
A Small Sugar Molecule with Huge Potential in Targeted Cancer Therapy
 , , and
, , and
Abstract
: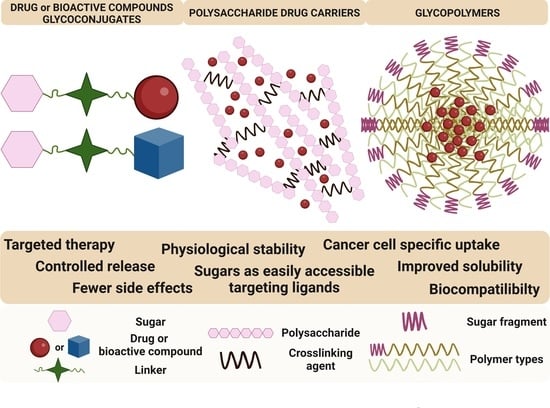
1. Introduction
2. Glycoconjugates
2.1. Anticancer Drug Glycoconjugates

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Conjugated Sugar | Type of Anticancer Activity Studies; Transportation Mode | Activity Compared to Glycone/Properties | Ref. |
|---|---|---|---|---|
| Ifosfamide | D-Glucose | Alkylating agent | Glufosfamide | [62] |
| Doxorubicin (DOX, ADM) | 2-amino-2-deoxy-D-glucose and succinic acid | Antitumor antibiotic GLUTs mediated | 2DG–SA–DOX
| [67] |
| Doxorubicin (DOX, ADM) | Galactose | Antitumor antibiotic | Gal-DOX1
| [70] |
| Doxorubicin (DOX, ADM) | Galactose | ASPG mediated | Gal-DOX2
| [71] |
| Chlorambucil (CLB) | Amino derivatives of glucose, mannose, galactose, xylose, lyxose, D-threoside | Alkylating and DNA-complexing agent | D-threoside-CLB
| [92] |
| Chlorambucil (CLB) | Peracetylated 2-fluorodeoxyglucose | FDG-CLB
| [93] | |
| Paclitaxel (PTX) | Glucose | Mitotic inhibitor | Glu-PTX
| [68,69,94] |
| Paclitaxel (PTX) | Glucose | 2FGlu-PTX/PTX
| [73] | |
| Paclitaxel (PTX) | Glucose | a single (GluSA-PTX) and double (bis-GluSA-PTX)
| [74] | |
| Azomycin | Glucose | GLUTs-mediated | Glucoazomycins
| [66] |
| Geldanamycin (GA) | Glucose | HSP90 inhibitor | Glu-GA
| [95] |
| Geldanamycin (GA) | Galactose Lactose | HSP90 inhibitor | Gal-GA and Lac-GA
| [95] |
| Emodin (EM) | D-rhamnose | Tyrosine kinase inhibitor | Rha-EM
| [96] |
| Platinum | Glucose | GLUTs mediated | Glucose-conjugated Pt(IV) complexes
| [77] |
| Oxaliplatin | Glucose, Mannose Galactose | GLUTs mediated |
| [75] |
2.2. Glycoconjugates of Biological Active Compounds
| Entry | Glycoconjugates | Attached Sugar | Tested Cell Line | Methodology | Best Research Effects IC50: Glycoconjugates/IC50 Precursor | Ref. |
|---|---|---|---|---|---|---|
| 1. |  | D-Glu, L-Rha D-Ara, D-Gal D-Man D-Xyl | A549 DLD-1 B16-F1 WS1 | resazurin reduction test (RTT), in vitro | L-Rha(OH): A549, IC50: 2.6/10.3 µM DLD-1, IC50: 3.9/15 µM B16-F1, IC50: 3.9/16.1 µM WS1, IC50: 31/12 µM D-Ara(OH): B16-F1, IC50: 11/16.1 µM WS1, IC50: 47/12 µM - improvement in hydrosolubility | [132,133] |
| 2. |  | D-Glu L-Rha D-Ara | A549 DLD-1 B16-F1 WS1 | resazurin reduction test (RTT), in vitro | D-Glc(OH): A549, IC50: 8.4/19 µM DLD-1, IC50: 3.9/25 µM B16-F1, IC50: 7.1/26 µM WS1, IC50: 9.3/19 µM | [133] |
| 3. |  | D-Glu, L-Rha D-Ara, D-Gal D-Man, D-Xyl | A549 DLD-1 WS1 | resazurin reduction test (RTT), in vitro | improvement in hydrosolubility | [132] |
| 4. |  | D-Glu L-Rha D-Ara | A549 DLD-1 B16-F1 WS1 | resazurin reduction test (RTT), in vitro | L-Glc(OH): A549, IC50: 14/165 µM DLD-1, IC50: 14/125 µM B16-F1, IC50: 15/104 µM WS1, IC50: 13.3/63 µM D-Ara(OH): A549, IC50: 28/165 µM DLD-1, IC50: 50/125 µM B16-F1, IC50: 27/104 µM WS1, IC50: 15.8/63 µM | [133] |
| 5. |  | D-Glu L-Rha D-Ara D-Gal D-Man D-Xyl | A549 DLD-1 WS1 | resazurin reduction test (RTT), in vitro | D-Glc(OH): A549, IC50: 31/>75 µM DLD-1, IC50: 40/>75 µM WS1, IC50: 40/>75 µM D-Gal(OH): A549, IC50: 30/>75 µM DLD-1, IC50: 40/>75 µM WS1, IC50: 30/>75 µM - improvement in hydrosolubility | [132] |
| 6. |  | A549 DLD-1 WS1 | - non-hemolytic, HD50 >100 µM - better hydrosolubility than BA - a good in vitro stability in phosphate buffer can be hydrolyzed in the presence of β-D-glucuronidase | [134] | ||
| 7. |  | D-Glu L-Rha D-Ara | A549 DLD-1 MCF-7 PC-3 WS1 | resazurin reduction test (RTT), in vitro | L-Rha(OH): A549, IC50: 1.9/3.8 µM DLD-1, IC50: 1.9/6.6 µM MCF-7, IC50: 1.7/23.3 µM PC-3, IC50: 1.8/17.9 µM WS1, IC50: 1.3/3.6 µM | [135] |
| 8. |  | chacotriosyl | A549 DLD-1 MCF-7 PC-3 WS1 | resazurin reduction test (RTT), in vitro | chacotriosyl: A549, IC50: 14/>50 µM DLA-1, IC50: 13/>50 µM MCF-7, IC50: 15/>50 µM PC-3, IC50: 13/>50 µM WS1, IC50: 9/>50 µM | [137] |
| 9. |  | D-Man | CEM, MCF-7 A549, HeLa BJ-H-tert RPMI 8226 G 361 | Calcein AM assay | D-Man(OH): CEM, IC50: 12.9/21.2 µM MCF-7, IC50: 35.5/>50 µM A549, IC50: 44.6/>50 µM HeLa, IC50: 42.8/>50 µM BJ-H-ter, IC50: 43.1/48.6 µM | [138] |
| 10. |  | D-Man tri-D-Man | CEM MCF-7 A549 HeLa BJ-H-tert RPMI 8226 G 361 | Calcein AM assay | D-Man(OH): MCF-7, IC50: 39.2/>50 µM A549, IC50: 44.6/>50 µM HeLa, IC50: 45.7/>50 µM BJ-H-tert, IC50: 35.6/48.6 µM | [138] |
| 11. |  | Glu | glucose-conjugated BN (B10) - apoptotic and non-apoptotic cell death coexist upon B10 treatment - it turns autophagy into a cell death mechanism | [141] | ||
| 12. |  | L-Rha di-L-Rha tri-L-Rha tetra-L-Rha | DLD-1 WS1 | Hoechst test in vitro | L-Rha(OH): DLAD-1, IC50: 4.0/20 µM WS1, IC50: 33.0/36 µM di-L-Rha(OH): DLAD-1, IC50: 5.0/20 µM WS1, IC50: >100/36 µM tri-L-Rha(OH): DLAD-1, IC50: >100/20 µM WS1, IC50: >100/36 µM - in most cases, increasing the number of sugar units leads to reduction of cytotoxity | [142] |
| 13. |  | L-Arap l-Rhap L-Manp D-Idop | CEM MCF-7 HeLa G-361 BJ | cytotoxicity compared to BA 3-O- L-Arap-28-O-L-Arap: CEM, IC50: 2.6/40 µM MCF-7, IC50: 1.6/>50 µM HeLa, IC50: 1.2/47.6 µM G-361, IC50: 0.9/>50 µM BJ, IC50: 1.3/>50 µM 3-O- L-Rhap-28-O-L-Arap: CEM, IC50: 2.4/40 µM MCF-7, IC50: 1.7/>50 µM HeLa, IC50: 1.5/47.6 µM G-361, IC50: 1.1/>50 µM BJ, IC50: 1.5/>50 µM | [143] | |
| 14. |  | A549, NCI-H2087, NCI-H522, NCI-H1993 NCI-H1755, and LLC1 | 3,28-bis-O-L-Rham: IC50: 2.9 -5.9 μM - significantly inhibited tumor growth - can induce apoptotic cell death via disturbance of the mitochondrial electron transfer chain, reduced ROS production, and decreased membrane potential | [147] | ||
| 15. |  | GalNAc | HepG2 Huh7 PC-3 A549 | MTT-based cell viability assay | - high affinity for the asialoglycoprotein receptor (ASGPR) of hepatocytes (in silico) - moderate cytotoxicity and selectivity against HepG2 (IC50: 25.9 µM, for BN IC50: 4.2 µM) | [150] |
3. Sugar-Containing Drug Carriers
3.1. Polysaccharide Drug Carriers
| Entry | Polysaccharide | Type of Drug Binding | Anticancer Drug | Type of Anticancer Activity Studies | Activity/Properties | Ref. |
|---|---|---|---|---|---|---|
| 1. | Chitosan (low molecular weight chitosan, LMWC) | conjugation via succinic anhydride | PTX | B16F10 female C57BL6 mice, melanoma cells; in vivo | IC50 values comparable to parent PTX | [198] |
| 2. | Chitosan/10% dextran sulfates | encapsulation | DOX | A375 and C26; in vitro | the presence of dextran sulfate allowed the DOX-loaded carrier to maintain cytotoxicity at a level comparable to free drug | [199] |
| 3. | N,O-carboxymethyl chitosan (N,O-CMCS)−guar gum (N,O-CMCS/MAGG) | pH-responsive swelling of hydrogels | DOX | MCF-7, in vitro | 67% DOX release after 5 days in pH of 5.5 32% DOX release at pH of 7.4 IC50: 98.45 μg/mL | [200] |
| 4. | Chitosan nanoparticles (CCNP) | encapsulation in nanoparticles using an ionic gelation | CDDP | MCF-7, in vitro | 43.80% CDDP release in 6 h IC50: 4.085 μg/mL | [201] |
| 5. | Chitosan nanoparticle surface linked to rituximab (mAbCCNP) | encapsulation in nanoparticles using an ionic gelation | CDDP | MCF-7, in vitro | 22.52% CDDP release in 4 h no cytotoxicity | [201] |
| 6. | Chitosan | encapsulation in nanoparticles using an ionic gelation | 5-FU | SGC-7901, in vitro pharmacokinetic studies; in vivo | 76% release in the first 0.7 h, sustained release 0.7 to 8.0 h the same inhibitory effect as 5-FU injection half-life increased after intravenous administration compared with 5-FU solution, in vitro | [202] |
| 7. | Chitsan (CS-NPs) | encapsulation in nanoparticles using an ionic gelation | GEM | OVCAR-8, in vitro | 77.27% drug release in 24 h cytotoxicity nanoparticles loaded with drug comparable to parent drug | [203] |
| 8. | Chitosan chemical conjugated with epidermal growth factor receptor variation III (CS-NPs-EGFRv) | encapsulation in nanoparticles using an ionic gelation | GEM | OVCAR-8, in vitro | the cytotoxicity of CS-NPs-EGFRv loaded with the drug is higher than parent drug | [203] |
| 9. | Chitosan (CHT) | conjugation via succinic anhydride (SA), nanoparticles prepared by the precipitation dialysis method | DTX | MDA-MB-231, in vitro | the release of the drug was pH dependent, higher in pH = 5.6 than in pH = 7.4 IC50 of DTX-SA-CHT: 4.68 μg/mL IC50 of DTX: 37.50 μg/mL pharmacokinetic studies show that bioavailability increases with increased half-life and decreased elimination of drug from the biological system | [204] |
| 10. | Pullulan/Chitosan 1:2 (NEPl2-Cs 1:2) | nano-emulsion | DOX | A375 BRAF and HaCaT; in vitro | increased induction of melanoma cell apoptosis and a definite increase in cytotoxicity against A375 cells in case of drug-loaded nano-emulsion application in comparison to free DOX | [205] |
| 11. | Alginate/Chitosan | encapsulation in nanoparticles using two-phase system | DOX | 4T1, in vitro | at pH 5.5, 70% of DOX has been released within 8 h time point, 90% of the drug was released within 24 h IC50 of nanoparticles with DOX: 0.15 μg/mL IC50 of DOX: 0.13 μg/mL | [206] |
| 12. | Alginate (ALG) | PTX -loaded nanoparticles prepared by the nano-emulsification polymer cross-linking method | PTX | Cell cycle analysis, breast cancer cells, in vitro | PTX -loaded nanoparticles inhibit cellular proliferation, block cell cycle progression, and induce apoptosis in cancer cells the percentage of apoptotic cells in untreated cells increased from 11% to 83% after treatment with PTX nanoparticles | [207] |
| 13. | Alginate (ALG) | co-loaded hydrogel (ACA) | CDDP and AuNPS | CT26, in vitro | the ACA nanocomplex is more effective than CDDP: the ACA nanocomplex at a concentration of 5 µg/mL (per cisplatin) and 20 µg/mL of free cisplatin resulted in the same cytotoxicity (survival rate: 66%) the ACA nanocomplex increased the brightness of computed tomography images and contrast to noise ratio | [195] |
| 14. | Dextran as a copolymer component DEX-P(OEGMA-co-MGMA) | DOX covalently decorated on the copolymer nanocarrier by conjugation via a pH-responsive hydrazone bond | DOX as conjugate (DOXDT) | 4T1, HeLa human cervical cancer cell line, in vitro Balb/C mice bearing 4T1 tumor, in vivo | pH-dependent drug release (higher in an acidic environment) cell viability of HeLa and 4T1 cells significantly decreased in the presence of DOXDT, in vitro the tumor volume of DOXDT treated mice was smaller than in control group (control group: increasing from 139.74 to 1376.35 mm3 after 14 days; DOXDT group: increasing to 296.63 mm3) | [193] |
| 15. | Dextran (DEX-SS) | dextran-based nanogels (DEX-SS) created by Schiff base formation between polyaldehyde dextran (DEX-CHO) and cystamine DOX conjugated into DEX-SS nanogels via Schiff base linkages | DOX as conjugate | H1299 and Hela, in vitro | DOX-loaded dextran nanogels penetrate cancer cells and, under the influence of both the environmental pH and the amount of GSH, release the drug | [194] |
| 16. | Dextran (DEX) | negatively charged dextran-based dual conjugates with different length linkers | DTX and DHA as conjugate | HTB-177, MCF-7, and 4T1 mouse breast cancer cells, in vitro 4T1 breast cancer cells in BALB/C mice, in vivo | in vitro: comparable activity of DTX and its conjugate (DEX-DHA-DTX) the conjugates improved drug solubility and increased the amount of drug within tumor cells, while its concentration in healthy cells was lower than that with free DTX in vivo: the conjugate caused tumor disappearance in mice, no side effects | [208] |
| 17. | Dextran oxidised to dicarboxydextran (DXA) | CDDP-crosslinked DXA nanogels | CDDP | A2780, A2780/CP CDDP-resistant subline, A549, 22Rv1, PC-3, in vitro | CDDP conjugates with high-Mw DXA showed up to four times increased anticancer efficacy against malignant prostatic cell lines than free CDDP, and significantly inhibited ovarian cancer cell migration | [209] |
| 18. | Hyaluronic acid (HA) | dual drug-loaded HA micelles (HA-DOX-CDDP) | DOX and CDDP | 4T1, NIH-3T3, in vitro 4T1-xenografted Balb/c mice, in vivo | HA-DOX-CDDP micelles exhibited in vitro: increased drug release at acidic pH, better drug uptake and increased antiproliferative activity than in case of free DOX in vivo: less systemic toxicity and greater efficacy than free DOX | [197] |
| 19. | Hyaluronic acid conjugated with casein (HA/casein 3:1) | hyaluronic acid -coated paclitaxel-loaded casein nanoparticles (HA-PTX-Cas NPs) | PTX | A375, in vitro male hairless mice HRS/J, in vivo | compared to uncoated PTX-Cas NPs, HA-PTX-Cas NPs reached a higher entrapment efficiency (93.1%) and exhibited satisfactory stability, HA-PTX-Cas exhibited a high efficiency (61.3%) in inhibiting A375 tumor mice experiments showed 74.6% tumor inhibition of HA-PTX-Cas by intravenously administration | [210] |
