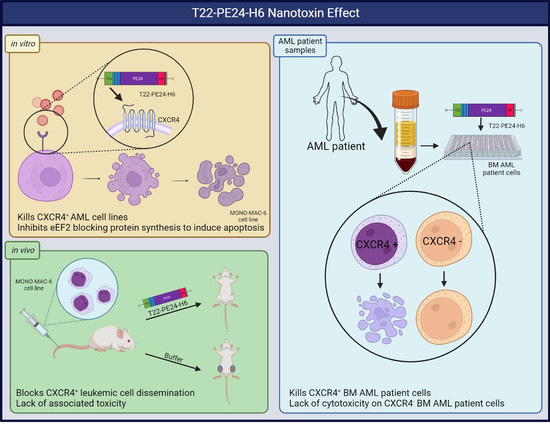
:
1. Introduction
2. Methods
2.1. AML Cell Lines and Patient Samples
2.2. Lentiviral Luciferase Transduction
2.3. Nanoparticle Design and Production
2.4. Detection of CXCR4 Expression in AML Cell Lines
2.5. Analysis of T22-PE24-H6 Antineoplastic Effect In Vitro
2.6. Evaluation of T22-PE24-H6 Antineoplastic Activity in a Disseminated AML Mouse Model
2.7. Immunohistochemical Analysis of CXCR4 and CD45 in AML Infiltrated Tissues
2.8. Toxicity Analysis in Mouse Tissues and Blood
2.9. Detection of CXCR4 Expression in AML Patients’ BM Cells
2.10. Evaluation of T22-PE24-H6 Antineoplastic Activity in Cultured AML Patients’ BM Cells
2.11. Statistical Analysis
3. Results
3.1. T22-PE24-H6 Antineoplastic Effect in CXCR4+ AML Cell Lines
3.2. T22-PE24-H6 Induced Apoptosis in MONO-MAC-6 Cells
3.3. Antineoplastic Effect of T22-PE24-H6 in a CXCR4+ AML Mouse Model
3.4. Toxicity Evaluation in Mouse Tissues
3.5. T22-PE24-H6 In Vitro Effect in AML Patients’ BM Samples
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hwang, S.M. Classification of acute myeloid leukemia. Blood Res. 2020, 55, S1–S4. [Google Scholar] [CrossRef]
- Fleischmann, M.; Schnetzke, U.; Hochhaus, A.; Scholl, S. Management of Acute Myeloid Leukemia: Current Treatment Options and Future Perspectives. Cancers 2021, 13, 5722. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [Green Version]
- Heuser, M.; Ofran, Y.; Boissel, N.; Mauri, S.B.; Craddock, C.; Janssen, J.; Wierzbowska, A.; Buske, C. Acute myeloid leukaemia in adult patients: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up†. Ann. Oncol. 2020, 31, 697–712. [Google Scholar] [CrossRef]
- Mrózek, K.; Marcucci, G.; Nicolet, D.; Maharry, K.S.; Becker, H.; Whitman, S.P.; Metzeler, K.H.; Schwind, S.; Wu, Y.-Z.; Kohlschmidt, J.; et al. Prognostic significance of the European LeukemiaNet standardized system for reporting cytogenetic and molecular alterations in adults with acute myeloid leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 4515–4523. [Google Scholar] [CrossRef]
- Verma, D.; Kantarjian, H.; Faderl, S.; O’Brien, S.; Pierce, S.; Vu, K.; Freireich, E.; Keating, M.; Cortes, J.; Ravandi, F. Late relapses in acute myeloid leukemia: Analysis of characteristics and outcome. Leuk. Lymphoma 2010, 51, 778–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, S.; Behnam Azad, B.; Nimmagadda, S. The intricate role of CXCR4 in cancer. Adv. Cancer Res. 2014, 124, 31–82. [Google Scholar] [CrossRef] [Green Version]
- Domanska, U.M.; Kruizinga, R.C.; Nagengast, W.B.; Timmer-Bosscha, H.; Huls, G.; de Vries, E.G.E.; Walenkamp, A.M.E. A review on CXCR4/CXCL12 axis in oncology: No place to hide. Eur. J. Cancer Oxf. Engl. 2013, 49, 219–230. [Google Scholar] [CrossRef]
- Balkwill, F. The significance of cancer cell expression of the chemokine receptor CXCR4. Semin. Cancer Biol. 2004, 14, 171–179. [Google Scholar] [CrossRef]
