
Association of Gut Hormones and Microbiota with Vascular Dysfunction in Obesity

 ,
,
Abstract
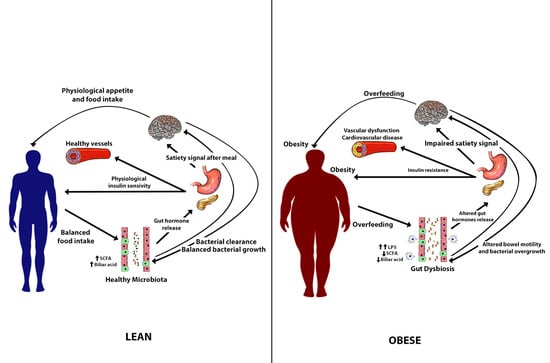
:
1. Introduction
2. Methods
3. Obesity and Vascular Complications
4. Obesity, Metabolic Diseases, and Endothelial Dysfunction
5. Perivascular Adipose Tissue (PVAT) and Fat Accumulation Mechanisms Linked with Endothelial Dysfunction and Insulin Resistance
6. Gut Hormones
6.1. Gut Hormones in Metabolic Disease and Obesity
6.2. GLP-1, GIP, and Oxyntomodulin
6.3. Ghrelin and Obestatin
6.4. PYY and Insulin like Peptide 5
6.5. Cholecystokinin
7. Obesity and Gut Microbiota
8. Gut Microbiota, Energy Harvest, and Storage
9. Gut Microbiota and Satiety Signaling
10. Gut Microbiota, Insulin Resistance, Vascular Disfunction, and Cardiovascular Disease
11. The Autonomic Nervous System (ANS)
12. The Brain-Gut-Microbiome Axis (BGM)
13. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F.; et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef] [Green Version]
- Finucane, M.M.; Stevens, G.A.; Cowan, M.J.; Danaei, G.; Lin, J.K.; Paciorek, C.J.; Singh, G.M.; Gutierrez, H.R.; Lu, Y.; Bahalim, A.N.; et al. National, regional, and global trends in body-mass index since 1980: Systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9·1 million participants. Lancet 2011, 377, 557–567. [Google Scholar] [CrossRef] [Green Version]
- Ahima, R.S.; Lazar, M.A. Physiology. The health risk of obesity—Better metrics imperative. Science 2013, 341, 856–858. [Google Scholar] [CrossRef] [PubMed]
- Purnell, J.Q. Definitions, Classification, and Epidemiology of Obesity. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., Kaltsas, G., et al., Eds.; Endotext: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Candi, E.; Tesauro, M.; Cardillo, C.; Lena, A.M.; Schinzari, F.; Rodia, G.; Sica, G.; Gentileschi, P.; Rovella, V.; Annicchiarico-Petruzzelli, M.; et al. Metabolic profiling of visceral adipose tissue from obese subjects with or without metabolic syndrome. Biochem. J. 2018, 475, 1019–1035. [Google Scholar] [CrossRef]
- Piro, M.C.; Tesauro, M.; Lena, A.M.; Gentileschi, P.; Sica, G.; Rodia, G.; Annicchiarico-Petruzzelli, M.; Rovella, V.; Cardillo, C.; Melino, G.; et al. Free-amino acid metabolic profiling of visceral adipose tissue from obese subjects. Amino Acids 2020, 52, 1125–1137. [Google Scholar] [CrossRef] [PubMed]
- Cercato, C.; Fonseca, F.A. Cardiovascular risk and obesity. Diabetol. Metab. Syndr. 2019, 11, 74. [Google Scholar] [CrossRef]
- Esper, R.J.; Nordaby, R.A.; Vilariño, J.O.; Paragano, A.; Cacharrón, J.L.; Machado, R.A. Endothelial dysfunction: A comprehensive appraisal. Cardiovasc. Diabetol. 2006, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Versari, D.; Daghini, E.; Virdis, A.; Ghiadoni, L.; Taddei, S. Endothelial dysfunction as a target for prevention of cardiovascular disease. Diabetes Care 2009, 32 (Suppl. 2), S314–S321. [Google Scholar] [CrossRef] [Green Version]
- Vita, J.A.; Keaney, J.F., Jr. Endothelial function: A barometer for cardiovascular risk? Circulation 2002, 106, 640–642. [Google Scholar] [CrossRef] [Green Version]