| 20. | Hyaluronic acid (HA) | HA conjugates of DOX and GEM with different linkers | DOX and GEM | MDA-MB-231, 4T1, in vitro BALB/c mice bearing 4T1 tumor, in vivo | polymer conjugates released GEM faster than DOX more effective in killing triple negative breast cancer cells in vitro, more effectively inhibited the growth of the 4T1 tumor model in vivo than free DOX and GEM after intravenous and subcutaneous injection | [211] |
| 21. | Hyaluronic acid coated B-mR9 | nanoparticles coated with HA branched modified nona-arginine (B-mR9), composed of redox-cleavable disulfide bonds and complexed with MTX (B-mR9-MTX/HA) | MTX | NCI-H460, MCF-7, NIH-3T3, in vitro female, 6 weeks old BALB/c nude mice, in vivo | B-mR9-MTX/HA in vitro: improve drug delivery to cancer cells in vivo: better biodistribution, long retention in the body, and high tumor inhibition ability | [212] |
| 22. | amine-functionalized nanocrystalline cellulose grafted folic acid/magnetic nanoparticles (AF-NCC/Fe3O4 NPs) | encapsulating DOX in AF-NCC/Fe3O4 NPs | DOX | Saos-2, in vitro | high encapsulation efficacy high stability at physiological pH high rate of drug release at low pH increased therapeutic effects compared to free DOX | [213] |
| 23. | Thiolated heparin | polyion complex crosslinking by oxidation under atmosphere | DOX | MDA-MB-231 and HUVEC, in vitro 4 weeks old female Balb/c nude mice, in vivo | pH and GSH dual-sensitive drug release behavior in vitro polyion complex showed improved, compared to free drugs, anti-tumor performance and lower side effect to normal tissue both in vitro and in vivo | [214] |
3.2. Glycopolymers
3.2.1. Glycopolymers with Encapsulated Drug
pH-Responsive Glycopolymers
Light-Responsive Glycopolymers
Thermoresponsive Glycopolymers
3.2.2. Glycopolymers with Bounded Drug
| Entry | Polymer | Attached Sugar | Linker (Binding Type) | Drug | Methodology | Results (IC50 or Percent Inhibition) | The Postulated Mechanism | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1. | PAG-b-PFMA | Glu | ester bond | DOX | MBA-MD-231 MTT assay, in vitro | Free DOX MBA-MD-231 IC50: 0.631 μM Glycopolymer MBA-MD-231 IC50: 0.908 μM | REDOX-responsive glycopolymer | [241] |
| 2. | PMAG-b-P(Lys-co-Phe) | Glu |  | PTX | MCF-7, A549 CTB assay, in vitro | PTX-LANS (commercially available formulation with PTX) MCF-7 IC50: 4 ± 1 ng/mL A549 IC50: 2.0 ± 0.3 ng/mL PMAG-b-P(Lys-co-Phe) MCF-7 IC50: 4.1 ± 0.5 ng/mL A549 IC50: 4.4 ± 0.6 ng/mL | pH sensitive | [231] |
| 3. | PMAG’-b-PFBEMA | Glu | ester bond | DOX AuNPs | MDA-MB-231 MTT assay, in vitro | - | GLUT transporters, pH sensitive | [232] |
| 4. | P(NIPAM-co-OVAG)-b-PNIPAM | Glu |  | ConA | SMMC-7721 MTT assay, in vitro | - | ConA receptor thermosensitive glycopolymer | [244] |
| 5. | PEG-b-DEA-b- GAMA PEG-b-PGAMA PEG-b-PS-b-PGAMA | Glu |  | BTZ | L929 | - | pH-responsive glycopolymer | [233] |
| 6. | PEG-b-PGAMA-b-PDEA | Glu |  | DOX | in vitro | - | pH-sensitive micelles | [234] |
| 7. | P(MAG-co-HEMA)-b-PBAE | Glu |  | DOX | U87-MG, MTT assay in vitro | - | pH-responsive glycopolymer | [235] |
| 8. | GP-Gluc-CDDP | Glu | ester bond | CDDP | OSC-19, U87MG, in vitro cytotoxicity assay | - | GLUT transporters | [248] |
| 9. | p(1-O-MAFru)-b-PMMA | Fruc | ester bond | curcumin | MCF-7, RAW 264.7, SRB assay, in vitro | glycopolymer MCF-7 IC50: 15.2 µM RAW 264.7 IC50: 5.7 µM | GLUT transporters | [249] |
| 10. | P(1-O-MAFru)-b-PMMA | Fruc | ester bond | PTX | MDA-MB-231, MCF-7, flow cytometry, in vitro | glycopolymer MDA-MB-231 IC50: 4.48 ± 0.10 µM MCF-7 IC50: 27.57 ± 0.50 µM | GLUT transporters | [250] |
| 11. | P(FrucMA-b-MAEBA)-Py | Fruc | ester bond- sugar | DOX (conjugate) | MCF-7, MDA-MB-231 MTT assay, in vitro | Apoptotic effect (%) for 24 h of glycopolymer: MCF-7: 85.00% MDA-MB-231: 81.24% glycopolymer with folic acid MCF-7: 87.46% MDA-MB-231: 96.58% free DOX: MCF-7: 42.68% MDA-MB-231: 72.80% | GLUT transporters | [251] |
| 12. | P(FrucMA-b-MAc)-GEM | Fruc | ester bond | CDDP, GEM | MDA-MB-231, CCD-1079Sk, in vitro | glycopolymer CCD-1079Sk IC50: 125.68 ± 0.011 μg/mL MDA-MB-231 IC50: 31.51 ± 0.021 μg/mL | pH-sensitive glycopolymer, GLUT transporters | [252] |
| 13. | P(MMA-b-MAEBA-b-FrucMA)-ZnPc/Dox | Fruc | ester bond | DOX | 3T3, MCF-7, MDA-MB-231, MTT assay, in vitro | Free DOX for 4h 3T3 IC50: 22.31 ± 3.39 μg/mL MDA-MB-231 IC50: 28.22 ± 3.55 μg/mL GNPs-ZnPc/Dox for 4 h 3T3 IC50: 13.21 ± 1.39 μg/mL MDA-MB-231 IC50: 10.57 ± 1.27 μg/mL with the presence of light irradiation 3T3 IC50: 3.502 ± 0.22 μg/mL MDA-MB-231 IC50: 1.43 ± 0.09 μg/mL | pH-sensitive glycopolymer, GLUT5 transporter (fructose transporter) | [236] |
| 14. | P(MAFruc)-b-P(3-VBA)-co- MMA | Fruc | ester bond imine linker-AMF  | AMF | MCF-7, and MDA-MB-231, SRB assay, in vitro | free amonafide: MCF-7 IC50: 11.23 μM MDA-MB-231 IC50: 13.98 μM Glycopolymer: MCF-7 IC50: 7.19 μM MDA-MB-231 IC50: 4.92 μM | GLUT transporters | [247] |
| 15. | P(1-O-MA’Fruc)-b-PMMA | Fruc | ester bond | DOX | MCF-7, MDA-MB-231, flow cytometry, in vitro | - | GLUT transporters | [253] |
| 16. | P(BOB-HA)-P(Fruc)-PDS/Vc | Fruc | ester bond | DOX | 4T1, MTT assay, in vitro | - | light-responsive glycopolymer (NIR) | [242] |
| 17. | PEG-b-PAEG-b-PAA cl-micelles/Cys | Gal | ester bond | DOX | HepG2, NIH3T3 MTT assay, in vitro | cell viability (%) for 24 h HepG2: 38% NIH3T2: 68% | ASGP-R receptors, redox-sensitive micelles | [254] |
| 18. | PMAGal- b -PMAChols | Gal | ester bond | DOX |
SK-Hep-1, MTT assay, in vitro | SK-Hep-1 IC50: 9.06 μM | receptor ASGP-R | [255] |
| 19. | p(IVDG-co-BMDO) | Gal |  | DOX | L929, HeLa, MTT assay, in vitro | free DOX: HeLa IC50: 0.8 mg/mL glycopolymer: HeLa IC50: 1.9 mg/mL, | pH-sensitive polymeric micelles | [237] |
| 20. | IGPC | Gal | ester bond | DOX |
HepG2, MTT assay, in vitro | free DOX: HepG2 IC50: 0.45μg/mL DOX-loaded glucose HepG2 IC50: 0.75 μg/mL galactose-containing micelles HepG2 IC50: 0.20μg/mL | uptake by ASGP-R | [256] |
| 21. | mPEG-b-PMAGal-co-DOX | Gal | ester bond | DOX | HepG2, MCF-7, MTT assay, in vitro | Free DOX: HepG2 IC50: 0.61 μM MCF-7 IC50: 0.70 μM Glycopolymer: HepG2 IC50: 1.22 μM MCF-7 IC50: 2.97 μM | receptor ASGP-R | [257] |
| 22. | (PCL)2−b-Pr-gly−b-GP | Gal |  | DOX | HepG2, MTT assay, in vitro | free Dox: HepG2 IC50: 2.2 μg/mL Dox-loaded UCL (uncross-linked) micelles: HepG2 IC50: 5.7 μg/mL Dox-loaded ICL (interface crosslinked) micelles: HepG2 IC50: 7.83 μg/mL | receptor ASGP-R | [258] |
| 23. | PMAG-b-PAA | Gal |  | PTX |
A549, MCF-7 in vitro | PTX-LANS® (commercially available formulation with PTX) A549 IC50: 2 ng/mL MCF-7 IC50: 4 ng/mL PMAG-b-PAA NPs A549 IC50: 1.8 ng/mL MCF-7 IC50: 8 ng/mL | - | [259] |
| 24. | PADGal | Gal | ester bond | DOX | HepG2 and HeLa, NIH3T3, MTT assay, in vitro | HepG2 IC50: 2.9 μg/mL HeLa IC50: 9.0 μg/mL NIH3T3 IC50: 12.5 μg/ml | uptake by ASGP-R | [260] |
| 25. | P(MAGal-co-DMAEMA)-b-PPDSMA | Gal | ester bond | DOX | HepG2, MTT assay, in vitro | - | receptor ASGP-R | [261] |
| 26. | pGal(Ac)-b-pNIPAA | Gal |  | DOX | HeLa, A549, HepG2, MTT assay, in vitro | - | uptake by ASGP-R | [262] |
| 27. | P(AzoMA)-b-P(GalEtMA)) | Gal |  | hydrophobic compound Nile red | A375, SRB assay, in vitro | - | light-responsive glycopolymer | [240] |
| 28. | P((DEGMA)-b-P(OVNG)) | Gal |  | AuNRs | L-929, MTT assay HepG2, flow cytometry, in vitro | - | thermoresponsive glycopolymer (photothermal treatment for the tumor- phototherapy) | [245,246] |
| 29. |
PHML-b-PMAGal P(HML-st-MAGal) | Gal | ester bond | pDNA | H1299, MTT assay, in vitro | - | pDNA binding affinities | [263] |
| 30. | PCL- b -PManEA | Man |  | DOX and ConA | UMUC3, MTT assay, in vitro | free DOX UMUC3 IC50: 0.79 µg/mL Glycopolymer UMUC3 IC50: 1.98 µg/mL | ConA receptor | [264] |
| 31. | P(ManMac)-r-(MAA) | Man |  | AuNPs with DOX | HeLa, A549, SH-SY5Y, MTT assay, in vitro | - | pH-sensitive drug | [238] |
| 32. | Man-GP-(PCL)2 | Man |  | Nile red or Rhodamine B | MDA-MB-231, MTT assay, in vitro | - | receptor MRC2 | [265] |
| 33. | PAAMAM- (FUDR+CARB)-MOF-808 | Man |  | FUDR and CARB | MCF-7, PANC-1, HepG2, cytotoxicity assay, in vitro | free CARB MCF-7 IC50: 59.4 μg/mL HepG2 IC50: 15.8 μg/mL free FUDR PANC-1 IC50: 20.0 μg/mL HepG2 IC50: 14.0 μg/mL PAAMAM- (FUDR+CARB)-MOF-808 HepG2 IC50: 0.13 (equiv. FUDR μg/mL); 2.4 μg/mL (equiv. CARB μg/mL) MCF-7 IC50: 0.46 μg/mL (equiv. FUDR μg/Ml); 8.3 equiv. CARB μg/mL) PANC-1 IC50: 0.38 μg/mL (equiv. FUDR μg/Ml); 6.8 μg/mL (equiv. CARB μg/mL) | mannose receptors CD206 | [266] |
| 34. | GP’-b-PCL | Lac |  | DOX | HepG2, MTT assay, in vitro | Glycopolymer HepG2 IC50:0.43 µg/mL DOX-loaded non-glycomicelles HepG2 IC50: 6.55 µg/mL | uptake by ASGP-R | [239] |
| 35. | pDMSN-pLAMA | Lac |  | MSNs | HepG2, NIH3T3, MTT assay, in vitro | pDMSN-pLAMA HepG2 IC50:0.43 µM | receptor ASGP-R | [267] |
| 36. | P(AcGlcMA-b-MAA) P(AcFrucMA-b-MAA) | Glu and Fruc | ester bond | DACP-Pt drug | A2780, MCF-7 and MB-MDA-231, flow cytometry, in vitro | DACP-Pt drug: A2780 IC50:5.8 µM MCF-7 IC50:5.08 µM MB-MDA-231 IC50:11 µM Glu polimer: A2780 IC50: 2.8 µM MCF-7 IC50: 20.1 µM MB-MDA-231 IC50: 15.9 µM Fruc polymer:A2780 IC50: 2.5 µM MCF-7 IC50:7.3 µM MB-MDA-231 IC50:4.8 µM | GLUT transporters | [268] |
| 37. | DEGMA-co-OVNGmix | Glu Gal |  | AuNPs and ConA | HepG2, L929, CCK8 assay, in vitro | - | ConA receptor | [269] |
| 38. | HA-(PEG-DNP) | glucuronic acid N-acetylglucosamine | - | - | MDA-MB-23, ADCC assay, in vitro | - | multivalent antibody recruiting glycopolymers (MARGs) | [270] |
| 39. | PLAMA-b-PSBMA-b-PNIPAM | Lac and Gal |  | DOX | HepG2,HeLa, CCK-8, in vitro | Nanomedicines with galactose HepG2 IC50: 0.31 µg/mL HeLa IC50: 1.21µg/mL Nanomedicines without galactose HepG2 IC50: 1.42 µg/mL HeLa IC50: 1.57 µg/mL | receptor ASGP-R, thermo- and redox-sensitive glycopolymer | [243] |
| 40. | BGP | Fuc, SialA, HD |  | no drug a | COS7, B16, MTT assay, in vitro | - | mimic natural glycosaminoglycan (heparin) | [271] |
3.2.3. Sugars Incorporated into Glycopolymers
Glucose Glycopolymers
Fructose Glycopolymers
Galactose Glycopolymers
Mannose Glycopolymers
Disaccharide Glycopolymers
Other Glycopolymers
4. Calculation Methods in Drug Design
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Anticancer drugs | |
| 5-FU | 5-fluorouracil |
| ADM | adriamycin |
| AMF | amonafide |
| BTZ | bortezomib |
| CARB | carboplatin |
| CDDP | cisplatin |
| CLB | chlorambucil |
| ConA | concanavalin A |
| DHA | docosahexaenoic acid |
| DOX | doxorubicin |
| DTX | docetaxel |
| GEM | gemcitabine |
| MTX | methotrexate |
| PDX | podophyllotoxin |
| PTX | paclitaxel |
| Cell lines | |
| 4T1 | murine breast cancer cell line |
| A375 | human melanoma cell line |
| A549 | human pulmonary adenocarcinoma cell line |
| A2780 | epithelial ovarian cancer cell line |
| A2780/CP | CDDP-resistant subline |
| AsPC-1 | human pancreas adenocarcinoma |
| B16-F1 | mouse melanoma cell line |
| B16-F10 | mouse melanoma cell line |
| BJ-H-tert RPMI | normal fibroblasts |
| C26 | murine colorectal carcinoma cell line |
| Calu-3 | human lung cancer cell line |
| C57BL6 | mice melanoma cell line |
| CEM | T-lymphoblastic leukemia cell line |
| Colo-205 | colorectal adenocarcinoma |
| CT26 | mouse colon adenocarcinoma cell line |
| DLD-1 | colorectal adenocarcinoma cell line |
| G 361 | malignant melanoma cell line |
| H1299 | human non-small lung carcinoma cell line |
| HaCaT | keratinocyte cancer cell line |
| HCT-116 | human colon carcinoma cell line |
| HeLa | human cervical cancer cell line |
| HepG2 | human hepatocellular carcinoma cell line |
| HL-60 | human promyelocytic leukemia cell line |
| HTB-177 | lung cancer cell line |
| HUVEC | human umbilical vein endothelial |
| Hs683 | human brain glioma cell line |
| K-562 | human chronic myelogenous leukaemia |
| MCF-7 | human breast cancer cell line |
| MDA-MB-231 | human breast cancer cell line |
| NCI-H460 | lung cancer cell line |
| CD44- | fibroblast cell line |
| NKE | normal kidney epithelial cell line |
| OVCAR-3 | ovarian carcinoma cell line |
| OVCAR-8 | ovarian carcinoma cell line |
| PANC-1 | human pancreas ductal adenocarcinoma |
| PC-3 | human prostate cancer cell line |
| RPMI 8226 | multiple myeloma cell line |
| 22Rv1 | human prostate carcinoma cell line |
| Saos-2 | human osteosarcoma cell line |
| SHY5Y | hyman neuroblastoma cell line |
| SK-Hep-1 | human hepatic adenocarcinoma |
| SGC-790 | endocervical adenocarcinoma |
| U-251 | human glioblastoma cell line |
| WS1 | normal skin fibroblasts |
| VMCF-7 | human breast cancer |
| XHepG2 | human liver cancer cell line |
References
- Bray, F.; Laversanne, M.; Weiderpass, E.; Soerjomataram, I. The Ever-increasing Importance of Cancer as a Leading Cause of Premature Death Worldwide. Cancer 2021, 127, 3029–3030. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Wild, C.P.; Weiderpass, E.; Steward, B.W. World Cancer Report: Cancer Research for Cancer Prevention; International Agency for Research on Cancer: Lyon, France, 2020; Available online: http://publications.iarc.fr/586 (accessed on 8 March 2023).