- Zlotnik, A. Chemokines and cancer. Int. J. Cancer 2006, 119, 2026–2029. [Google Scholar] [CrossRef]
- Cho, B.-S.; Kim, H.-J.; Konopleva, M. Targeting the CXCL12/CXCR4 axis in acute myeloid leukemia: From bench to bedside. Korean J. Intern. Med. 2017, 32, 248–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konoplev, S.; Rassidakis, G.Z.; Estey, E.; Kantarjian, H.; Liakou, C.I.; Huang, X.; Xiao, L.; Andreeff, M.; Konopleva, M.; Medeiros, L.J. Overexpression of CXCR4 predicts adverse overall and event-free survival in patients with unmutated FLT3 acute myeloid leukemia with normal karyotype. Cancer 2007, 109, 1152–1156. [Google Scholar] [CrossRef]
- Zhang, Y.; Saavedra, E.; Tang, R.; Gu, Y.; Lappin, P.; Trajkovic, D.; Liu, S.-H.; Smeal, T.; Fantin, V.; De Botton, S.; et al. Targeting primary acute myeloid leukemia with a new CXCR4 antagonist IgG1 antibody (PF-06747143). Sci. Rep. 2017, 7, 7305. [Google Scholar] [CrossRef] [Green Version]
- Cao, T.; Ye, Y.; Liao, H.; Shuai, X.; Jin, Y.; Su, J.; Zheng, Q. Relationship between CXC chemokine receptor 4 expression and prognostic significance in acute myeloid leukemia. Medicine 2019, 98, e15948. [Google Scholar] [CrossRef]
- Ahn, J.Y.; Seo, K.; Weinberg, O.K.; Arber, D.A. The Prognostic Value of CXCR4 in Acute Myeloid Leukemia. Appl. Immunohistochem. Mol. Morphol. 2013, 21, 79–84. [Google Scholar] [CrossRef]
- Konoplev, S.; Lin, P.; Yin, C.C.; Lin, E.; González, G.M.N.; Kantarjian, H.M.; Andreeff, M.; Medeiros, L.J.; Konopleva, M. CXCR4 Expression, CXCR4 Activation, and Wild Type NPM1 Are Independently Associated with Unfavorable Prognosis in Patients with Acute Myeloid Leukemia. Clin. Lymphoma Myeloma Leuk. 2013, 13, 686–692. [Google Scholar] [CrossRef] [Green Version]
- Rombouts, E.J.C.; Pavic, B.; Löwenberg, B.; Ploemacher, R.E. Relation between CXCR-4 expression, Flt3 mutations, and unfavorable prognosis of adult acute myeloid leukemia. Blood 2004, 104, 550–557. [Google Scholar] [CrossRef] [Green Version]
- Spoo, A.C.; Lübbert, M.; Wierda, W.G.; Burger, J.A. CXCR4 is a prognostic marker in acute myelogenous leukemia. Blood 2007, 109, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Su, L.; Hu, Z.; Yang, Y. Role of CXCR4 in the progression and therapy of acute leukaemia. Cell Prolif. 2021, 54, e13076. [Google Scholar] [CrossRef]
- Unzueta, U.; Céspedes, M.V.; Ferrer-Miralles, N.; Casanova, I.; Cedano, J.; Corchero, J.L.; Domingo-Espín, J.; Villaverde, A.; Mangues, R.; Vázquez, E. Intracellular CXCR4+ cell targeting with T22-empowered protein-only nanoparticles. Int. J. Nanomedicine 2012, 7, 4533–4544. [Google Scholar] [CrossRef] [Green Version]
- Michalska, M.; Wolf, P. Pseudomonas Exotoxin A: Optimized by evolution for effective killing. Front. Microbiol. 2015, 6. Available online: https://www.frontiersin.org/article/10.3389/fmicb.2015.00963 (accessed on 28 January 2022). [CrossRef] [PubMed] [Green Version]