- Marti, C.N.; Gheorghiade, M.; Kalogeropoulos, A.P.; Georgiopoulou, V.V.; Quyyumi, A.A.; Butler, J. Endothelial dysfunction, arterial stiffness, and heart failure. J. Am. Coll. Cardiol. 2012, 60, 1455–1469. [Google Scholar] [CrossRef] [Green Version]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [Green Version]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Daiber, A.; Steven, S.; Weber, A.; Shuvaev, V.V.; Muzykantov, V.R.; Laher, I.; Li, H.; Lamas, S.; Münzel, T. Targeting vascular (endothelial) dysfunction. Br. J. Pharmacol. 2017, 174, 1591–1619. [Google Scholar] [CrossRef]
- Pi, X.; Xie, L.; Patterson, C. Emerging Roles of Vascular Endothelium in Metabolic Homeostasis. Circ. Res. 2018, 123, 477–494. [Google Scholar] [CrossRef]
- Daiber, A.; Di Lisa, F.; Oelze, M.; Kröller-Schön, S.; Steven, S.; Schulz, E.; Münzel, T. Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function. Br. J. Pharmacol. 2017, 174, 1670–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daiber, A.; Oelze, M.; Daub, S.; Steven, S.; Schuff, A.; Kröller-Schön, S.; Hausding, M.; Wenzel, P.; Schulz, E.; Gori, T.; et al. Vascular Redox Signaling, Redox Switches in Endothelial Nitric Oxide Synthase (eNOS Uncoupling), and Endothelial Dysfunction. In Systems Biology of Free Radicals and Antioxidants; Laher, I., Ed.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 1177–1211. [Google Scholar] [CrossRef]
- Förstermann, U.; Münzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schinzari, F.; Iantorno, M.; Campia, U.; Mores, N.; Rovella, V.; Tesauro, M.; Di Daniele, N.; Cardillo, C. Vasodilator responses and endothelin-dependent vasoconstriction in metabolically healthy obesity and the metabolic syndrome. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E787–E792. [Google Scholar] [CrossRef] [Green Version]
- Campia, U.; Tesauro, M.; Di Daniele, N.; Cardillo, C. The vascular endothelin system in obesity and type 2 diabetes: Pathophysiology and therapeutic implications. Life Sci. 2014, 118, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Bigornia, S.J.; Mott, M.M.; Hess, D.T.; Apovian, C.M.; McDonnell, M.E.; Duess, M.A.; Kluge, M.A.; Fiscale, A.J.; Vita, J.A.; Gokce, N. Long-term successful weight loss improves vascular endothelial function in severely obese individuals. Obesity (Silver Spring) 2010, 18, 754–759. [Google Scholar] [CrossRef]
- Levin, E.R. Endothelins. N. Engl. J. Med. 1995, 333, 356–363. [Google Scholar] [CrossRef]
- Mather, K.J.; Lteif, A.; Steinberg, H.O.; Baron, A.D. Interactions between endothelin and nitric oxide in the regulation of vascular tone in obesity and diabetes. Diabetes 2004, 53, 2060–2066. [Google Scholar] [CrossRef] [Green Version]
- Tesauro, M.; Schinzari, F.; Rovella, V.; Di Daniele, N.; Lauro, D.; Mores, N.; Veneziani, A.; Cardillo, C. Ghrelin restores the endothelin 1/nitric oxide balance in patients with obesity-related metabolic syndrome. Hypertension 2009, 54, 995–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzuca, M.Q.; Khalil, R.A. Vascular endothelin receptor type B: Structure, function and dysregulation in vascular disease. Biochem. Pharmacol. 2012, 84, 147–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberti, K.G.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.; Loria, C.M.; Smith, S.C., Jr. Harmonizing the metabolic syndrome: A joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120, 1640–1645. [Google Scholar] [CrossRef] [Green Version]