- Batist, G. Systemic Cancer Therapy: Achievements and Challenges That Lie Ahead. Front. Pharmacol. 2013, 4, 57. [Google Scholar] [CrossRef] [Green Version]
- Zugazagoitia, J.; Guedes, C.; Ponce, S.; Ferrer, I.; Molina-Pinelo, S.; Paz-Ares, L. Current Challenges in Cancer Treatment. Clin. Ther. 2016, 38, 1551–1566. [Google Scholar] [CrossRef] [Green Version]
- Maeda, H.; Khatami, M. Analyses of Repeated Failures in Cancer Therapy for Solid Tumors: Poor Tumor-selective Drug Delivery, Low Therapeutic Efficacy and Unsustainable Costs. Clin. Transl. Med. 2018, 7, e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyman, D.M.; Vasan, N.; Baselga, J. A View on Drug Resistance in Cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Zhong, L. Small Molecules in Targeted Cancer Therapy: Advances, Challenges, and Future Perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef]
- Souza, C. Prodrugs for Targeted Cancer Therapy. Expert Rev. Anticancer Ther. 2019, 19, 483–502. [Google Scholar] [CrossRef]
- Patel, T.K.; Adhikari, N.; Amin, S.A.; Biswas, S.; Jha, T.; Ghosh, B. Small Molecule Drug Conjugates (SMDCs): An Emerging Strategy for Anticancer Drug Design and Discovery. New J. Chem. 2021, 45, 5291–5321. [Google Scholar] [CrossRef]
- Low, P.S. Ligand-Targeted Drug Delivery. Chem. Rev. 2017, 117, 12133–12164. [Google Scholar] [CrossRef]
- Srinivasarao, M.; Galliford, C.V.; Low, P. Principles in the Design of Ligand-Targeted Cancer Therapeutics and Imaging Agents. Nat. Rev. Drug Discov. 2015, 14, 203–2019. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, L.; Li, X.-F. Hypoxia and the Tumor Microenvironment. Technol. Cancer Res. Treat. 2021, 20, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Boedtkjer, E.; Pedersen, S.F. The Acidic Tumor Microenvironment as a Driver of Cancer. Annu. Rev. Physiol. 2020, 82, 103–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, F.; Ramnath, N.; Nagrath, D. Reactive Oxygen Species in the Tumor Microenvironment: An Overview. Cancers 2019, 11, 1191. [Google Scholar] [CrossRef] [Green Version]
- Bobko, A.A.; Eubank, T.D.; Driesschaert, B.; Khramtsov, V.V. In Vivo EPR Assessment of PH, PO2, Redox Status, and Concentrations of Phosphate and Glutathione in the Tumor Microenvironment. J. Vis. Exp. 2018, 133, e56624. [Google Scholar] [CrossRef] [Green Version]
- Lossow, K.; Schwarz, M.; Kipp, A.P. Are Trace Element Concentrations Suitable Biomarkers for the Diagnosis of Cancer? Redox Biol. 2021, 42, 101900. [Google Scholar] [CrossRef]
- Itoh, Y. Proteolytic Modulation of Tumor Microenvironment Signals during Cancer Progression. Front. Oncol. 2022, 12, 935231. [Google Scholar] [CrossRef]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.; Fernandes, A. Targeting Tumor Microenvironment for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. We Need to Talk about the Warburg Effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullapudi, S.S.; Mitra, D.; Li, M.; Kang, E.-T.; Chiong, E.; Neoh, K.G. Potentiating Anti-Cancer Chemotherapeutics and Antimicrobials via Sugar-Mediated Strategies. Mol. Syst. Des. Eng. 2020, 5, 772–791. [Google Scholar] [CrossRef]
- Mueckler, M.; Thorens, B. The SLC2 (GLUT) Family of Membrane Transporters. Mol. Aspects Med. 2013, 34, 121–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barron, C.C.; Bilan, P.J.; Tsakiridis, T.; Tsiani, E. Facilitative Glucose Transporters: Implications for Cancer Detection, Prognosis and Treatment. Metabolism 2016, 65, 124–139. [Google Scholar] [CrossRef]
- Tanasova, M.; Fedie, J.R. Molecular Tools for Facilitative Carbohydrate Transporters (Gluts). ChemBioChem 2017, 18, 1774–1788. [Google Scholar] [CrossRef] [PubMed]
- Szablewski, L. Expression of Glucose Transporters in Cancers. Biochim. Biophys. Acta BBA Rev. Cancer 2013, 1835, 164–169. [Google Scholar] [CrossRef]
- Carvalho, K.C.; Cunha, I.W.; Rocha, R.M.; Ayala, F.R.; Cajaíba, M.M.; Begnami, M.D.; Vilela, R.S.; Paiva, G.R.; Andrade, R.G.; Soares, F.A. GLUT1 Expression in Malignant Tumors and Its Use as an Immunodiagnostic Marker. Clinics 2011, 66, 965–972. [Google Scholar] [CrossRef] [Green Version]
- Ben-Haim, S.; Ell, P. 18F-FDG PET and PET/CT in the Evaluation of Cancer Treatment Response. J. Nucl. Med. 2009, 50, 88–99. [Google Scholar] [CrossRef] [Green Version]
- Molejon, M.I.; Weiz, G.; Breccia, J.D.; Vaccaro, M.I. Glycoconjugation: An Approach to Cancer Therapeutics. World J. Clin. Oncol. 2020, 11, 110–120. [Google Scholar] [CrossRef]
- Fadaka, A.; Ajiboye, B.; Ojo, O.; Adewale, O.; Olayide, I.; Emuowhochere, R. Biology of Glucose Metabolization in Cancer Cells. J. Oncol. Sci. 2017, 3, 45–51. [Google Scholar] [CrossRef]
- Kim, S.-H.; Baek, K.-H. Regulation of Cancer Metabolism by Deubiquitinating Enzymes: The Warburg Effect. Int. J. Mol. Sci. 2021, 22, 6173. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Herrero, E.; Fernández-Medarde, A. Advanced Targeted Therapies in Cancer: Drug Nanocarriers, the Future of Chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef] [Green Version]
- Senapati, S.; Mahanta, A.K.; Kumar, S.; Maiti, P. Controlled Drug Delivery Vehicles for Cancer Treatment and Their Performance. Signal Transduct. Target. Ther. 2018, 3, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenzel, M.H. Glycopolymers for Drug Delivery: Opportunities and Challenges. Macromolecules 2022, 55, 4867–4890. [Google Scholar] [CrossRef]
- Abasalizadeh, F.; Moghaddam, S.V.; Alizadeh, E.; Akbari, E.; Kashani, E.; Fazljou, S.M.B.; Torbati, M.; Akbarzadeh, A. Alginate-Based Hydrogels as Drug Delivery Vehicles in Cancer Treatment and Their Applications in Wound Dressing and 3D Bioprinting. J. Biol. Eng. 2020, 14, 8. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Liu, X.-R.; Chen, Q.-B.; Li, Y.; Zhou, J.-L.; Zhou, L.-Y.; Zou, T. Hyaluronic Acid and Albumin Based Nanoparticles for Drug Delivery. J. Control. Release 2021, 331, 416–433. [Google Scholar] [CrossRef]
- Wang, Y.; Xin, D.; Liu, K.; Zhu, M.; Xiang, J. Heparin−Paclitaxel Conjugates as Drug Delivery System: Synthesis, Self-Assembly Property, Drug Release, and Antitumor Activity. Bioconjug. Chem. 2009, 20, 2214–2221. [Google Scholar] [CrossRef]
- Kuo, P.-H.; Teng, Y.-H.; Cin, A.-L.; Han, W.; Huang, P.-W.; Wang, L.H.-C.; Chou, Y.-T.; Yang, J.-L.; Tseng, Y.-L.; Kao, M.; et al. Heparan Sulfate Targeting Strategy for Enhancing Liposomal Drug Accumulation and Facilitating Deep Distribution in Tumors. Drug Deliv. 2020, 27, 542–555. [Google Scholar] [CrossRef]
- Li, L.; Ni, R.; Shao, Y.; Mao, S. Carrageenan and Its Applications in Drug Delivery. Carbohydr. Polym. 2014, 103, 1–11. [Google Scholar] [CrossRef]
- Varshosaz, J. Dextran Conjugates in Drug Delivery. Expert Opin. Drug Deliv. 2012, 9, 509–523. [Google Scholar] [CrossRef]
- Bhattarai, N.; Gunn, J.; Zhang, M. Chitosan-Based Hydrogels for Controlled, Localized Drug Delivery. Adv. Drug Deliv. Rev. 2010, 62, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Miao, T.; Wang, J.; Zeng, Y.; Liu, G.; Chen, X. Polysaccharide-Based Controlled Release Systems for Therapeutics Delivery and Tissue Engineering: From Bench to Bedside. Adv. Sci. 2018, 5, 1700513. [Google Scholar] [CrossRef] [Green Version]
- Varki, A. Biological Roles of Glycans. Glycobiology 2017, 27, 3–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraser-Reid, B.O.; Tatsuta, K.; Thiem, J. (Eds.) Glycoscience: Chemistry and Chemical Biology; Springer: Berlin/Heidelberg, Germany, 2008; ISBN 978-3-540-36154-1. [Google Scholar]
- Pereira, D.M.; Valentao, P.; Correia-Da-Silva, G.; Teixeira, N.; Andrade, P. Plant Secondary Metabolites in Cancer Chemotherapy: Where Are We? Curr. Pharm. Biotechnol. 2012, 13, 632–650. [Google Scholar] [CrossRef]
- Cragg, G.M. Paclitaxel (Taxol®): A Success Story with Valuable Lessons for Natural Product Drug Discovery and Development. Med. Res. Rev. 1998, 18, 315–331. [Google Scholar] [CrossRef]
- Cseke, L.J.; Kirakosyan, A.; Kaufman, P.B.; Warber, S.; Duke, J.A.; Brielmann, H.L. Natural Products from Plants; CRC Press: Boca Raton, FL, USA, 2016; ISBN 978-0-429-12563-8. [Google Scholar]
- Mustacchi, G.; De Laurentiis, M. The Role of Taxanes in Triple-Negative Breast Cancer: Literature Review. Drug Des. Devel. Ther. 2015, 2015, 4303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuwayama, T.; Nakamura, S.; Hayashi, N.; Takano, T.; Tsugawa, K.; Sato, T.; Kitani, A.; Okuyama, H.; Yamauchi, H. Randomized Multicenter Phase II Trial of Neoadjuvant Therapy Comparing Weekly Nab-Paclitaxel Followed by FEC with Docetaxel Followed by FEC in HER2− Early-Stage Breast Cancer. Clin. Breast Cancer 2018, 18, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-J.; Keam, B.; Ock, C.-Y.; Song, S.; Kim, M.; Kim, S.H.; Kim, K.H.; Kim, J.-S.; Kim, T.M.; Kim, D.-W.; et al. A Phase II Study of Pembrolizumab and Paclitaxel in Patients with Relapsed or Refractory Small-Cell Lung Cancer. Lung Cancer 2019, 136, 122–128. [Google Scholar] [CrossRef]
- Kizek, R.; Adam, V.; Hrabeta, J.; Eckschlager, T.; Smutny, S.; Burda, J.V.; Frei, E.; Stiborova, M. Anthracyclines and Ellipticines as DNA-Damaging Anticancer Drugs: Recent Advances. Pharmacol. Ther. 2012, 133, 26–39. [Google Scholar] [CrossRef]
- de Bono, J.S.; Ashworth, A. Translating Cancer Research into Targeted Therapeutics. Nature 2010, 467, 543–549. [Google Scholar] [CrossRef]
- Florea, A.-M.; Büsselberg, D. Cisplatin as an Anti-Tumor Drug: Cellular Mechanisms of Activity, Drug Resistance and Induced Side Effects. Cancers 2011, 3, 1351–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, J.; Muhsin, M.; Kirkpatrick, P. Oxaliplatin. Nat. Rev. Drug Discov. 2004, 3, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Oda, H.; Sugawara, Y.; Masuya, M.; Nakase, K.; Fujioka, M.; Imai, H.; Katayama, N. Oxaliplatin-Induced Acute Thrombocytopenia: A Case Report and Review of the Literature. Intern. Med. 2013, 52, 611–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, J.N.; Tazawa, M.J. Glucose–Aspirin: Synthesis and In Vitro Anti-Cancer Activity Studies. Bioorg. Med. Chem. Lett. 2012, 22, 3168–3171. [Google Scholar] [CrossRef] [PubMed]
- Peltier-Pain, P.; Timmons, S.C.; Grandemange, A.; Benoit, E.; Thorson, J.S. Warfarin Glycosylation Invokes a Switch from Anticoagulant to Anticancer Activity. ChemMedChem 2011, 6, 1347–1350. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Lu, Y.; Gao, X.; Liu, R.; Zhang-Negrerie, D.; Shi, Y.; Wang, Y.; Wang, S.; Gao, Q. Highly Water-Soluble Platinum(Ii) Complexes as GLUT Substrates for Targeted Therapy: Improved Anticancer Efficacy and Transporter-Mediated Cytotoxic Properties. Chem. Commun. 2013, 49, 2421. [Google Scholar] [CrossRef]
- Cao, X.; Du, X.; Jiao, H.; An, Q.; Chen, R.; Fang, P.; Wang, J.; Yu, B. Carbohydrate-Based Drugs Launched during 2000−2021. Acta Pharm. Sin. B 2022, 12, 3783–3821. [Google Scholar] [CrossRef]
- Stüben, J.; Port, R.; Bertram, B.; Bollow, U.; Hull, W.E.; Schaper, M.; Pohl, J.; Wiessler, M. Pharmacokinetics and Whole-Body Distribution of the New Chemotherapeutic Agent β- D -Glucosylisophosphoramide Mustard and Its Effects on the Incorporation of [Methyl- 3 H]-Thymidine in Various Tissues of the Rat. Cancer Chemother. Pharmacol. 1996, 38, 355–365. [Google Scholar] [CrossRef]
