Plasma Metabolites Related to Peripheral and Hepatic Insulin Sensitivity Are Not Directly Linked to Gut Microbiota Composition

 , , ,
, , ,
Abstract
:
1. Introduction
2. Materials and Methods
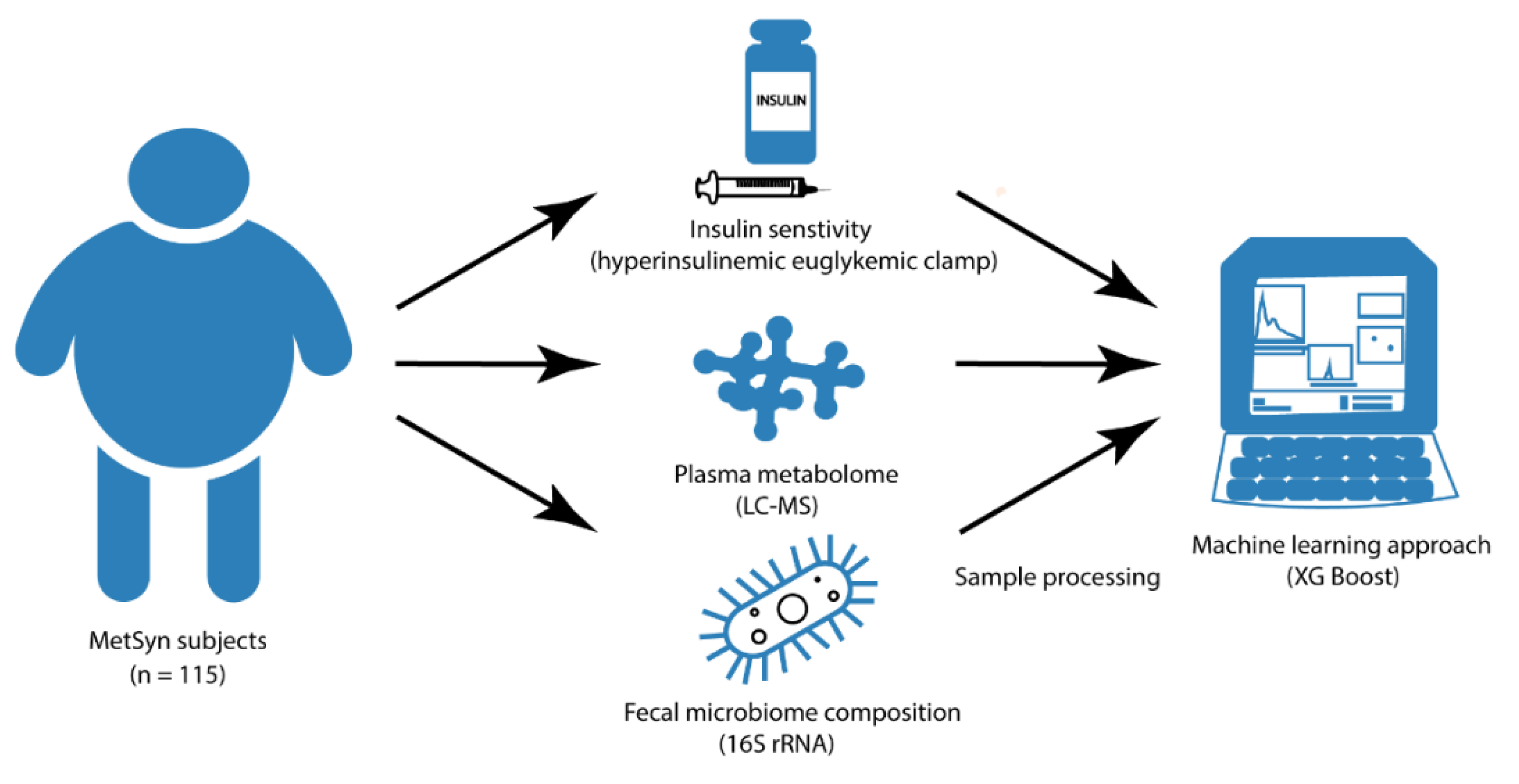
2.1. Study Design and Participants
- fasting plasma glucose ≥ 5.6 mmol/L
- triglycerides ≥ 1.7 mmol/L
- waist-circumference ≥ 102 cm for men and ≥ 88 cm for women
- high-density lipoprotein (HDL-) cholesterol ≤ 1.04 mmol/L
- blood pressure ≥ 130/85 mmHg.