- Sánchez-García, L.; Serna, N.; Álamo, P.; Sala, R.; Céspedes, M.V.; Roldan, M.; Sánchez-Chardi, A.; Unzueta, U.; Casanova, I.; Mangues, R.; et al. Self-assembling toxin-based nanoparticles as self-delivered antitumoral drugs. J. Control. Release Off. J. Control. Release Soc. 2018, 274, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Falgàs, A.; Pallarès, V.; Serna, N.; Sánchez-García, L.; Sierra, J.; Gallardo, A.; Alba-Castellón, L.; Álamo, P.; Unzueta, U.; Villaverde, A.; et al. Selective delivery of T22-PE24-H6 to CXCR4+ diffuse large B-cell lymphoma cells leads to wide therapeutic index in a disseminated mouse model. Theranostics 2020, 10, 5169–5180. [Google Scholar] [CrossRef]
- Rioja-Blanco, E.; Arroyo-Solera, I.; Álamo, P.; Casanova, I.; Gallardo, A.; Unzueta, U.; Serna, N.; Sánchez-García, L.; Quer, M.; Villaverde, A.; et al. Self-assembling protein nanocarrier for selective delivery of cytotoxic polypeptides to CXCR4+ head and neck squamous cell carcinoma tumors. Acta Pharm. Sin. B 2021, 12, 2578–2591. [Google Scholar] [CrossRef] [PubMed]
- Rioja-Blanco, E.; Arroyo-Solera, I.; Álamo, P.; Casanova, I.; Gallardo, A.; Unzueta, U.; Serna, N.; Sánchez-García, L.; Quer, M.; Villaverde, A.; et al. CXCR4-targeted nanotoxins induce GSDME-dependent pyroptosis in head and neck squamous cell carcinoma. J. Exp. Clin. Cancer Res. CR 2022, 41, 49. [Google Scholar] [CrossRef]
- Medina-Gutiérrez, E.; Céspedes, M.V.; Gallardo, A.; Rioja-Blanco, E.; Pavón, M.À.; Asensio-Puig, L.; Farré, L.; Alba-Castellón, L.; Unzueta, U.; Villaverde, A.; et al. Novel Endometrial Cancer Models Using Sensitive Metastasis Tracing for CXCR4-Targeted Therapy in Advanced Disease. Biomedicines 2022, 10, 1680. [Google Scholar] [CrossRef]
- Céspedes, M.V.; Unzueta, U.; Tatkiewicz, W.; Sánchez-Chardi, A.; Conchillo-Solé, O.; Álamo, P.; Xu, Z.; Casanova, I.; Corchero, J.L.; Pesarrodona, M.; et al. In vivo architectonic stability of fully de novo designed protein-only nanoparticles. ACS Nano 2014, 8, 4166–4176. [Google Scholar] [CrossRef] [Green Version]
- Pallarès, V.; Núñez, Y.; Sánchez-García, L.; Falgàs, A.; Serna, N.; Unzueta, U.; Gallardo, A.; Alba-Castellón, L.; Álamo, P.; Sierra, J.; et al. Antineoplastic effect of a diphtheria toxin-based nanoparticle targeting acute myeloid leukemia cells overexpressing CXCR4. J. Controlled Release 2021, 335, 117–129. [Google Scholar] [CrossRef]
- Falgàs, A.; Pallarès, V.; Unzueta, U.; Núñez, Y.; Sierra, J.; Gallardo, A.; Alba-Castellón, L.; Mangues, M.A.; Álamo, P.; Villaverde, A.; et al. Specific Cytotoxic Effect of an Auristatin Nanoconjugate Towards CXCR4+ Diffuse Large B-Cell Lymphoma Cells. Int. J. Nanomedicine 2021, 16, 1869–1888. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Chang, J.-H.; Kwon, H.-Y. Expression of 14-3-3delta, cdc2 and cyclin B proteins related to exotoxin A-induced apoptosis in HeLa S3 cells. Int. Immunopharmacol. 2007, 7, 1185–1191. [Google Scholar] [CrossRef]