- Pohl, J.; Bertram, B.; Hilgard, P.; Nowrousian, M.R.; Stüben, J.; Wießler, M. D-19575—A Sugar-Linked Isophosphoramide Mustard Derivative Exploiting Transmembrane Glucose Transport. Cancer Chemother. Pharmacol. 1995, 35, 364–370. [Google Scholar] [CrossRef]
- Briasoulis, E.; Judson, I.; Pavlidis, N.; Beale, P.; Wanders, J.; Groot, Y.; Veerman, G.; Schuessler, M.; Niebch, G.; Siamopoulos, K.; et al. Phase I Trial of 6-Hour Infusion of Glufosfamide, a New Alkylating Agent with Potentially Enhanced Selectivity for Tumors That Overexpress Transmembrane Glucose Transporters: A Study of the European Organization for Research and Treatment of Cancer Early Clinical Studies Group. J. Clin. Oncol. 2000, 18, 3535–3544. [Google Scholar] [CrossRef]
- Calvaresi, E.C.; Hergenrother, P.J. Glucose Conjugation for the Specific Targeting and Treatment of Cancer. Chem. Sci. 2013, 4, 2319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahrjou, N.; Ghosh, A.; Tanasova, M. Targeting of GLUT5 for Transporter-Mediated Drug-Delivery Is Contingent upon Substrate Hydrophilicity. Int. J. Mol. Sci. 2021, 22, 5073. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Shustov, G.; Liang, H.; Khlebnikov, V.; Zheng, W.; Yang, X.-H.; Cheeseman, C.; Wiebe, L.I. Design, Synthesis, and Preliminary Biological Evaluation of 6- O -Glucose–Azomycin Adducts for Diagnosis and Therapy of Hypoxic Tumors. J. Med. Chem. 2012, 55, 6033–6046. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Cui, S.; Li, S.; Du, C.; Tian, J.; Wan, S.; Qian, Z.; Gu, Y.; Chen, W.R.; Wang, G. Targeted Cancer Therapy with a 2-Deoxyglucose–Based Adriamycin Complex. Cancer Res. 2013, 73, 1362–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.-Z.; Sinchaikul, S.; Reddy, P.V.G.; Chang, M.-Y.; Chen, S.-T. Synthesis of 2′-Paclitaxel Methyl 2-Glucopyranosyl Succinate for Specific Targeted Delivery to Cancer Cells. Bioorg. Med. Chem. Lett. 2007, 17, 617–620. [Google Scholar] [CrossRef]
- Lin, Y.-S.; Tungpradit, R.; Sinchaikul, S.; An, F.-M.; Liu, D.-Z.; Phutrakul, S.; Chen, S.-T. Targeting the Delivery of Glycan-Based Paclitaxel Prodrugs to Cancer Cells via Glucose Transporters. J. Med. Chem. 2008, 51, 7428–7441. [Google Scholar] [CrossRef]
- Ma, Y.; Chen, H.; Su, S.; Wang, T.; Zhang, C.; Fida, G.; Cui, S.; Zhao, J.; Gu, Y. Galactose as Broad Ligand for Multiple Tumor Imaging and Therapy. J. Cancer 2015, 6, 658–670. [Google Scholar] [CrossRef]
- Sharma, A.; Kim, E.-J.; Shi, H.; Lee, J.Y.; Chung, B.G.; Kim, J.S. Development of a Theranostic Prodrug for Colon Cancer Therapy by Combining Ligand-Targeted Delivery and Enzyme-Stimulated Activation. Biomaterials 2018, 155, 145–151. [Google Scholar] [CrossRef]
- Mielczarek-Puta, M.; Struga, M.; Roszkowski, P. Synthesis and Anticancer Effects of Conjugates of Doxorubicin and Unsaturated Fatty Acids (LNA and DHA). Med. Chem. Res. 2019, 28, 2153–2164. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Lian, X.; Li, X.; Ya, Q.; Li, T.; Zhang, Y.; Yang, Y.; Zhang, Y. Synthesis of 2′-Paclitaxel 2-Deoxy-2-Fluoro-Glucopyranosyl Carbonate for Specific Targeted Delivery to Cancer Cells. Carbohydr. Res. 2020, 493, 108034. [Google Scholar] [CrossRef]
- Mao, Y.; Zhang, Y.; Luo, Z.; Zhan, R.; Xu, H.; Chen, W.; Huang, H. Synthesis, Biological Evaluation and Low-Toxic Formulation Development of Glycosylated Paclitaxel Prodrugs. Molecules 2018, 23, 3211. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Gao, X.; Liu, R.; Yang, J.; Zhang, M.; Mi, Y.; Shi, Y.; Gao, Q. Design, Synthesis of Novel Platinum(II) Glycoconjugates, and Evaluation of Their Antitumor Effects. Chem. Biol. Drug Des. 2016, 87, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Patra, M.; Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. A Potent Glucose-Platinum Conjugate Exploits Glucose Transporters and Preferentially Accumulates in Cancer Cells. Angew. Chem. Int. Ed. 2016, 55, 2550–2554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Yang, X.; Zhao, C.; Wang, P.G.; Wang, X. Glucose-Conjugated Platinum(IV) Complexes as Tumor-Targeting Agents: Design, Synthesis and Biological Evaluation. Bioorg. Med. Chem. 2019, 27, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, S.P.; Patra, M. Platinum Glycoconjugates: “Sweet Bullets” for Targeted Cancer Therapy? Curr. Opin. Chem. Biol. 2023, 72, 102236. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Yang, J.; Seeberger, P.H.; Yin, J. Glycoconjugates for Glucose Transporter-Mediated Cancer-Specific Targeting and Treatment. Carbohydr. Res. 2020, 498, 108195. [Google Scholar] [CrossRef] [PubMed]
- Martin, H.; Lázaro, L.R.; Gunnlaugsson, T.; Scanlan, E.M. Glycosidase Activated Prodrugs for Targeted Cancer Therapy. Chem. Soc. Rev. 2022, 51, 9694–9716. [Google Scholar] [CrossRef]
- Koźmiński, P.; Halik, P.K.; Chesori, R.; Gniazdowska, E. Overview of Dual-Acting Drug Methotrexate in Different Neurological Diseases, Autoimmune Pathologies and Cancers. Int. J. Mol. Sci. 2020, 21, 3483. [Google Scholar] [CrossRef]
- Maksimovic, V.; Pavlovic-Popovic, Z.; Vukmirovic, S.; Cvejic, J.; Mooranian, A.; Al-Salami, H.; Mikov, M.; Golocorbin-Kon, S. Molecular Mechanism of Action and Pharmacokinetic Properties of Methotrexate. Mol. Biol. Rep. 2020, 47, 4699–4708. [Google Scholar] [CrossRef]
- Abolmaali, S.S.; Tamaddon, A.M.; Dinarvand, R. A Review of Therapeutic Challenges and Achievements of Methotrexate Delivery Systems for Treatment of Cancer and Rheumatoid Arthritis. Cancer Chemother. Pharmacol. 2013, 71, 1115–1130. [Google Scholar] [CrossRef]
- Abdulrahman, L.; Bakare, O.; Abdulrahman, M. The Chemical Approach of Methotrexate Targeting. Front. Biomed. Sci. 2016, 1, 50–73. [Google Scholar]
- Li, S.; Sun, Z.; Meng, X.; Deng, G.; Zhang, J.; Zhou, K.; Li, W.; Zhou, L.; Gong, P.; Cai, L. Targeted Methotrexate Prodrug Conjugated with Heptamethine Cyanine Dye Improving Chemotherapy and Monitoring Itself Activating by Dual-Modal Imaging. Front. Mater. 2018, 5, 35. [Google Scholar] [CrossRef]
- Pignatello, R.; Vicari, L.; Sorrenti, V.; Di Giacomo, C.; Spampinato, G.; Puglisi, G.; Toth, I. Lipophilic Methotrexate Conjugates with Glucose-Lipoamino Acid Moieties: Synthesis and In Vitro Antitumor Activity. Drug Dev. Res. 2001, 52, 454–461. [Google Scholar] [CrossRef]
- Barnett, J.E.G.; Holman, G.D.; Munday, K.A. Structural Requirements for Binding to the Sugar-Transport System of the Human Erythrocyte. Biochem. J. 1973, 131, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woźniak, M.; Pastuch-Gawołek, G.; Makuch, S.; Wiśniewski, J.; Ziółkowski, P.; Szeja, W.; Krawczyk, M.; Agrawal, S. Overcoming Hypoxia-Induced Chemoresistance in Cancer Using a Novel Glycoconjugate of Methotrexate. Pharmaceuticals 2020, 14, 13. [Google Scholar] [CrossRef]
- Woźniak, M.; Pastuch-Gawołek, G.; Makuch, S.; Wiśniewski, J.; Krenács, T.; Hamar, P.; Gamian, A.; Szeja, W.; Szkudlarek, D.; Krawczyk, M.; et al. In Vitro and In Vivo Efficacy of a Novel Glucose–Methotrexate Conjugate in Targeted Cancer Treatment. Int. J. Mol. Sci. 2021, 22, 1748. [Google Scholar] [CrossRef]
- Woźniak, M.; Makuch, S.; Pastuch-Gawołek, G.; Wiśniewski, J.; Szeja, W.; Nowak, M.; Krawczyk, M.; Agrawal, S. The Effect of a New Glucose–Methotrexate Conjugate on Acute Lymphoblastic Leukemia and Non-Hodgkin’s Lymphoma Cell Lines. Molecules 2021, 26, 2547. [Google Scholar] [CrossRef]
- Chiorean, E.G.; Dragovich, T.; Hamm, J.; Barrios, C.H.; Gorini, C.F.; Langmuir, V.K.; Kroll, S.; Jung, D.T.; Tidmarsh, G.T.; Loehrer, P.J. A Phase 2 Trial of Glufosfamide in Combination with Gemcitabine in Chemotherapy-Naive Pancreatic Adenocarcinoma. Am. J. Clin. Oncol. 2010, 33, 111–116. [Google Scholar] [CrossRef]
- Goff, R.D.; Thorson, J.S. Assessment of Chemoselective Neoglycosylation Methods Using Chlorambucil as a Model. J. Med. Chem. 2010, 53, 8129–8139. [Google Scholar] [CrossRef] [Green Version]
- Miot-Noirault, E.; Reux, B.; Debiton, E.; Madelmont, J.-C.; Chezal, J.-M.; Coudert, P.; Weber, V. Preclinical Investigation of Tolerance and Antitumour Activity of New Fluorodeoxyglucose-Coupled Chlorambucil Alkylating Agents. Investig. New Drugs 2011, 29, 424–433. [Google Scholar] [CrossRef]
- Wu, J.; Liu, Q.; Lee, R.J. A Folate Receptor-Targeted Liposomal Formulation for Paclitaxel. Int. J. Pharm. 2006, 316, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Cao, X.; Xian, M.; Fang, L.; Cai, T.B.; Ji, J.J.; Tunac, J.B.; Sun, D.; Wang, P.G. Synthesis and Enzyme-Specific Activation of Carbohydrate−Geldanamycin Conjugates with Potent Anticancer Activity. J. Med. Chem. 2005, 48, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Song, G.; Deng, J.; Jiang, L.; Xiong, P.; Yang, B.; Liu, S. Antitumor Effects and Mechanism of Novel Emodin Rhamnoside Derivatives against Human Cancer Cells In Vitro. PLoS ONE 2015, 10, e0144781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, D.J.; Cragg, G.M.; Snader, K.M. Natural Products as Sources of New Drugs over the Period 1981−2002. J. Nat. Prod. 2003, 66, 1022–1037. [Google Scholar] [CrossRef]
- Amiri, S.; Dastghaib, S.; Ahmadi, M.; Mehrbod, P.; Khadem, F.; Behrouj, H.; Aghanoori, M.-R.; Machaj, F.; Ghamsari, M.; Rosik, J.; et al. Betulin and Its Derivatives as Novel Compounds with Different Pharmacological Effects. Biotechnol. Adv. 2020, 38, 107409. [Google Scholar] [CrossRef]
- Sousa, J.L.C.; Freire, C.S.R.; Silvestre, A.J.D.; Silva, A.M.S. Recent Developments in the Functionalization of Betulinic Acid and Its Natural Analogues: A Route to New Bioactive Compounds. Molecules 2019, 24, 355. [Google Scholar] [CrossRef]
- Csuk, R. Betulinic Acid and Its Derivatives: A Patent Review (2008–2013). Expert Opin. Ther. Pat. 2014, 24, 913–923. [Google Scholar] [CrossRef]
- Bhat, S.S.; Prasad, S.K.; Shivamallu, C.; Prasad, K.S.; Syed, A.; Reddy, P.; Cull, C.A.; Amachawadi, R.G. Genistein: A Potent Anti-Breast Cancer Agent. Curr. Issues Mol. Biol. 2021, 43, 1502–1517. [Google Scholar] [CrossRef]
- Szeja, W.; Grynkiewicz, G.; Rusin, A. Isoflavones, Their Glycosides and Glycoconjugates. Synthesis and Biological Activity. Curr. Org. Chem. 2016, 21, 218–235. [Google Scholar] [CrossRef]
- Jain, S.; Chandra, V.; Jain, P.K.; Pathak, K.; Pathak, D.; Vaidya, A. Comprehensive Review on Current Developments of Quinoline-Based Anticancer Agents. Arab. J. Chem. 2019, 12, 4920–4946. [Google Scholar] [CrossRef] [Green Version]
- Matada, B.S.; Pattanashettar, R.; Yernale, N.G. A Comprehensive Review on the Biological Interest of Quinoline and Its Derivatives. Bioorg. Med. Chem. 2021, 32, 115973. [Google Scholar] [CrossRef]
- Afzal, O.; Kumar, S.; Haider, M.R.; Ali, M.R.; Kumar, R.; Jaggi, M.; Bawa, S. A Review on Anticancer Potential of Bioactive Heterocycle Quinoline. Eur. J. Med. Chem. 2015, 97, 871–910. [Google Scholar] [CrossRef] [PubMed]
- Solomon, V.R.; Lee, H. Quinoline as a Privileged Scaffold in Cancer Drug Discovery. Curr. Med. Chem. 2011, 18, 1488–1508. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Xu, H.; Chen, W.; Zhan, P.; Liu, X. 8-Hydroxyquinoline: A Privileged Structure with a Broad-Ranging Pharmacological Potential. MedChemComm 2015, 6, 61–74. [Google Scholar] [CrossRef]