2.2. Outcomes
2.2.1. Metabolic Profiling
2.2.2. Hyperinsulinemic Euglycemic Clamp
2.2.3. Fecal Microbiota and Dietary Intake
2.2.4. Machine Learning Models and Statistical Analysis
3. Results
3.1. Participants Characteristics
3.2. Plasma Metabolites and Insulin Sensitivity
3.3. Gut Microbiota and Insulin Sensitivity
3.4. Interrelation between Plasma Metabolites, Gut Microbiota, and Insulin Sensitivity
3.5. Plasma Metabolites and Gut Microbiota Alpha-Diversity
4. Discussion
4.1. Plasma Metabolites and Insulin Sensitivity
4.2. Gut Microbiota Composition and Insulin Sensitivity
4.3. Interrelation of Plasma Metabolites with Gut Microbiota and Insulin Sensitivity
4.4. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- NCD Risk Factor Collaboration. Worldwide trends in diabetes since 1980: A pooled analysis of 751 population-based studies with 4.4 million participants. Lancet 2016, 387, 1513–1530. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.J.; Larson, M.G.; Vasan, R.S.; Cheng, S.; Rhee, E.P.; McCabe, E.; Lewis, G.D.; Fox, C.S.; Jacques, P.F.; Fernandez, C.; et al. Metabolite profiles and the risk of developing diabetes. Nat. Med. 2011, 17, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Natividad, J.M.; Agus, A.; Planchais, J.; Lamas, B.; Jarry, A.C.; Martin, R.; Michel, M.L.; Chong-Nguyen, C.; Roussel, R.; Straube, M.; et al. Impaired Aryl Hydrocarbon Receptor Ligand Production by the Gut Microbiota Is a Key Factor in Metabolic Syndrome. Cell Metab. 2018, 28, 737–749 e734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kootte, R.S.; Levin, E.; Salojarvi, J.; Smits, L.P.; Hartstra, A.V.; Udayappan, S.D.; Hermes, G.; Bouter, K.E.; Koopen, A.M.; Holst, J.J.; et al. Improvement of Insulin Sensitivity after Lean Donor Feces in Metabolic Syndrome Is Driven by Baseline Intestinal Microbiota Composition. Cell Metab. 2017, 26, 611–619 e616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Org, E.; Blum, Y.; Kasela, S.; Mehrabian, M.; Kuusisto, J.; Kangas, A.J.; Soininen, P.; Wang, Z.; Ala-Korpela, M.; Hazen, S.L.; et al. Relationships between gut microbiota, plasma metabolites, and metabolic syndrome traits in the METSIM cohort. Genome Biol. 2017, 18, 70. [Google Scholar] [CrossRef]
- Guasch-Ferre, M.; Hruby, A.; Toledo, E.; Clish, C.B.; Martinez-Gonzalez, M.A.; Salas-Salvado, J.; Hu, F.B. Metabolomics in Prediabetes and Diabetes: A Systematic Review and Meta-analysis. Diabetes Care 2016, 39, 833–846. [Google Scholar] [CrossRef] [Green Version]
- Koh, A.; Molinaro, A.; Stahlman, M.; Khan, M.T.; Schmidt, C.; Manneras-Holm, L.; Wu, H.; Carreras, A.; Jeong, H.; Olofsson, L.E.; et al. Microbially Produced Imidazole Propionate Impairs Insulin Signaling through mTORC1. Cell 2018, 175, 947–961 e917. [Google Scholar] [CrossRef] [Green Version]
- De Vadder, F.; Kovatcheva-Datchary, P.; Goncalves, D.; Vinera, J.; Zitoun, C.; Duchampt, A.; Backhed, F.; Mithieux, G. Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell 2014, 156, 84–96. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Brial, F.; Alzaid, F.; Sonomura, K.; Kamatani, Y.; Meneyrol, K.; Le Lay, A.; Pean, N.; Hedjazi, L.; Sato, T.A.; Venteclef, N.; et al. The Natural Metabolite 4-Cresol Improves Glucose Homeostasis and Enhances beta-Cell Function. Cell Rep. 2020, 30, 2306–2320 e2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noecker, C.; Chiu, H.C.; McNally, C.P.; Borenstein, E. Defining and Evaluating Microbial Contributions to Metabolite Variation in Microbiome-Metabolome Association Studies. mSystems 