- Andersson, Y.; Juell, S.; Fodstad, Ø. Downregulation of the antiapoptotic MCL-1 protein and apoptosis in MA-11 breast cancer cells induced by an anti-epidermal growth factor receptor-Pseudomonas exotoxin a immunotoxin. Int. J. Cancer 2004, 112, 475–483. [Google Scholar] [CrossRef]
- Du, X.; Youle, R.J.; FitzGerald, D.J.; Pastan, I. Pseudomonas exotoxin A-mediated apoptosis is Bak dependent and preceded by the degradation of Mcl-1. Mol. Cell. Biol. 2010, 30, 3444–3452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Hao, S. A-1210477, a selective MCL-1 inhibitor, overcomes ABT-737 resistance in AML. Oncol. Lett. 2019, 18, 5481–5489. [Google Scholar] [CrossRef] [Green Version]
- Ewald, L.; Dittmann, J.; Vogler, M.; Fulda, S. Side-by-side comparison of BH3-mimetics identifies MCL-1 as a key therapeutic target in AML. Cell Death Dis. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, F.; He, H.; Bai, H.; Yang, F.; Ma, M.; Gu, N.; Zhang, Y. A biomimetic nanocomposite with enzyme-like activities and CXCR4 antagonism efficiently enhances the therapeutic efficacy of acute myeloid leukemia. Bioact. Mater. 2022, 18, 526–538. [Google Scholar] [CrossRef]
- Ren, X.-H.; Xu, C.; Li, L.-L.; Zuo, Y.; Han, D.; He, X.-Y.; Cheng, S.-X. A targeting delivery system for effective genome editing in leukemia cells to reverse malignancy. J. Control. Release Off. J. Control. Release Soc. 2022, 343, 645–656. [Google Scholar] [CrossRef]
- Wang, Y.; Xie, Y.; Williams, J.; Hang, Y.; Richter, L.; Becker, M.; Amador, C.; Oupický, D.; Hyde, R.K. Use of polymeric CXCR4 inhibitors as siRNA delivery vehicles for the treatment of acute myeloid leukemia. Cancer Gene Ther. 2020, 27, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.-Z.; Ye, J.C.; Pu, J.J.; Liu, H.; Ding, W.; Zheng, H.; Liu, D. Novel agents and regimens for hematological malignancies: Recent updates from 2020 ASH annual meeting. J. Hematol. Oncol. 2021, 14, 66. [Google Scholar] [CrossRef]
- Costa, M.J.; Kudaravalli, J.; Ma, J.-T.; Ho, W.-H.; Delaria, K.; Holz, C.; Stauffer, A.; Chunyk, A.G.; Zong, Q.; Blasi, E.; et al. Optimal design, anti-tumour efficacy and tolerability of anti-CXCR4 antibody drug conjugates. Sci. Rep. 2019, 9, 2443. [Google Scholar] [CrossRef] [Green Version]
- Kularatne, S.A.; Deshmukh, V.; Ma, J.; Tardif, V.; Lim, R.K.V.; Pugh, H.M.; Sun, Y.; Manibusan, A.; Sellers, A.J.; Barnett, R.S.; et al. A CXCR4-targeted site-specific antibody-drug conjugate. Angew. Chem. Int. Ed. Engl. 2014, 53, 11863–11867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-García, L.; Sala, R.; Serna, N.; Álamo, P.; Parladé, E.; Alba-Castellón, L.; Voltà-Durán, E.; Sánchez-Chardi, A.; Unzueta, U.; Vázquez, E.; et al. A refined cocktailing of pro-apoptotic nanoparticles boosts anti-tumor activity. Acta Biomater. 2020, 113, 584–596. [Google Scholar] [CrossRef]
- Díaz, R.; Sánchez-García, L.; Serna, N.; Sánchez-Chardi, A.; Cano-Garrido, O.; Sánchez, J.M.; Unzueta, U.; Vazquez, E.; Villaverde, A. Engineering a recombinant chlorotoxin as cell-targeted cytotoxic nanoparticles. Sci. China Mater. 2019, 62, 892–898. [Google Scholar] [CrossRef] [Green Version]