- Gupta, R.; Luxami, V.; Paul, K. Insights of 8-Hydroxyquinolines: A Novel Target in Medicinal Chemistry. Bioorg. Chem. 2021, 108, 104633. [Google Scholar] [CrossRef]
- Oliveri, V.; Vecchio, G. 8-Hydroxyquinolines in Medicinal Chemistry: A Structural Perspective. Eur. J. Med. Chem. 2016, 120, 252–274. [Google Scholar] [CrossRef]
- Prachayasittikul, V.; Prachayasittikul, V.; Prachayasittikul, S.; Ruchirawat, S. 8-Hydroxyquinolines: A Review of Their Metal Chelating Properties and Medicinal Applications. Drug Des. Devel. Ther. 2013, 7, 1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savić-Gajić, I.M.; Savić, I.M. Drug Design Strategies with Metal-Hydroxyquinoline Complexes. Expert Opin. Drug Discov. 2020, 15, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Lauria, A.; La Monica, G.; Bono, A.; Martorana, A. Quinoline Anticancer Agents Active on DNA and DNA-Interacting Proteins: From Classical to Emerging Therapeutic Targets. Eur. J. Med. Chem. 2021, 220, 113555. [Google Scholar] [CrossRef]
- Gaur, K.; Vázquez-Salgado, A.; Duran-Camacho, G.; Dominguez-Martinez, I.; Benjamín-Rivera, J.; Fernández-Vega, L.; Sarabia, L.C.; García, A.C.; Pérez-Deliz, F.; Román, J.M.; et al. Iron and Copper Intracellular Chelation as an Anticancer Drug Strategy. Inorganics 2018, 6, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupte, A.; Mumper, R.J. Elevated Copper and Oxidative Stress in Cancer Cells as a Target for Cancer Treatment. Cancer Treat. Rev. 2009, 35, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Oliveri, V.; Giuffrida, M.L.; Vecchio, G.; Aiello, C.; Viale, M. Gluconjugates of 8-Hydroxyquinolines as Potential Anti-Cancer Prodrugs. Dalton Trans. 2012, 41, 4530. [Google Scholar] [CrossRef] [PubMed]
- Oliveri, V.; Viale, M.; Caron, G.; Aiello, C.; Gangemi, R.; Vecchio, G. Glycosylated Copper(II) Ionophores as Prodrugs for β-Glucosidase Activation in Targeted Cancer Therapy. Dalton Trans. 2013, 42, 2023–2034. [Google Scholar] [CrossRef] [PubMed]
- Oliveri, V.; Viale, M.; Aiello, C.; Vecchio, G. New 8-Hydroxyquinoline Galactosides. The Role of the Sugar in the Antiproliferative Activity of Copper(II) Ionophores. J. Inorg. Biochem. 2015, 142, 101–108. [Google Scholar] [CrossRef]
- Oliveri, V.; Grasso, G.I.; Bellia, F.; Attanasio, F.; Viale, M.; Vecchio, G. Soluble Sugar-Based Quinoline Derivatives as New Antioxidant Modulators of Metal-Induced Amyloid Aggregation. Inorg. Chem. 2015, 54, 2591–2602. [Google Scholar] [CrossRef]
- Li, W.; Zhang, Z.-W.; Wang, S.-X.; Ren, S.-M.; Jiang, T. Synthesis and Analysis of Potential DNA Intercalators Containing Quinoline-Glucose Hybrids. Chem. Biol. Drug Des. 2009, 74, 80–86. [Google Scholar] [CrossRef]
- Rashad, A.E.; El-Sayed, W.A.; Mohamed, A.M.; Ali, M.M. Synthesis of New Quinoline Derivatives as Inhibitors of Human Tumor Cells Growth. Arch. Pharm. 2010, 343, 440–448. [Google Scholar] [CrossRef]
- Freitas, L.B.D.O.; Borgati, T.F.; de Freitas, R.P.; Ruiz, A.L.T.G.; Marchetti, G.M.; de Carvalho, J.E.; da Cunha, E.F.F.; Ramalho, T.C.; Alves, R.B. Synthesis and Antiproliferative Activity of 8-Hydroxyquinoline Derivatives Containing a 1,2,3-Triazole Moiety. Eur. J. Med. Chem. 2014, 84, 595–604. [Google Scholar] [CrossRef]
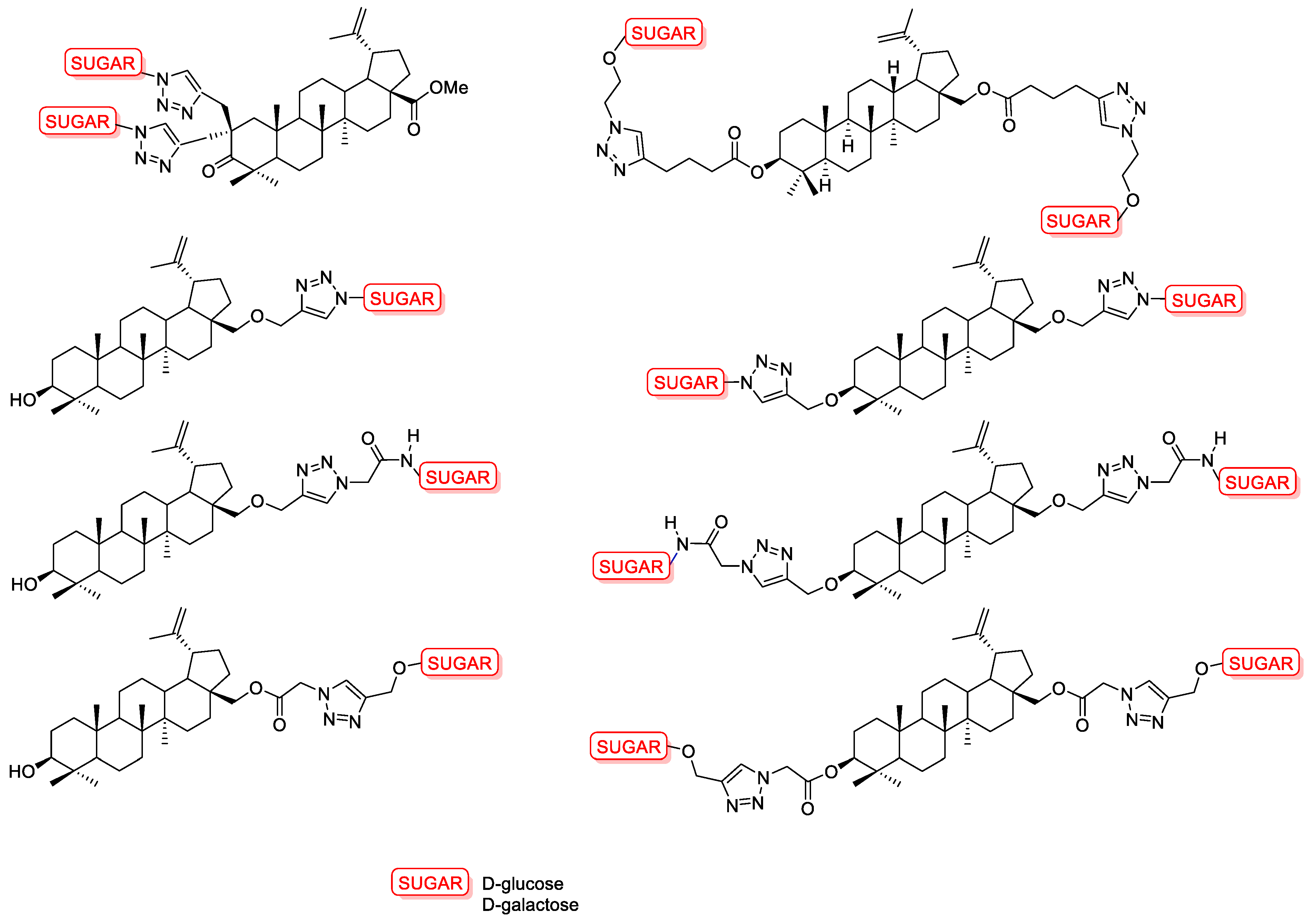
- Pastuch-Gawołek, G.; Malarz, K.; Mrozek-Wilczkiewicz, A.; Musioł, M.; Serda, M.; Czaplinska, B.; Musiol, R. Small Molecule Glycoconjugates with Anticancer Activity. Eur. J. Med. Chem. 2016, 112, 130–144. [Google Scholar] [CrossRef]
- Krawczyk, M.; Pastuch-Gawolek, G.; Mrozek-Wilczkiewicz, A.; Kuczak, M.; Skonieczna, M.; Musiol, R. Synthesis of 8-Hydroxyquinoline Glycoconjugates and Preliminary Assay of Their Β1,4-GalT Inhibitory and Anti-Cancer Properties. Bioorg. Chem. 2019, 84, 326–338. [Google Scholar] [CrossRef]
- Krawczyk, M.; Pastuch-Gawołek, G.; Pluta, A.; Erfurt, K.; Domiński, A.; Kurcok, P. 8-Hydroxyquinoline Glycoconjugates: Modifications in the Linker Structure and Their Effect on the Cytotoxicity of the Obtained Compounds. Molecules 2019, 24, 4181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krawczyk, M.; Pastuch-Gawołek, G.; Hadasik, A.; Erfurt, K. 8-Hydroxyquinoline Glycoconjugates Containing Sulfur at the Sugar Anomeric Position—Synthesis and Preliminary Evaluation of Their Cytotoxicity. Molecules 2020, 25, 4174. [Google Scholar] [CrossRef] [PubMed]
- Domińska, M.; Pastuch-Gawołek, G.; Skonieczna, M.; Szeja, W.; Domiński, A.; Kurcok, P. Glycoconjugation of Quinoline Derivatives Using the C-6 Position in Sugars as a Strategy for Improving the Selectivity and Cytotoxicity of Functionalized Compounds. Molecules 2022, 27, 6918. [Google Scholar] [CrossRef] [PubMed]
- Juang, Y.-P.; Liang, P.-H. Biological and Pharmacological Effects of Synthetic Saponins. Molecules 2020, 25, 4974. [Google Scholar] [CrossRef] [PubMed]
- Grymel, M.; Zawojak, M.; Adamek, J. Triphenylphosphonium Analogues of Betulin and Betulinic Acid with Biological Activity: A Comprehensive Review. J. Nat. Prod. 2019, 82, 1719–1730. [Google Scholar] [CrossRef] [PubMed]
- Jonnalagadda, S.C.; Suman, P.; Morgan, D.C.; Seay, J.N. Recent Developments on the Synthesis and Applications of Betulin and Betulinic Acid Derivatives as Therapeutic Agents. In Studies in Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 2017; Volume 53, pp. 45–84. ISBN 978-0-444-63930-1. [Google Scholar]
- Hordyjewska, A.; Ostapiuk, A.; Horecka, A. Betulin and Betulinic Acid in Cancer Research. J. Pre-Clin. Clin. Res. 2018, 12, 72–75. [Google Scholar] [CrossRef]
- Zhang, D.-M.; Xu, H.-G.; Wang, L.; Li, Y.-J.; Sun, P.-H.; Wu, X.-M.; Wang, G.-J.; Chen, W.-M.; Ye, W.-C. Betulinic Acid and Its Derivatives as Potential Antitumor Agents: Betulinic Acid and Its Derivatives with Antitumor Activities. Med. Res. Rev. 2015, 35, 1127–1155. [Google Scholar] [CrossRef] [PubMed]
- Thibeault, D.; Gauthier, C.; Legault, J.; Bouchard, J.; Dufour, P.; Pichette, A. Synthesis and Structure–Activity Relationship Study of Cytotoxic Germanicane- and Lupane-Type 3β-O-Monodesmosidic Saponins Starting from Betulin. Bioorg. Med. Chem. 2007, 15, 6144–6157. [Google Scholar] [CrossRef]
- Gauthier, C.; Legault, J.; Lebrun, M.; Dufour, P.; Pichette, A. Glycosidation of Lupane-Type Triterpenoids as Potent In Vitro Cytotoxic Agents. Bioorg. Med. Chem. 2006, 14, 6713–6725. [Google Scholar] [CrossRef]
- Gauthier, C.; Legault, J.; Rondeau, S.; Pichette, A. Synthesis of Betulinic Acid Acyl Glucuronide for Application in Anticancer Prodrug Monotherapy. Tetrahedron Lett. 2009, 50, 988–991. [Google Scholar] [CrossRef]
- Gauthier, C.; Legault, J.; Lavoie, S.; Rondeau, S.; Tremblay, S.; Pichette, A. Synthesis and Cytotoxicity of Bidesmosidic Betulin and Betulinic Acid Saponins. J. Nat. Prod. 2009, 72, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, C.; Legault, J.; Piochon-Gauthier, M.; Pichette, A. Advances in the Synthesis and Pharmacological Activity of Lupane-Type Triterpenoid Saponins. Phytochem. Rev. 2011, 10, 521–544. [Google Scholar] [CrossRef]
- Gauthier, C.; Legault, J.; Piochon, M.; Lavoie, S.; Tremblay, S.; Pichette, A. Synthesis, Cytotoxicity, and Haemolytic Activity of Chacotrioside Lupane-Type Neosaponins and Their Germanicane-Type Rearrangement Products. Bioorg. Med. Chem. Lett. 2009, 19, 2310–2314. [Google Scholar] [CrossRef] [PubMed]
- Cmoch, P.; Pakulski, Z.; Swaczynová, J.; Strnad, M. Synthesis of Lupane-Type Saponins Bearing Mannosyl and 3,6-Branched Trimannosyl Residues and Their Evaluation as Anticancer Agents. Carbohydr. Res. 2008, 343, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, C.; Legault, J.; Girard-Lalancette, K.; Mshvildadze, V.; Pichette, A. Haemolytic Activity, Cytotoxicity and Membrane Cell Permeabilization of Semi-Synthetic and Natural Lupane- and Oleanane-Type Saponins. Bioorg. Med. Chem. 2009, 17, 2002–2008. [Google Scholar] [CrossRef]
- Kommera, H.; Kaluđerović, G.N.; Bette, M.; Kalbitz, J.; Fuchs, P.; Fulda, S.; Mier, W.; Paschke, R. In Vitro Anticancer Studies of α- and β-d-Glucopyranose Betulin Anomers. Chem. Biol. Interact. 2010, 185, 128–136. [Google Scholar] [CrossRef]
- Gonzalez, P.; Mader, I.; Tchoghandjian, A.; Enzenmüller, S.; Cristofanon, S.; Basit, F.; Debatin, K.-M.; Fulda, S. Impairment of Lysosomal Integrity by B10, a Glycosylated Derivative of Betulinic Acid, Leads to Lysosomal Cell Death and Converts Autophagy into a Detrimental Process. Cell Death Differ. 2012, 19, 1337–1346. [Google Scholar] [CrossRef] [Green Version]
- Sylla, B.; Lavoie, S.; Legault, J.; Gauthier, C.; Pichette, A. Synthesis, Cytotoxicity and Anti-Inflammatory Activity of Rhamnose-Containing Ursolic and Betulinic Acid Saponins. RSC Adv. 2019, 9, 39743–39757. [Google Scholar] [CrossRef] [Green Version]
- Korda, A.; Rárová, L.; Pakulski, Z.; Strnad, M.; Oklešťková, J.; Kuczynska, K.; Cmoch, P.; Gwardiak, K.; Karczewski, R. New Lupane Bidesmosides Exhibiting Strong Cytotoxic Activities In Vitro. Bioorg. Chem. 2020, 100, 103868. [Google Scholar] [CrossRef]
- Cmoch, P.; Korda, A.; Rárová, L.; Oklešťková, J.; Strnad, M.; Luboradzki, R.; Pakulski, Z. Synthesis and Structure–Activity Relationship Study of Cytotoxic Lupane-Type 3β-O-Monodesmosidic Saponins with an Extended C-28 Side Chain. Tetrahedron 2014, 70, 2717–2730. [Google Scholar] [CrossRef]
- Sidoryk, K.; Korda, A.; Rárová, L.; Oklešťková, J.; Strnad, M.; Cmoch, P.; Pakulski, Z.; Gwardiak, K.; Karczewski, R.; Luboradzki, R. Synthesis and Biological Activity of New Homolupanes and Homolupane Saponins. Tetrahedron 2015, 71, 2004–2012. [Google Scholar] [CrossRef]
- Sidoryk, K.; Korda, A.; Rárová, L.; Oklešťková, J.; Pakulski, Z.; Strnad, M.; Cmoch, P.; Gwardiak, K.; Karczewski, R. Synthesis and Cytotoxicity of 28a-Homothiolupanes and 28a-Homothiolupane Saponins: 28a-Homothiolupanes and 28a-Homothiolupane Saponins. Eur. J. Org. Chem. 2016, 2016, 373–383. [Google Scholar] [CrossRef]
- Mihoub, M.; Pichette, A.; Sylla, B.; Gauthier, C.; Legault, J. Bidesmosidic Betulin Saponin Bearing L-Rhamnopyranoside Moieties Induces Apoptosis and Inhibition of Lung Cancer Cells Growth In Vitro and In Vivo. PLoS ONE 2018, 13, e0193386. [Google Scholar] [CrossRef] [Green Version]
- Spivak, A.Y.; Gubaidullin, R.R.; Galimshina, Z.R.; Nedopekina, D.A.; Odinokov, V.N. Effective Synthesis of Novel C(2)-Propargyl Derivatives of Betulinic and Ursolic Acids and Their Conjugation with β-d-Glucopyranoside Azides via Click Chemistry. Tetrahedron 2016, 72, 1249–1256. [Google Scholar] [CrossRef]
- Grymel, M.; Pastuch-Gawołek, G.; Lalik, A.; Zawojak, M.; Boczek, S.; Krawczyk, M.; Erfurt, K. Glycoconjugation of Betulin Derivatives Using Copper-Catalyzed 1,3-Dipolar Azido-Alkyne Cycloaddition Reaction and a Preliminary Assay of Cytotoxicity of the Obtained Compounds. Molecules 2020, 25, 6019. [Google Scholar] [CrossRef] [PubMed]
- Yamansarov, E.Y.; Lopatukhina, E.V.; Evteev, S.A.; Skvortsov, D.A.; Lopukhov, A.V.; Kovalev, S.V.; Vaneev, A.N.; Shkil’, D.O.; Akasov, R.A.; Lobov, A.N.; et al. Discovery of Bivalent GalNAc-Conjugated Betulin as a Potent ASGPR-Directed Agent against Hepatocellular Carcinoma. Bioconjug. Chem. 2021, 32, 763–781. [Google Scholar] [CrossRef]