- Grundy, S.M.; Brewer, H.B., Jr.; Cleeman, J.I.; Smith, S.C., Jr.; Lenfant, C. Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation 2004, 109, 433–438. [Google Scholar] [CrossRef] [Green Version]
- Kahn, B.B.; Flier, J.S. Obesity and insulin resistance. J. Clin. Investig. 2000, 106, 473–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, M.; Yanagisawa, M. Endothelin: 20 years from discovery to therapy. Can. J. Physiol. Pharmacol. 2008, 86, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Cardillo, C.; Kilcoyne, C.M.; Waclawiw, M.; Cannon, R.O., 3rd; Panza, J.A. Role of endothelin in the increased vascular tone of patients with essential hypertension. Hypertension 1999, 33, 753–758. [Google Scholar] [CrossRef] [Green Version]
- Cardillo, C.; Kilcoyne, C.M.; Cannon, R.O., 3rd; Panza, J.A. Increased activity of endogenous endothelin in patients with hypercholesterolemia. J. Am. Coll. Cardiol. 2000, 36, 1483–1488. [Google Scholar] [CrossRef] [Green Version]
- Cardillo, C.; Campia, U.; Bryant, M.B.; Panza, J.A. Increased activity of endogenous endothelin in patients with type II diabetes mellitus. Circulation 2002, 106, 1783–1787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mather, K.J.; Mirzamohammadi, B.; Lteif, A.; Steinberg, H.O.; Baron, A.D. Endothelin contributes to basal vascular tone and endothelial dysfunction in human obesity and type 2 diabetes. Diabetes 2002, 51, 3517–3523. [Google Scholar] [CrossRef] [Green Version]
- Ohkita, M.; Takaoka, M.; Shiota, Y.; Nojiri, R.; Matsumura, Y. Nitric oxide inhibits endothelin-1 production through the suppression of nuclear factor kappa B. Clin. Sci. 2002, 103 (Suppl. 48), 68s–71s. [Google Scholar] [CrossRef] [PubMed]
- Vicent, D.; Ilany, J.; Kondo, T.; Naruse, K.; Fisher, S.J.; Kisanuki, Y.Y.; Bursell, S.; Yanagisawa, M.; King, G.L.; Kahn, C.R. The role of endothelial insulin signaling in the regulation of vascular tone and insulin resistance. J. Clin. Investig. 2003, 111, 1373–1380. [Google Scholar] [CrossRef] [Green Version]
- Cardillo, C.; Nambi, S.S.; Kilcoyne, C.M.; Choucair, W.K.; Katz, A.; Quon, M.J.; Panza, J.A. Insulin stimulates both endothelin and nitric oxide activity in the human forearm. Circulation 1999, 100, 820–825. [Google Scholar] [CrossRef] [Green Version]
- Muniyappa, R.; Montagnani, M.; Koh, K.K.; Quon, M.J. Cardiovascular actions of insulin. Endocr. Rev. 2007, 28, 463–491. [Google Scholar] [CrossRef]
- Zeng, G.; Nystrom, F.H.; Ravichandran, L.V.; Cong, L.N.; Kirby, M.; Mostowski, H.; Quon, M.J. Roles for insulin receptor, PI3-kinase, and Akt in insulin-signaling pathways related to production of nitric oxide in human vascular endothelial cells. Circulation 2000, 101, 1539–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, M.G.; Wallis, M.G.; Barrett, E.J.; Vincent, M.A.; Richards, S.M.; Clerk, L.H.; Rattigan, S. Blood flow and muscle metabolism: A focus on insulin action. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E241–E258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anfossi, G.; Russo, I.; Doronzo, G.; Trovati, M. Relevance of the vascular effects of insulin in the rationale of its therapeutical use. Cardiovasc. Hematol. Disord. Drug Targets 2007, 7, 228–249. [Google Scholar] [CrossRef] [PubMed]