- Díaz, R.; Pallarès, V.; Cano-Garrido, O.; Serna, N.; Sánchez-García, L.; Falgàs, A.; Pesarrodona, M.; Unzueta, U.; Sánchez-Chardi, A.; Sánchez, J.M.; et al. Selective CXCR4+ Cancer Cell Targeting and Potent Antineoplastic Effect by a Nanostructured Version of Recombinant Ricin. Small Weinh. Bergstr. Ger. 2018, 14, e1800665. [Google Scholar] [CrossRef]
- Khirehgesh, M.R.; Sharifi, J.; Safari, F.; Akbari, B. Immunotoxins and nanobody-based immunotoxins: Review and update. J. Drug Target. 2021, 29, 848–862. [Google Scholar] [CrossRef]
- Nejadmoghaddam, M.-R.; Minai-Tehrani, A.; Ghahremanzadeh, R.; Mahmoudi, M.; Dinarvand, R.; Zarnani, A.-H. Antibody-Drug Conjugates: Possibilities and Challenges. Avicenna J. Med. Biotechnol. 2019, 11, 3–23. [Google Scholar]
- Nobre, C.F.; Newman, M.J.; DeLisa, A.; Newman, P. Moxetumomab pasudotox-tdfk for relapsed/refractory hairy cell leukemia: A review of clinical considerations. Cancer Chemother. Pharmacol. 2019, 84, 255–263. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-S.; Jun, S.-Y.; Kim, Y.-S. Critical Issues in the Development of Immunotoxins for Anticancer Therapy. J. Pharm. Sci. 2020, 109, 104–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazor, R.; Pastan, I. Immunogenicity of Immunotoxins Containing Pseudomonas Exotoxin A: Causes, Consequences, and Mitigation. Front. Immunol. 2020, 11, 1261. [Google Scholar] [CrossRef]
- López-Laguna, H.; Unzueta, U.; Conchillo-Solé, O.; Sánchez-Chardi, A.; Pesarrodona, M.; Cano-Garrido, O.; Voltà, E.; Sánchez-García, L.; Serna, N.; Saccardo, P.; et al. Assembly of histidine-rich protein materials controlled through divalent cations. Acta Biomater. 2019, 83, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Falgàs, A.; Pallarès, V.; Unzueta, U.; Céspedes, M.V.; Arroyo-Solera, I.; Moreno, M.J.; Sierra, J.; Gallardo, A.; Mangues, M.A.; Vázquez, E.; et al. A CXCR4-targeted nanocarrier achieves highly selective tumor uptake in diffuse large B-cell lymphoma mouse models. Haematologica 2020, 105, 741–753. [Google Scholar] [CrossRef] [Green Version]
- Céspedes, M.V.; Unzueta, U.; Aviñó, A.; Gallardo, A.; Álamo, P.; Sala, R.; Sánchez-Chardi, A.; Casanova, I.; Mangues, M.A.; Lopez-Pousa, A.; et al. Selective depletion of metastatic stem cells as therapy for human colorectal cancer. EMBO Mol. Med. 2018, 10, e8772. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M. Antibody-Drug Conjugates (ADCs): Magic Bullets at Last! Mol. Pharm. 2015, 12, 1701–1702. [Google Scholar] [CrossRef]
- Peled, A.; Tavor, S. Role of CXCR4 in the Pathogenesis of Acute Myeloid Leukemia. Theranostics 2013, 3, 34–39. [Google Scholar] [CrossRef]
- Ramakrishnan, R.; Peña-Martínez, P.; Agarwal, P.; Rodriguez-Zabala, M.; Chapellier, M.; Högberg, C.; Eriksson, M.; Yudovich, D.; Shah, M.; Ehinger, M.; et al. CXCR4 Signaling Has a CXCL12-Independent Essential Role in Murine MLL-AF9-Driven Acute Myeloid Leukemia. Cell Rep. 2020, 31, 107684. [Google Scholar] [CrossRef]