- Moon, Y.J.; Wang, X.; Morris, M.E. Dietary Flavonoids: Effects on Xenobiotic and Carcinogen Metabolism. Toxicol. Vitr. 2006, 20, 187–210. [Google Scholar] [CrossRef] [PubMed]
- Mukund, V. Genistein: Its Role in Breast Cancer Growth and Metastasis. Curr. Drug Metab. 2020, 21, 6–10. [Google Scholar] [CrossRef]
- Sharifi-Rad, J.; Quispe, C.; Imran, M.; Rauf, A.; Nadeem, M.; Gondal, T.A.; Ahmad, B.; Atif, M.; Mubarak, M.S.; Sytar, O.; et al. Genistein: An Integrative Overview of Its Mode of Action, Pharmacological Properties, and Health Benefits. Oxid. Med. Cell. Longev. 2021, 2021, 3268136. [Google Scholar] [CrossRef]
- Kim, S.-H.; Kim, C.-W.; Jeon, S.-Y.; Go, R.-E.; Hwang, K.-A.; Choi, K.-C. Chemopreventive and Chemotherapeutic Effects of Genistein, a Soy Isoflavone, upon Cancer Development and Progression in Preclinical Animal Models. Lab. Anim. Res. 2014, 30, 143. [Google Scholar] [CrossRef] [Green Version]
- Spagnuolo, C.; Russo, G.L.; Orhan, I.E.; Habtemariam, S.; Daglia, M.; Sureda, A.; Nabavi, S.F.; Devi, K.P.; Loizzo, M.R.; Tundis, R.; et al. Genistein and Cancer: Current Status, Challenges, and Future Directions. Adv. Nutr. 2015, 6, 408–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuli, H.S.; Tuorkey, M.J.; Thakral, F.; Sak, K.; Kumar, M.; Sharma, A.K.; Sharma, U.; Jain, A.; Aggarwal, V.; Bishayee, A. Molecular Mechanisms of Action of Genistein in Cancer: Recent Advances. Front. Pharmacol. 2019, 10, 1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polkowski, K.; Popiołkiewicz, J.; Krzeczyński, P.; Ramza, J.; Pucko, W.; Zegrocka-Stendel, O.; Boryski, J.; Skierski, J.S.; Mazurek, A.P.; Grynkiewicz, G. Cytostatic and Cytotoxic Activity of Synthetic Genistein Glycosides against Human Cancer Cell Lines. Cancer Lett. 2004, 203, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Wang, S.; Li, X.; Zou, T.; Huang, X.; Zhang, W.; Chen, Y.; Yang, C.; Pan, Q.; Liu, H.-F. Prospects of and Limitations to the Clinical Applications of Genistein. Discov. Med. 2019, 27, 177–188. [Google Scholar] [PubMed]
- Masilamani, M.; Wei, J.; Sampson, H.A. Regulation of the Immune Response by Soybean Isoflavones. Immunol. Res. 2012, 54, 95–110. [Google Scholar] [CrossRef]
- Nemeth, K.; Plumb, G.W.; Berrin, J.-G.; Juge, N.; Jacob, R.; Naim, H.Y.; Williamson, G.; Swallow, D.M.; Kroon, P.A. Deglycosylation by Small Intestinal Epithelial Cell?-Glucosidases Is a Critical Step in the Absorption and Metabolism of Dietary Flavonoid Glycosides in Humans. Eur. J. Nutr. 2003, 42, 29–42. [Google Scholar] [CrossRef]
- Rusin, A.; Gogler, A.; Głowala-Kosińska, M.; Bochenek, D.; Gruca, A.; Grynkiewicz, G.; Zawisza, J.; Szeja, W.; Krawczyk, Z. Unsaturated Genistein Disaccharide Glycoside as a Novel Agent Affecting Microtubules. Bioorg. Med. Chem. Lett. 2009, 19, 4939–4943. [Google Scholar] [CrossRef]
- Ahmed, A.A.; Goldsmith, J.; Fokt, I.; Le, X.-F.; Krzysko, K.A.; Lesyng, B.; Bast, R.C.; Priebe, W. A Genistein Derivative, ITB-301, Induces Microtubule Depolymerization and Mitotic Arrest in Multidrug-Resistant Ovarian Cancer. Cancer Chemother. Pharmacol. 2011, 68, 1033–1044. [Google Scholar] [CrossRef] [Green Version]
- Gogler-Pigłowska, A.; Rusin, A.; Bochenek, D.; Krawczyk, Z. Aneugenic Effects of the Genistein Glycosidic Derivative Substituted at C7 with the Unsaturated Disaccharide. Cell Biol. Toxicol. 2012, 28, 331–342. [Google Scholar] [CrossRef]
- Rusin, A.; Zawisza-Puchałka, J.; Kujawa, K.; Gogler-Pigłowska, A.; Wietrzyk, J.; Świtalska, M.; Głowala-Kosińska, M.; Gruca, A.; Szeja, W.; Krawczyk, Z.; et al. Synthetic Conjugates of Genistein Affecting Proliferation and Mitosis of Cancer Cells. Bioorg. Med. Chem. 2011, 19, 295–305. [Google Scholar] [CrossRef]
- Gruca, A.; Krawczyk, Z.; Szeja, W.; Grynkiewicz, G.; Rusin, A. Synthetic Genistein Glycosides Inhibiting EGFR Phosphorylation Enhance the Effect of Radiation in HCT 116 Colon Cancer Cells. Molecules 2014, 19, 18558–18573. [Google Scholar] [CrossRef] [Green Version]
- Goj, K.; Rusin, A.; Szeja, W.; Kitel, R.; Komor, R.; Grynkiewicz, G. Synthesis of Genistein 2,3-anhydroglycoconjugates—Potential Antiproliferative Agents. Acta Pol. Pharm. Drug Res. 2012, 69, 1239–1247. [Google Scholar]
- Szeja, W.; Grynkiewicz, G.; Bieg, T.; Swierk, P.; Byczek, A.; Papaj, K.; Kitel, R.; Rusin, A. Synthesis and Cytotoxicity of 2,3-Enopyranosyl C-Linked Conjugates of Genistein. Molecules 2014, 19, 7072–7093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaj, K.; Kasprzycka, A.; Góra, A.; Grajoszek, A.; Rzepecka, G.; Stojko, J.; Barski, J.-J.; Szeja, W.; Rusin, A. Structure–Bioavailability Relationship Study of Genistein Derivatives with Antiproliferative Activity on Human Cancer Cell. J. Pharm. Biomed. Anal. 2020, 185, 113216. [Google Scholar] [CrossRef]
- Antosiak, A.; Milowska, K.; Maczynska, K.; Rozalska, S.; Gabryelak, T. Cytotoxic Activity of Genistein-8-C-Glucoside Form Lupinus Luteus, L. and Genistein against Human SK-OV-3 Ovarian Carcinoma Cell Line. Med. Chem. Res. 2017, 26, 64–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Y.; Zhang, Y.; Zou, J.; Huang, L.-P.; Chordia, M.D.; Yue, W.; Wu, J.-J.; Pan, D.-F. Synthesis and Biological Evaluation of Genistein-IR783 Conjugate: Cancer Cell Targeted Delivery in MCF-7 for Superior Anti-Cancer Therapy. Molecules 2019, 24, 4120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.; Sawa, T.; Maeda, H. Factors and Mechanism of “EPR” Effect and the Enhanced Antitumor Effects of Macromolecular Drugs Including SMANCS. In Polymer Drugs in the Clinical Stage; Advances in Experimental Medicine and Biology; Maeda, H., Kabanov, A., Kataoka, K., Okano, T., Eds.; Kluwer Academic Publishers: Boston, MA, USA, 2004; Volume 519, pp. 29–49. ISBN 978-0-306-47471-2. [Google Scholar]
- Stuurman, F.E.; Nuijen, B.; Beijnen, J.H.; Schellens, J.H.M. Oral Anticancer Drugs: Mechanisms of Low Bioavailability and Strategies for Improvement. Clin. Pharmacokinet. 2013, 52, 399–414. [Google Scholar] [CrossRef]
- Muller, C. Prodrug Approaches for Enhancing the Bioavailability of Drugs with Low Solubility. Chem. Biodivers. 2009, 6, 2071–2083. [Google Scholar] [CrossRef]
- Broxterman, H.J.; Gotink, K.J.; Verheul, H.M.W. Understanding the Causes of Multidrug Resistance in Cancer: A Comparison of Doxorubicin and Sunitinib. Drug Resist. Updates 2009, 12, 114–126. [Google Scholar] [CrossRef]
- Xia, W.; Low, P.S. Folate-Targeted Therapies for Cancer. J. Med. Chem. 2010, 53, 6811–6824. [Google Scholar] [CrossRef]
- Akhtar, M.J.; Ahamed, M.; Alhadlaq, H.A.; Alrokayan, S.A.; Kumar, S. Targeted Anticancer Therapy: Overexpressed Receptors and Nanotechnology. Clin. Chim. Acta 2014, 436, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Fernández, M.; Javaid, F.; Chudasama, V. Advances in Targeting the Folate Receptor in the Treatment/Imaging of Cancers. Chem. Sci. 2018, 9, 790–810. [Google Scholar] [CrossRef] [Green Version]
- Pranatharthiharan, S.; Patel, M.D.; Malshe, V.C.; Pujari, V.; Gorakshakar, A.; Madkaikar, M.; Ghosh, K.; Devarajan, P.V. Asialoglycoprotein Receptor Targeted Delivery of Doxorubicin Nanoparticles for Hepatocellular Carcinoma. Drug Deliv. 2017, 24, 20–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, N.; Iqbal, N. Human Epidermal Growth Factor Receptor 2 (HER2) in Cancers: Overexpression and Therapeutic Implications. Mol. Biol. Int. 2014, 2014, 852748. [Google Scholar] [CrossRef]
- Chen, C.; Zhao, S.; Karnad, A.; Freeman, J.W. The Biology and Role of CD44 in Cancer Progression: Therapeutic Implications. J. Hematol. Oncol. 2018, 11, 64. [Google Scholar] [CrossRef] [Green Version]
- Pliszka, M.; Szablewski, L. Glucose Transporters as a Target for Anticancer Therapy. Cancers 2021, 13, 4184. [Google Scholar] [CrossRef] [PubMed]
- Dumitriu, S. (Ed.) Polysaccharides: Structural Diversity and Functional Versatility, 2nd ed.; Marcel Dekker: New York, NY, USA, 2005; ISBN 978-0-8247-5480-8. [Google Scholar]
- Alvarez-Lorenzo, C.; Blanco-Fernandez, B.; Puga, A.M.; Concheiro, A. Crosslinked Ionic Polysaccharides for Stimuli-Sensitive Drug Delivery. Adv. Drug Deliv. Rev. 2013, 65, 1148–1171. [Google Scholar] [CrossRef]
- Pushpamalar, J.; Veeramachineni, A.K.; Owh, C.; Loh, X.J. Biodegradable Polysaccharides for Controlled Drug Delivery. ChemPlusChem 2016, 81, 504–514. [Google Scholar] [CrossRef]
- Yadav, N.; Francis, A.P.; Priya, V.V.; Patil, S.; Mustaq, S.; Khan, S.S.; Alzahrani, K.J.; Banjer, H.J.; Mohan, S.K.; Mony, U.; et al. Polysaccharide-Drug Conjugates: A Tool for Enhanced Cancer Therapy. Polymers 2022, 14, 950. [Google Scholar] [CrossRef]
- Rodrigues, S.; Cardoso, L.; da Costa, A.; Grenha, A. Biocompatibility and Stability of Polysaccharide Polyelectrolyte Complexes Aimed at Respiratory Delivery. Materials 2015, 8, 5647–5670. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Jiao, Y.; Wang, Y.; Zhou, C.; Zhang, Z. Polysaccharides-Based Nanoparticles as Drug Delivery Systems. Adv. Drug Deliv. Rev. 2008, 60, 1650–1662. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Chen, M.; Ding, Y.; Yang, P.; Wang, M.; Zhang, H.; He, Y.; Ma, H. Polysaccharides as Potential Anti-Tumor Biomacromolecules—A Review. Front. Nutr. 2022, 9, 838179. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Xiong, S.-B.; Yang, H.; Yin, X.-D.; Zhao, R.-B. Mitoxantrone-Loaded BSA Nanospheres and Chitosan Nanospheres for Local Injection against Breast Cancer and Its Lymph Node Metastases. Int. J. Pharm. 2006, 307, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Jeong, Y.; Choi, C.; Roh, S.; Kang, S.; Jang, M.; Nah, J. Retinol-Encapsulated Low Molecular Water-Soluble Chitosan Nanoparticles. Int. J. Pharm. 2006, 319, 130–138. [Google Scholar] [CrossRef]
- Wurm, F.; Rietzler, B.; Pham, T.; Bechtold, T. Multivalent Ions as Reactive Crosslinkers for Biopolymers—A Review. Molecules 2020, 25, 1840. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Li, H.; Zhao, X.; Ye, F.; Zhao, G. Hydrophobically Modified Polysaccharides and Their Self-Assembled Systems: A Review on Structures and Food Applications. Carbohydr. Polym. 2022, 284, 119182. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, T.; Ma, X.; Wang, Y.; Lu, Y.; Jia, D.; Huang, X.; Chen, J.; Xu, Z.; Wen, F. The Design and Synthesis of Dextran-Doxorubicin Prodrug-Based PH-Sensitive Drug Delivery System for Improving Chemotherapy Efficacy. Asian J. Pharm. Sci. 2020, 15, 605–616. [Google Scholar] [CrossRef]
- Yu, K.; Yang, X.; He, L.; Zheng, R.; Min, J.; Su, H.; Shan, S.; Jia, Q. Facile Preparation of PH/Reduction Dual-Stimuli Responsive Dextran Nanogel as Environment-Sensitive Carrier of Doxorubicin. Polymer 2020, 200, 122585. [Google Scholar] [CrossRef]
- Keshavarz, M.; Moloudi, K.; Paydar, R.; Abed, Z.; Beik, J.; Ghaznavi, H.; Shakeri-Zadeh, A. Alginate Hydrogel Co-Loaded with Cisplatin and Gold Nanoparticles for Computed Tomography Image-Guided Chemotherapy. J. Biomater. Appl. 2018, 33, 161–169. [Google Scholar] [CrossRef]