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wurtz, P.; Makinen, V.P.; Soininen, P.; Kangas, A.J.; Tukiainen, T.; Kettunen, J.; Savolainen, M.J.; Tammelin, T.; Viikari, J.S.; Ronnemaa, T.; et al. Metabolic signatures of insulin resistance in 7,098 young adults. Diabetes 2012, 61, 1372–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiehn, O.; Garvey, W.T.; Newman, J.W.; Lok, K.H.; Hoppel, C.L.; Adams, S.H. Plasma metabolomic profiles reflective of glucose homeostasis in non-diabetic and type 2 diabetic obese African-American women. PLoS ONE 2010, 5, e15234. [Google Scholar] [CrossRef] [Green Version]
- Rizza, R.A. Pathogenesis of fasting and postprandial hyperglycemia in type 2 diabetes: Implications for therapy. Diabetes 2010, 59, 2697–2707. [Google Scholar] [CrossRef] [Green Version]
- DeFronzo, R.A.; Tobin, J.D.; Andres, R. Glucose clamp technique: A method for quantifying insulin secretion and resistance. Am. J. Physiol. 1979, 237, E214–E223. [Google Scholar] [CrossRef]
- Tam, C.S.; Xie, W.; Johnson, W.D.; Cefalu, W.T.; Redman, L.M.; Ravussin, E. Defining insulin resistance from hyperinsulinemic-euglycemic clamps. Diabetes Care 2012, 35, 1605–1610. [Google Scholar] [CrossRef] [Green Version]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojarvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916 e917. [Google Scholar] [CrossRef]
- Langeveld, M.; Ghauharali, K.J.; Sauerwein, H.P.; Ackermans, M.T.; Groener, J.E.; Hollak, C.E.; Aerts, J.M.; Serlie, M.J. Type I Gaucher disease, a glycosphingolipid storage disorder, is associated with insulin resistance. J. Clin. Endocrinol. Metab. 2008, 93, 845–851. [Google Scholar] [CrossRef] [Green Version]
- Finegood, D.T.; Bergman, R.N.; Vranic, M. Estimation of endogenous glucose production during hyperinsulinemic-euglycemic glucose clamps. Comparison of unlabeled and labeled exogenous glucose infusates. Diabetes 1987, 36, 914–924. [Google Scholar] [CrossRef]
- Costea, P.I.; Zeller, G.; Sunagawa, S.; Pelletier, E.; Alberti, A.; Levenez, F.; Tramontano, M.; Driessen, M.; Hercog, R.; Jung, F.E.; et al. Towards standards for human fecal sample processing in metagenomic studies. Nat. Biotechnol. 2017, 35, 1069–1076. [Google Scholar] [CrossRef]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Yu, C.; Chen, Y.; Cline, G.W.; Zhang, D.; Zong, H.; Wang, Y.; Bergeron, R.; Kim, J.K.; Cushman, S.W.; Cooney, G.J. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J. Biol. Chem. 2002, 277, 50230–50236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavez, J.A.; Summers, S.A. Characterizing the effects of saturated fatty acids on insulin signaling and ceramide and diacylglycerol accumulation in 3T3-L1 adipocytes and C2C12 myotubes. Arch. Biochem. Biophys. 2003, 419, 101–109. [Google Scholar] [CrossRef]
- Ruderman, N.B.; Saha, A.K.; Vavvas, D.; Witters, L.A. Malonyl-CoA, fuel sensing, and insulin resistance. Am. J. Physiol.-Endocrinol. Metab. 1999, 276, E1–E18. [Google Scholar] [CrossRef]
- Morino, K.; Petersen, K.F.; Shulman, G.I. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Am. Diabetes Assoc. 2006. [Google Scholar] [CrossRef] [Green Version]
- Schrauwen, P.; Hesselink, M.K. Oxidative capacity, lipotoxicity, and mitochondrial damage in type 2 diabetes. Diabetes 2004, 53, 1412–1417. [Google Scholar] [CrossRef] [Green Version]