- Yao, Y.; Li, F.; Huang, J.; Jin, J.; Wang, H. Leukemia stem cell-bone marrow microenvironment interplay in acute myeloid leukemia development. Exp. Hematol. Oncol. 2021, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Alewine, C.; Hassan, R.; Pastan, I. Advances in anticancer immunotoxin therapy. The Oncologist 2015, 20, 176–185. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient No. | Sex | Age (yr) | FAB | 2016 WHO Classification | Cytogenetics | FLT3 Mutation | NPM1 Mutation | CEBPA Mutation | CXCR4 MFIR |
|---|---|---|---|---|---|---|---|---|---|
| #100 | M | 18 | M1 | AML with biallelic mutation of CEBPA | Normal | Negative | Negative | Positive | 1.08 |
| #24 | M | 38 | M5 | AML with t(8;21)(q22;q22.1); RUNX1-RUNX1T1 | 45,X,Y,t(8;21)(q22;q22) [17]/46,XY [3] | Negative | Negative | Negative | 1.13 |
| #72 | M | 26 | M4 | AML with biallelic mutation of CEBPA | Normal | Negative | Negative | Positive | 1.53 |
| #17 | F | 71 | M2 | AML with mutated NPM1 | Normal | ITD | Positive | Negative | 1.75 |
| #79 | F | 61 | M4 | AML with mutated NPM1 | Normal | ITD | Positive | Negative | 2.56 |
| #36 | F | 45 | M5 | AML with t(9;11)(p21.3;q23.3; MLLT3-KMT2A | 46,XX,t(9;11) (p22;q23) [20] | Negative | Negative | Negative | 6.78 |
| #77 | M | 71 | M5 | AML with myelodysplasia-related changes | Normal | Negative | Negative | Negative | 7.77 |
| #23 | F | 80 | M4 | AML with mutated NPM1 | Normal | Negative | Positive | Negative | 11.40 |
| #42 | F | 66 | M4 | AML with mutated RUNX1 | Normal | TKD | Negative | Negative | 32.23 |
| #48 | M | 79 | M5 | AML with myelodysplasia-related changes | Normal | Negative | Positive | negative | 53.17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Núñez, Y.; Garcia-León, A.; Falgàs, A.; Serna, N.; Sánchez-García, L.; Garrido, A.; Sierra, J.; Gallardo, A.; Unzueta, U.; Vázquez, E.; et al. T22-PE24-H6 Nanotoxin Selectively Kills CXCR4-High Expressing AML Patient Cells In Vitro and Potently Blocks Dissemination In Vivo. Pharmaceutics 2023, 15, 727. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics15030727
Núñez Y, Garcia-León A, Falgàs A, Serna N, Sánchez-García L, Garrido A, Sierra J, Gallardo A, Unzueta U, Vázquez E, et al. T22-PE24-H6 Nanotoxin Selectively Kills CXCR4-High Expressing AML Patient Cells In Vitro and Potently Blocks Dissemination In Vivo. Pharmaceutics. 2023; 15(3):727. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics15030727
Chicago/Turabian StyleNúñez, Yáiza, Annabel Garcia-León, Aïda Falgàs, Naroa Serna, Laura Sánchez-García, Ana Garrido, Jorge Sierra, Alberto Gallardo, Ugutz Unzueta, Esther Vázquez, and et al. 2023. "T22-PE24-H6 Nanotoxin Selectively Kills CXCR4-High Expressing AML Patient Cells In Vitro and Potently Blocks Dissemination In Vivo" Pharmaceutics 15, no. 3: 727. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics15030727