- Senbanjo, L.T.; Chellaiah, M.A. CD44: A Multifunctional Cell Surface Adhesion Receptor Is a Regulator of Progression and Metastasis of Cancer Cells. Front. Cell Dev. Biol. 2017, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Yu, T.; Li, Y.; Gu, X. Development of a Hyaluronic Acid-Based Nanocarrier Incorporating Doxorubicin and Cisplatin as a PH-Sensitive and CD44-Targeted Anti-Breast Cancer Drug Delivery System. Front. Pharmacol. 2020, 11, 532457. [Google Scholar] [CrossRef]
- Lee, E.; Lee, J.; Lee, I.-H.; Yu, M.; Kim, H.; Chae, S.Y.; Jon, S. Conjugated Chitosan as a Novel Platform for Oral Delivery of Paclitaxel. J. Med. Chem. 2008, 51, 6442–6449. [Google Scholar] [CrossRef]
- Janes, K.A.; Fresneau, M.P.; Marazuela, A.; Fabra, A.; Alonso, M.J. Chitosan Nanoparticles as Delivery Systems for Doxorubicin. J. Control. Release 2001, 73, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Pandit, A.H.; Nisar, S.; Imtiyaz, K.; Nadeem, M.; Mazumdar, N.; Rizvi, M.M.A.; Ahmad, S. Injectable, Self-Healing, and Biocompatible N,O-Carboxymethyl Chitosan/Multialdehyde Guar Gum Hydrogels for Sustained Anticancer Drug Delivery. Biomacromolecules 2021, 22, 3731–3745. [Google Scholar] [CrossRef]
- Sultan, M.H.; Moni, S.S.; Madkhali, O.A.; Bakkari, M.A.; Alshahrani, S.; Alqahtani, S.S.; Alhakamy, N.A.; Mohan, S.; Ghazwani, M.; Bukhary, H.A.; et al. Characterization of Cisplatin-Loaded Chitosan Nanoparticles and Rituximab-Linked Surfaces as Target-Specific Injectable Nano-Formulations for Combating Cancer. Sci. Rep. 2022, 12, 468. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Chen, Y.; Zhou, Y.; Guo, D.; Fan, Y.; Guo, F.; Zheng, Y.; Chen, W. Preparation of 5-Fluorouracil-Loaded Chitosan Nanoparticles and Study of the Sustained Release In Vitro and In Vivo. Asian J. Pharm. Sci. 2017, 12, 418–423. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Anjum, M.M.; Patel, K.K. Gemcitabine Cationic Polymeric Nanoparticles against Ovarian Cancer: Formulation, Characterization, and Targeted Drug Delivery. Drug Deliv. 2022, 29, 1060–1074. [Google Scholar] [CrossRef]
- Raza, K.; Thakur, G.S.; Misra, C.; Thotakura, N.; Al Saqr, A.; Almawash, S.; Preet, S. Chitosan-Based Nanoconjugate for Safe and Effective Delivery of Docetaxel to Cancer Cells: An Explorative Study. J. Drug Deliv. Sci. Technol. 2021, 64, 102653. [Google Scholar] [CrossRef]
- Fard, G.H.; Moinipoor, Z.; Anastasova-Ivanova, S.; Iqbal, H.M.N.; Dwek, M.V.; Getting, S.; Keshavarz, T. Development of Chitosan, Pullulan, and Alginate Based Drug-Loaded Nano-Emulsions as a Potential Malignant Melanoma Delivery Platform. Carbohydr. Polym. Technol. Appl. 2022, 4, 100250. [Google Scholar] [CrossRef]
- Rosch, J.G.; Winter, H.; DuRoss, A.N.; Sun, C.; Sahay, G. Inverse-Micelle Synthesis of Doxorubicin-Loaded Alginate/Chitosan Nanoparticles and In Vitro Assessment of Breast Cancer Cytotoxicity. Colloid Interface Sci. Commun. 2019, 28, 69–74. [Google Scholar] [CrossRef]
- Markeb, A.A.; El-Maali, N.A.; Sayed, D.M.; Osama, A.; Abdel-Malek, M.A.Y.; Zaki, A.H.; Elwanis, M.E.A.; Driscoll, J.J. Synthesis, Structural Characterization, and Preclinical Efficacy of a Novel Paclitaxel-Loaded Alginate Nanoparticle for Breast Cancer Treatment. Int. J. Breast Cancer 2016, 2016, 7549372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Zhang, N.; Wu, J.; Dong, P.; Lv, H.; Wang, Q.; Wang, S.; Yang, H.; Wang, S.; Li, X.; et al. A Novel Dextran-Based Dual Drug Conjugate Targeted Tumors with High Biodistribution Ratio of Tumors to Normal Tissues. Int. J. Nanomed. 2022, 17, 4895–4910. [Google Scholar] [CrossRef]
- Münster, L.; Fojtů, M.; Muchová, M.; Latečka, F.; Káčerová, S.; Capáková, Z.; Juriňáková, T.; Kuřitka, I.; Masařík, M.; Vícha, J. Enhancing Cisplatin Anticancer Effectivity and Migrastatic Potential by Modulation of Molecular Weight of Oxidized Dextran Carrier. Carbohydr. Polym. 2021, 272, 118461. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Wang, M.; Zhang, Y.; Fei, Z.; Xie, D.; Zhang, H.; Du, Q. Hyaluronan Oligosaccharides-Coated Paclitaxel-Casein Nanoparticles with Enhanced Stability and Antitumor Activity. Nutrients 2022, 14, 3888. [Google Scholar] [CrossRef]
- Mitragotri, S.; Vogus, D.R.; Evans, M.A.; Pusuluri, A.; Barajas, A.; Zhang, M.; Krishnan, V.; Nowak, M.; Menegatti, S.; Helgeson, M.E.; et al. A Hyaluronic Acid Conjugate Engineered to Synergistically and Sequentially Deliver Gemcitabine and Doxorubicin to Treat Triple Negative Breast Cancer. J. Control. Release 2017, 267, 191–202. [Google Scholar] [CrossRef]
- Kim, Y.-C.; Yoo, J.; Rejinold, N.S.; Lee, D.; Noh, I.; Koh, W.-G.; Jon, S. CD44-Mediated Methotrexate Delivery by Hyaluronan-Coated Nanoparticles Composed of a Branched Cell-Penetrating Peptide. ACS Biomater. Sci. Eng. 2020, 6, 494–504. [Google Scholar] [CrossRef]
- Karimian, A.; Yousefi, B.; Sadeghi, F.; Feizi, F.; Najafzadehvarzi, H.; Parsian, H. Synthesis of Biocompatible Nanocrystalline Cellulose against Folate Receptors as a Novel Carrier for Targeted Delivery of Doxorubicin. Chem. Biol. Interact. 2022, 351, 109731. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Chen, Q.; Ye, L.; Zhang, H.; Zhang, A.; Feng, Z. The Preparation of PH and GSH Dual Responsive Thiolated Heparin/DOX Complex and Its Application as Drug Carrier. Carbohydr. Polym. 2020, 230, 115592. [Google Scholar] [CrossRef]
- Carpino, L.A.; Ringsdorf, H.; Ritter, H. Pharmacologically Active Polymers, 10. Preparation and Polymerization of 1-O-(4-Methacryloylaminophenyl)-β-D-Glucopyranoside. Makromol. Chem. 1976, 177, 1631–1635. [Google Scholar] [CrossRef]
- Ting, S.R.S.; Chen, G.; Stenzel, M.H. Synthesis of Glycopolymers and Their Multivalent Recognitions with Lectins. Polym. Chem. 2010, 1, 1392. [Google Scholar] [CrossRef]
- Silsirivanit, A. Glycosylation Markers in Cancer. In Advances in Clinical Chemistry; Elsevier: Amsterdam, The Netherlands, 2019; Volume 89, pp. 189–213. ISBN 978-0-12-817145-5. [Google Scholar]
- Nangia-Makker, P.; Conklin, J.; Hogan, V.; Raz, A. Carbohydrate-Binding Proteins in Cancer, and Their Ligands as Therapeutic Agents. Trends Mol. Med. 2002, 8, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Holgersson, J.; Gustafsson, A.; Breimer, M.E. Characteristics of Protein–Carbohydrate Interactions as a Basis for Developing Novel Carbohydrate-based Antirejection Therapies. Immunol. Cell Biol. 2005, 83, 694–708. [Google Scholar] [CrossRef] [PubMed]
- Becer, C.R. The Glycopolymer Code: Synthesis of Glycopolymers and Multivalent Carbohydrate-Lectin Interactions. Macromol. Rapid Commun. 2012, 33, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Jafari, F.; Yilmaz, G.; Becer, C.R. Stimuli-Responsive Glycopolymers and Their Biological Applications. Eur. Polym. J. 2021, 142, 110147. [Google Scholar] [CrossRef]
- Deirram, N.; Zhang, C.; Kermaniyan, S.S.; Johnston, A.P.R.; Such, G.K. PH-Responsive Polymer Nanoparticles for Drug Delivery. Macromol. Rapid Commun. 2019, 40, 1800917. [Google Scholar] [CrossRef] [Green Version]
- Kanamala, M.; Wilson, W.R.; Yang, M.; Palmer, B.D.; Wu, Z. Mechanisms and Biomaterials in PH-Responsive Tumour Targeted Drug Delivery: A Review. Biomaterials 2016, 85, 152–167. [Google Scholar] [CrossRef]
- Cheng, R.; Meng, F.; Deng, C.; Zhong, Z. Bioresponsive Polymeric Nanotherapeutics for Targeted Cancer Chemotherapy. Nano Today 2015, 10, 656–670. [Google Scholar] [CrossRef]
- Wang, Z.; Deng, X.; Ding, J.; Zhou, W.; Zheng, X.; Tang, G. Mechanisms of Drug Release in PH-Sensitive Micelles for Tumour Targeted Drug Delivery System: A Review. Int. J. Pharm. 2018, 535, 253–260. [Google Scholar] [CrossRef]
- Pang, X.; Jiang, Y.; Xiao, Q.; Leung, A.W.; Hua, H.; Xu, C. PH-Responsive Polymer–Drug Conjugates: Design and Progress. J. Control. Release 2016, 222, 116–129. [Google Scholar] [CrossRef]
- He, X.; Li, J.; An, S.; Jiang, C. PH-Sensitive Drug-Delivery Systems for Tumor Targeting. Ther. Deliv. 2013, 4, 1499–1510. [Google Scholar] [CrossRef]
- Sonawane, S.J.; Kalhapure, R.S.; Govender, T. Hydrazone Linkages in PH Responsive Drug Delivery Systems. Eur. J. Pharm. Sci. 2017, 99, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Zhong, Y.; Cheng, R.; Deng, C.; Zhong, Z. PH-Sensitive Polymeric Nanoparticles for Tumor-Targeting Doxorubicin Delivery: Concept and Recent Advances. Nanomedicine 2014, 9, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Chan, J.M.; Farokhzad, O.C. PH-Responsive Nanoparticles for Drug Delivery. Mol. Pharm. 2010, 7, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Zashikhina, N.; Levit, M.; Dobrodumov, A.; Gladnev, S.; Lavrentieva, A.; Tennikova, T.; Korzhikova-Vlakh, E. Biocompatible Nanoparticles Based on Amphiphilic Random Polypeptides and Glycopolymers as Drug Delivery Systems. Polymers 2022, 14, 1677. [Google Scholar] [CrossRef]
- Bhattacharya, K.; Das, S.; Kundu, M.; Singh, S.; Kalita, U.; Mandal, M.; Singha, N.K. Gold Nanoparticle Embedded Stimuli-Responsive Functional Glycopolymer: A Potential Material for Synergistic Chemo-Photodynamic Therapy of Cancer Cells. Macromol. Biosci. 2022, 22, 2200069. [Google Scholar] [CrossRef]
- Zhang, X.; Yuan, T.; Dong, H.; Xu, J.; Wang, D.; Tong, H.; Ji, X.; Sun, B.; Zhu, M.; Jiang, X. Novel Block Glycopolymers Prepared as Delivery Nanocarriers for Controlled Release of Bortezomib. Colloid Polym. Sci. 2018, 296, 1827–1839. [Google Scholar] [CrossRef] [Green Version]
- Abdalla, I.; Xu, J.; Wang, D.; Tong, H.; Sun, B.; Ding, B.; Jiang, X.; Zhu, M. Investigation of PH-Responsive Block Glycopolymers with Different Structures for the Delivery of Doxorubicin. RSC Adv. 2019, 9, 1814–1821. [Google Scholar] [CrossRef] [Green Version]
- Kahveci, E.L.S.; Kahveci, M.U.; Celebi, A.; Avsar, T.; Derman, S. Glycopolymer and Poly(β-Amino Ester)-Based Amphiphilic Block Copolymer as a Drug Carrier. Biomacromolecules 2022, 23, 4896–4908. [Google Scholar] [CrossRef]
- Dag, A.; Cakilkaya, E.; Ozgen, P.S.O.; Atasoy, S.; Erdem, G.Y.; Cetin, B.; Kokuroǧlu, A.Ç.; Gürek, A.G. Phthalocyanine-Conjugated Glyconanoparticles for Chemo-Photodynamic Combination Therapy. Biomacromolecules 2021, 22, 1555–1567. [Google Scholar] [CrossRef]
- Xiao, N.; Liang, H.; Lu, J. Degradable and Biocompatible Aldehyde-Functionalized Glycopolymer Conjugated with Doxorubicin via Acid-Labile Schiff Base Linkage for PH-Triggered Drug Release. Soft Matter 2011, 7, 10834. [Google Scholar] [CrossRef]
- Yilmaz, G.; Guler, E.; Geyik, C.; Demir, B.; Ozkan, M.; Demirkol, D.O.; Ozcelik, S.; Timur, S.; Becer, C.R. PH Responsive Glycopolymer Nanoparticles for Targeted Delivery of Anti-Cancer Drugs. Mol. Syst. Des. Eng. 2018, 3, 150–158. [Google Scholar] [CrossRef]
- Chen, W.; Meng, F.; Cheng, R.; Deng, C.; Feijen, J.; Zhong, Z. Biodegradable Glycopolymer-b-Poly(ε-Caprolactone) Block Copolymer Micelles: Versatile Construction, Tailored Lactose Functionality, and Hepatoma-Targeted Drug Delivery. J. Mater. Chem. B 2015, 3, 2308–2317. [Google Scholar] [CrossRef] [PubMed]
- Pearson, S.; Vitucci, D.; Khine, Y.Y.; Dag, A.; Lu, H.; Save, M.; Billon, L.; Stenzel, M.H. Light-Responsive Azobenzene-Based Glycopolymer Micelles for Targeted Drug Delivery to Melanoma Cells. Eur. Polym. J. 2015, 69, 616–627. [Google Scholar] [CrossRef]