- Mihalik, S.J.; Goodpaster, B.H.; Kelley, D.E.; Chace, D.H.; Vockley, J.; Toledo, F.G.; DeLany, J.P. Increased levels of plasma acylcarnitines in obesity and type 2 diabetes and identification of a marker of glucolipotoxicity. Obesity (Silver Spring) 2010, 18, 1695–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villarreal-Perez, J.Z.; Villarreal-Martinez, J.Z.; Lavalle-Gonzalez, F.J.; Torres-Sepulveda Mdel, R.; Ruiz-Herrera, C.; Cerda-Flores, R.M.; Castillo-Garcia, E.R.; Rodriguez-Sanchez, I.P.; Martinez de Villarreal, L.E. Plasma and urine metabolic profiles are reflective of altered beta-oxidation in non-diabetic obese subjects and patients with type 2 diabetes mellitus. Diabetol. Metab. Syndr. 2014, 6, 129. [Google Scholar] [CrossRef] [Green Version]
- Bene, J.; Marton, M.; Mohas, M.; Bagosi, Z.; Bujtor, Z.; Oroszlan, T.; Gasztonyi, B.; Wittmann, I.; Melegh, B. Similarities in serum acylcarnitine patterns in type 1 and type 2 diabetes mellitus and in metabolic syndrome. Ann. Nutr. Metab. 2013, 62, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Barber, M.N.; Risis, S.; Yang, C.; Meikle, P.J.; Staples, M.; Febbraio, M.A.; Bruce, C.R. Plasma lysophosphatidylcholine levels are reduced in obesity and type 2 diabetes. PLoS ONE 2012, 7, e41456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Arrigo, P.; Servi, S. Synthesis of lysophospholipids. Molecules 2010, 15, 1354–1377. [Google Scholar] [CrossRef] [Green Version]
- Soga, T.; Ohishi, T.; Matsui, T.; Saito, T.; Matsumoto, M.; Takasaki, J.; Matsumoto, S.; Kamohara, M.; Hiyama, H.; Yoshida, S.; et al. Lysophosphatidylcholine enhances glucose-dependent insulin secretion via an orphan G-protein-coupled receptor. Biochem. Biophys. Res. Commun. 2005, 326, 744–751. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergstrom, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Backhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Arora, T.; Backhed, F. The gut microbiota and metabolic disease: Current understanding and future perspectives. J. Intern. Med. 2016, 280, 339–349. [Google Scholar] [CrossRef]
- Koopen, A.M.; Groen, A.K.; Nieuwdorp, M. Human microbiome as therapeutic intervention target to reduce cardiovascular disease risk. Curr. Opin. Lipidol. 2016, 27, 615–622. [Google Scholar] [CrossRef]
- Geurts, L.; Lazarevic, V.; Derrien, M.; Everard, A.; Van Roye, M.; Knauf, C.; Valet, P.; Girard, M.; Muccioli, G.G.; François, P. Altered gut microbiota and endocannabinoid system tone in obese and diabetic leptin-resistant mice: Impact on apelin regulation in adipose tissue. Front. Microbiol. 2011, 2, 149. [Google Scholar] [CrossRef] [Green Version]
- Clavel, T.; Desmarchelier, C.; Haller, D.; Gerard, P.; Rohn, S.; Lepage, P.; Daniel, H. Intestinal microbiota in metabolic diseases: From bacterial community structure and functions to species of pathophysiological relevance. Gut Microbes 2014, 5, 544–551. [Google Scholar] [CrossRef] [Green Version]
- Lippert, K.; Kedenko, L.; Antonielli, L.; Kedenko, I.; Gemeier, C.; Leitner, M.; Kautzky-Willer, A.; Paulweber, B.; Hackl, E. Gut microbiota dysbiosis associated with glucose metabolism disorders and the metabolic syndrome in older adults. Benef. Microbes 2017, 8, 545–556. [Google Scholar] [CrossRef]
- Zhang, X.; Shen, D.; Fang, Z.; Jie, Z.; Qiu, X.; Zhang, C.; Chen, Y.; Ji, L. Human gut microbiota changes reveal the progression of glucose intolerance. PLoS ONE 2013, 8, e71108. [Google Scholar] [CrossRef]
- Kameyama, K.; Itoh, K. Intestinal colonization by a Lachnospiraceae bacterium contributes to the development of diabetes in obese mice. Microbes Environ. 2014, 29, 427–430. [Google Scholar] [CrossRef] [Green Version]
- Gilijamse, P.W.; Hartstra, A.V.; Levin, E.; Wortelboer, K.; Serlie, M.J.; Ackermans, M.T.; Herrema, H.; Nederveen, A.J.; Imangaliyev, S.; Aalvink, S.; et al. Treatment with Anaerobutyricum soehngenii: A pilot study of safety and dose-response effects on glucose metabolism in human subjects with metabolic syndrome. NPJ Biofilms Microbiomes 2020, 6, 16. [Google Scholar] [CrossRef]