- Bhattacharya, K.; Banerjee, S.L.; Das, S.; Samanta, S.; Mandal, M.; Singha, N.K. REDOX Responsive Fluorescence Active Glycopolymer Based Nanogel: A Potential Material for Targeted Anticancer Drug Delivery. ACS Appl. Bio Mater. 2019, 2, 2587–2599. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Huang, T.; Wang, X.; Wang, G.; Liu, Z.; Chen, G.; Fan, Q. Dynamic-Covalent Hydrogel with NIR-Triggered Drug Delivery for Localized Chemo-Photothermal Combination Therapy. Biomacromolecules 2020, 21, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Lou, S.; Zhang, X.; Zhang, M.; Ji, S.; Wang, W.; Zhang, J.; Li, C.; Kong, D. Preparation of a Dual Cored Hepatoma-Specific Star Glycopolymer Nanogel via Arm-First ATRP Approach. Int. J. Nanomed. 2017, 12, 3653–3664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, K.; Bligh, S.W.A.; Nie, H.; Quan, J.; Zhu, L. Lectin Recognizing Thermoresponsive Double Hydrophilic Glycopolymer Micelles by RAFT Polymerization. RSC Adv. 2014, 4, 34912–34921. [Google Scholar] [CrossRef]
- Quan, J.; Shen, F.-W.; Cai, H.; Zhang, Y.-N.; Wu, H. Galactose-Functionalized Double-Hydrophilic Block Glycopolymers and Their Thermoresponsive Self-Assembly Dynamics. Langmuir 2018, 34, 10721–10731. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, Y.; Wu, H.; Shao, D.; Shen, F.; Cai, H.; Quan, J. Galactose-Functionalized GlycoAuNR as a Photothermal Conversion Complex: Its Binding to Lectin RCA 120 and Hepatoma-Targeting Therapy. J. Biomater. Appl. 2020, 34, 1300–1314. [Google Scholar] [CrossRef]
- Zhao, J.; Lu, M.; Lai, H.; Lu, H.; Lalevée, J.; Barner-Kowollik, C.; Stenzel, M.H.; Xiao, P. Delivery of Amonafide from Fructose-Coated Nanodiamonds by Oxime Ligation for the Treatment of Human Breast Cancer. Biomacromolecules 2018, 19, 481–489. [Google Scholar] [CrossRef]
- Suzuki, K.; Miura, Y.; Mochida, Y.; Miyazaki, T.; Toh, K.; Anraku, Y.; Melo, V.; Liu, X.; Ishii, T.; Nagano, O.; et al. Glucose Transporter 1-Mediated Vascular Translocation of Nanomedicines Enhances Accumulation and Efficacy in Solid Tumors. J. Control. Release 2019, 301, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Zhao, J.; Chen, F.; Lu, M.; Khine, Y.Y.; Macmillan, A.; Garvey, C.J.; Stenzel, M.H. Drug-Induced Morphology Transition of Self-Assembled Glycopolymers: Insight into the Drug–Polymer Interaction. Chem. Mater. 2018, 30, 5227–5236. [Google Scholar] [CrossRef]
- Cao, C.; Zhao, J.; Lu, M.; Garvey, C.J.; Stenzel, M.H. Correlation between Drug Loading Content and Biological Activity: The Complexity Demonstrated in Paclitaxel-Loaded Glycopolymer Micelle System. Biomacromolecules 2019, 20, 1545–1554. [Google Scholar] [CrossRef]
- Ozgen, P.S.O.; Atasoy, S.; Kurt, B.Z.; Durmus, Z.; Yigit, G.; Dag, A. Glycopolymer Decorated Multiwalled Carbon Nanotubes for Dual Targeted Breast Cancer Therapy. J. Mater. Chem. B 2020, 8, 3123–3137. [Google Scholar] [CrossRef] [PubMed]
- Katmerlikaya, T.G.; Dag, A.; Ozgen, P.S.O.; Ersen, B.C. Dual-Drug Conjugated Glyco-Nanoassemblies for Tumor-Triggered Targeting and Synergistic Cancer Therapy. ACS Appl. Bio Mater. 2022, 5, 5356–5364. [Google Scholar] [CrossRef]
- Zhao, J.; Lu, H.; Yao, Y.; Ganda, S.; Stenzel, M.H. Length vs. Stiffness: Which Plays a Dominant Role in the Cellular Uptake of Fructose-Based Rod-like Micelles by Breast Cancer Cells in 2D and 3D Cell Culture Models? J. Mater. Chem. B 2018, 6, 4223–4231. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, X.; Yu, P.; Li, C. Glycopolymer Micelles with Reducible Ionic Cores for Hepatocytes-Targeting Delivery of DOX. Int. J. Pharm. 2013, 441, 170–180. [Google Scholar] [CrossRef]
- Wang, Z.; Luo, T.; Cao, A.; Sun, J.; Jia, L.; Sheng, R. Morphology-Variable Aggregates Prepared from Cholesterol-Containing Amphiphilic Glycopolymers: Their Protein Recognition/Adsorption and Drug Delivery Applications. Nanomaterials 2018, 8, 136. [Google Scholar] [CrossRef] [Green Version]
- Suriano, F.; Pratt, R.; Tan, J.P.K.; Wiradharma, N.; Nelson, A.; Yang, Y.-Y.; Dubois, P.; Hedrick, J.L. Synthesis of a Family of Amphiphilic Glycopolymers via Controlled Ring-Opening Polymerization of Functionalized Cyclic Carbonates and Their Application in Drug Delivery. Biomaterials 2010, 31, 2637–2645. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, J.; Han, J.; Tian, B.; Shi, Y.; Ding, Y.; Wang, L.; Han, J. Galactose-Containing Polymer-DOX Conjugates for Targeting Drug Delivery. AAPS PharmSciTech 2017, 18, 749–758. [Google Scholar] [CrossRef]
- Pandey, B.; Patil, N.G.; Bhosle, G.S.; Ambade, A.V.; Gupta, S.S. Amphiphilic Glycopolypeptide Star Copolymer-Based Cross-Linked Nanocarriers for Targeted and Dual-Stimuli-Responsive Drug Delivery. Bioconjug. Chem. 2019, 30, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Levit, M.; Zashikhina, N.; Vdovchenko, A.; Dobrodumov, A.; Zakharova, N.; Kashina, A.; Rühl, E.; Lavrentieva, A.; Scheper, T.; Tennikova, T.; et al. Bio-Inspired Amphiphilic Block-Copolymers Based on Synthetic Glycopolymer and Poly(Amino Acid) as Potential Drug Delivery Systems. Polymers 2020, 12, 183. [Google Scholar] [CrossRef] [Green Version]
- Qu, J.; Li, G.; Li, Y.; Sun, Y.; Ma, X.; Che, H.; Li, Y.; Sui, X.; Wang, X. Synthesis of Inherently Fluorescent Core–Shell Nanoparticles for Cell Imaging and Targeting Therapy: An In Vitro Evaluation. Macromol. Mater. Eng. 2022, 307, 2100961. [Google Scholar] [CrossRef]
- Wang, Y.; Hong, C.-Y.; Pan, C.-Y. Galactose-Based Amphiphilic Block Copolymers: Synthesis, Micellization, and Bioapplication. Biomacromolecules 2013, 14, 1444–1451. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, Y.; Cai, C.; Rong, X.; Shao, M.; Li, J.; Yang, C.; Yu, G. Collaborative Assembly of Doxorubicin and Galactosyl Diblock Glycopolymers for Targeted Drug Delivery of Hepatocellular Carcinoma. Biomater. Sci. 2020, 8, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Sheng, R.; Luo, T.; Wang, Z.; Li, H.; Cao, A. Synthesis of Diblock/Statistical Cationic Glycopolymers with Pendant Galactose and Lysine Moieties: Gene Delivery Application and Intracellular Behaviors. J. Mater. Chem. B 2016, 4, 4696–4706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Cai, X.; Chen, J.; Hu, X.; Xu, L. Conjugation of Lectin to Poly(ε-Caprolactone)-Block-Glycopolymer Micelles for In Vitro Intravesical Drug Delivery. Polymers 2016, 8, 379. [Google Scholar] [CrossRef] [Green Version]
- Pati, D.; Das, S.; Patil, N.G.; Parekh, N.; Anjum, D.H.; Dhaware, V.; Ambade, A.V.; Gupta, S.S. Tunable Nanocarrier Morphologies from Glycopolypeptide-Based Amphiphilic Biocompatible Star Copolymers and Their Carbohydrate Specific Intracellular Delivery. Biomacromolecules 2016, 17, 466–475. [Google Scholar] [CrossRef] [Green Version]
- Duman, F.D.; Monaco, A.; Foulkes, R.; Becer, C.R.; Forgan, R.S. Glycopolymer-Functionalized MOF-808 Nanoparticles as a Cancer-Targeted Dual Drug Delivery System for Carboplatin and Floxuridine. ACS Appl. Nano Mater. 2022, 5, 13862–13873. [Google Scholar] [CrossRef]
- An, J.; Zhang, X.; Guo, Q.; Zhao, Y.; Wu, Z.; Li, C. Glycopolymer Modified Magnetic Mesoporous Silica Nanoparticles for MR Imaging and Targeted Drug Delivery. Colloids Surf. Physicochem. Eng. Asp. 2015, 482, 98–108. [Google Scholar] [CrossRef]
- Dag, A.; Callari, M.; Lu, H.; Stenzel, M.H. Modulating the Cellular Uptake of Platinum Drugs with Glycopolymers. Polym. Chem. 2016, 7, 1031–1036. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, Y.; Zhang, T.; Cai, H.; Xie, X.; Yang, Y.; Quan, J.; Wu, H. AuNSs@Glycopolymer-ConA Hybrid Nanoplatform for Photothermal Therapy of Hepatoma Cells. Chem. Eng. J. 2020, 389, 124460. [Google Scholar] [CrossRef]
- Lin, H.; Zhou, K.; Li, D.; Hong, H.; Xie, Y.; Gong, L.; Shen, Y.; Zhou, Z.; Shi, J.; Wu, Z. Dinitrophenol-Hyaluronan Conjugates as Multivalent Antibody-Recruiting Glycopolymers for Targeted Cancer Immunotherapy. ChemMedChem 2021, 16, 2960–2968. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Yan, Y.; Zhou, J.; Yang, Y.; Zhang, Y.; Chen, J. Multifunctionalized Brush-Like Glycopolymers with High Affinity to P-Selectin and Antitumor Metastasis Activity. Biomacromolecules 2021, 22, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Yongzhi, H.; Chen, S.; Luo, X.; Lin, Y.; Zhou, Y.; Jin, H.; Hou, B.; Deng, Y.; Tu, L.; et al. The Prognostic Value of GLUT1 in Cancers: A Systematic Review and Meta-Analysis. Oncotarget 2017, 8, 43356–43367. [Google Scholar] [CrossRef] [Green Version]
- Leeson, P.D.; Springthorpe, B. The Influence of Drug-like Concepts on Decision-Making in Medicinal Chemistry. Nat. Rev. Drug Discov. 2007, 6, 881–890. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Yusof, I.; Segall, M.D. Considering the Impact Drug-like Properties Have on the Chance of Success. Drug Discov. Today 2013, 18, 659–666. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in Cancer Therapy: Molecular Mechanisms of Action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Li, S.; Zu, Y.; Yang, G.; Yang, Z.; Luo, M.; Jiang, S.; Wink, M.; Efferth, T. Medicinal Chemistry of Paclitaxel and Its Analogues. Curr. Med. Chem. 2009, 16, 3966–3985. [Google Scholar] [CrossRef]
- Fang, W.-S.; Liang, X.-T. Recent Progress in Structure Activity Relationship and Mechanistic Studies of Taxol Analogues. Mini-Rev. Med. Chem. 2005, 5, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, T.; Yang, C.; Norris, A.; Glass, T.; Bane, S.; Ravindra, R.; Banerjee, A.; Metaferia, B.; Thomas, S.L.; Giannakakou, P.; et al. Evaluation of the Tubulin-Bound Paclitaxel Conformation: Synthesis, Biology, and SAR Studies of C-4 to C-3’ Bridged Paclitaxel Analogues. J. Med. Chem. 2007, 50, 713–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin, M.M.; Abuo-Rahma, G.E.-D.A.; Shaykoon, M.S.A.; Marzouk, A.A.; Abourehab, M.A.S.; Saraya, R.E.; Badr, M.; Sayed, A.M.; Beshr, E.A.M. Design, Synthesis, Cytotoxic Activities, and Molecular Docking of Chalcone Hybrids Bearing 8-Hydroxyquinoline Moiety with Dual Tubulin/EGFR Kinase Inhibition. Bioorg. Chem. 2023, 134, 106444. [Google Scholar] [CrossRef] [PubMed]
- Vale, N.; Pereira, M.; Santos, J.; Moura, C.; Marques, L.; Duarte, D. Prediction of Drug Synergism between Peptides and Antineoplastic Drugs Paclitaxel, 5-Fluorouracil, and Doxorubicin Using In Silico Approaches. Int. J. Mol. Sci. 2022, 24, 69. [Google Scholar] [CrossRef] [PubMed]

















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pastuch-Gawołek, G.; Szreder, J.; Domińska, M.; Pielok, M.; Cichy, P.; Grymel, M. A Small Sugar Molecule with Huge Potential in Targeted Cancer Therapy. Pharmaceutics 2023, 15, 913. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics15030913
Pastuch-Gawołek G, Szreder J, Domińska M, Pielok M, Cichy P, Grymel M. A Small Sugar Molecule with Huge Potential in Targeted Cancer Therapy. Pharmaceutics. 2023; 15(3):913. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics15030913
Chicago/Turabian StylePastuch-Gawołek, Gabriela, Julia Szreder, Monika Domińska, Mateusz Pielok, Piotr Cichy, and Mirosława Grymel. 2023. "A Small Sugar Molecule with Huge Potential in Targeted Cancer Therapy" Pharmaceutics 15, no. 3: 913. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics15030913