- Wilmanski, T.; Rappaport, N.; Earls, J.C.; Magis, A.T.; Manor, O.; Lovejoy, J.; Omenn, G.S.; Hood, L.; Gibbons, S.M.; Price, N.D. Blood metabolome predicts gut microbiome alpha-diversity in humans. Nat. Biotechnol. 2019, 37, 1217–1228. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, F.; Ding, X.; Wu, G.; Lam, Y.Y.; Wang, X.; Fu, H.; Xue, X.; Lu, C.; Ma, J.; et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science 2018, 359, 1151–1156. [Google Scholar] [CrossRef] [Green Version]
- Wen, L.; Duffy, A. Factors Influencing the Gut Microbiota, Inflammation, and Type 2 Diabetes. J. Nutr. 2017, 147, 1468S–1475S. [Google Scholar] [CrossRef] [Green Version]
- Haro, C.; Rangel-Zuniga, O.A.; Alcala-Diaz, J.F.; Gomez-Delgado, F.; Perez-Martinez, P.; Delgado-Lista, J.; Quintana-Navarro, G.M.; Landa, B.B.; Navas-Cortes, J.A.; Tena-Sempere, M.; et al. Intestinal Microbiota Is Influenced by Gender and Body Mass Index. PLoS ONE 2016, 11, e0154090. [Google Scholar] [CrossRef] [Green Version]
- Mueller, S.; Saunier, K.; Hanisch, C.; Norin, E.; Alm, L.; Midtvedt, T.; Cresci, A.; Silvi, S.; Orpianesi, C.; Verdenelli, M.C.; et al. Differences in fecal microbiota in different European study populations in relation to age, gender, and country: A cross-sectional study. Appl. Environ. Microbiol. 2006, 72, 1027–1033. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Group (n = 115) | |
|---|---|
| Male gender (%) | 90 |
| Age (years) | 55.9 ± 8.1 |
| Weight (kg) | 111.0 ± 15.5 |
| BMI (kg/m2) | 34.2 ± 3.9 |
| Blood pressure: systolic (mmHg) | 143 ± 18 |
| Blood pressure: diastolic (mmHg) | 89 ± 11 |
| Fasting glucose (mmol/L) | 5.8 ± 0.6 |
| Insulin (pmol/L) | 112 ± 51 |
| HOMA-IR | 4.0 ± 2.0 |
| Cholesterol: total (mmol/L) | 5.5 ± 1.1 |
| Cholesterol: HDL (mmol/L) | 1.2 ± 0.3 |
| Cholesterol: LDL (mmol/L) | 3.6 ± 0.9 |
| Cholesterol: triglycerides (mmol/L) | 1.5 ± 0.6 |
| ALT (U/L) | 31 (24–39) |
| CRP (mg/mL) | 2.0 (1.1–4.2) |
| Rd (μmol kg−1min−1) | 33.3 ± 13.2 |
| EGP suppression (%) | 68.7 ± 16.0 |
| Energy intake (kcal/day) | 1936 ± 449 |
| Fat intake (gram/day) | 73 ± 22 |
| Carbohydrate intake (gram/day) | 199 ± 64 |
| Protein intake (gram/day) | 88 ± 19 |
| Fiber intake (gram/day) | 18 ± 5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koopen, A.M.; de Clercq, N.C.; Warmbrunn, M.V.; Herrema, H.; Davids, M.; de Groot, P.F.; Kootte, R.S.; Bouter, K.E.C.; Nieuwdorp, M.; Groen, A.K.; et al. Plasma Metabolites Related to Peripheral and Hepatic Insulin Sensitivity Are Not Directly Linked to Gut Microbiota Composition. Nutrients 2020, 12, 2308. https://0-doi-org.brum.beds.ac.uk/10.3390/nu12082308
Koopen AM, de Clercq NC, Warmbrunn MV, Herrema H, Davids M, de Groot PF, Kootte RS, Bouter KEC, Nieuwdorp M, Groen AK, et al. Plasma Metabolites Related to Peripheral and Hepatic Insulin Sensitivity Are Not Directly Linked to Gut Microbiota Composition. Nutrients. 2020; 12(8):2308. https://0-doi-org.brum.beds.ac.uk/10.3390/nu12082308
Chicago/Turabian StyleKoopen, Annefleur M., Nicolien C. de Clercq, Moritz V. Warmbrunn, Hilde Herrema, Mark Davids, Pieter F. de Groot, Ruud S. Kootte, Kristien E. C. Bouter, Max Nieuwdorp, Albert K. Groen, and et al. 2020. "Plasma Metabolites Related to Peripheral and Hepatic Insulin Sensitivity Are Not Directly Linked to Gut Microbiota Composition" Nutrients 12, no. 8: 2308. https://0-doi-org.brum.beds.ac.uk/10.3390/nu12082308