- Rajapakse, N.W.; Chong, A.L.; Zhang, W.Z.; Kaye, D.M. Insulin-mediated activation of the L-arginine nitric oxide pathway in man, and its impairment in diabetes. PLoS ONE 2013, 8, e61840. [Google Scholar] [CrossRef] [Green Version]
- Contreras, C.; Sanchez, A.; Garcia-Sacristan, A.; Martinez, M.C.; Andriantsitohaina, R.; Prieto, D. Preserved insulin vasorelaxation and up-regulation of the Akt/eNOS pathway in coronary arteries from insulin resistant obese Zucker rats. Atherosclerosis 2011, 217, 331–339. [Google Scholar] [CrossRef] [Green Version]
- Cusi, K.; Maezono, K.; Osman, A.; Pendergrass, M.; Patti, M.E.; Pratipanawatr, T.; DeFronzo, R.A.; Kahn, C.R.; Mandarino, L.J. Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. J. Clin. Investig. 2000, 105, 311–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.A.; Montagnani, M.; Koh, K.K.; Quon, M.J. Reciprocal relationships between insulin resistance and endothelial dysfunction: Molecular and pathophysiological mechanisms. Circulation 2006, 113, 1888–1904. [Google Scholar] [CrossRef]
- Ottosson-Seeberger, A.; Lundberg, J.M.; Alvestrand, A.; Ahlborg, G. Exogenous endothelin-1 causes peripheral insulin resistance in healthy humans. Acta Physiol. Scand. 1997, 161, 211–220. [Google Scholar] [CrossRef]
- Lteif, A.; Vaishnava, P.; Baron, A.D.; Mather, K.J. Endothelin limits insulin action in obese/insulin-resistant humans. Diabetes 2007, 56, 728–734. [Google Scholar] [CrossRef] [Green Version]
- Shemyakin, A.; Salehzadeh, F.; Böhm, F.; Al-Khalili, L.; Gonon, A.; Wagner, H.; Efendic, S.; Krook, A.; Pernow, J. Regulation of glucose uptake by endothelin-1 in human skeletal muscle in vivo and in vitro. J. Clin. Endocrinol. Metab. 2010, 95, 2359–2366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busija, D.W.; Miller, A.W.; Katakam, P.; Erdos, B. Adverse effects of reactive oxygen species on vascular reactivity in insulin resistance. Antioxid. Redox Signal. 2006, 8, 1131–1140. [Google Scholar] [CrossRef]
- Sandu, O.A.; Ito, M.; Begum, N. Selected contribution: Insulin utilizes NO/cGMP pathway to activate myosin phosphatase via Rho inhibition in vascular smooth muscle. J. Appl. Physiol. 2001, 91, 1475–1482. [Google Scholar] [CrossRef]
- Baltieri, N.; Guizoni, D.M.; Victorio, J.A.; Davel, A.P. Protective Role of Perivascular Adipose Tissue in Endothelial Dysfunction and Insulin-Induced Vasodilatation of Hypercholesterolemic LDL Receptor-Deficient Mice. Front. Physiol. 2018, 9, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed-Ali, V.; Pinkney, J.H.; Coppack, S.W. Adipose tissue as an endocrine and paracrine organ. Int. J. Obes. Relat. Metab. Disord. 1998, 22, 1145–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weyer, C.; Funahashi, T.; Tanaka, S.; Hotta, K.; Matsuzawa, Y.; Pratley, R.E.; Tataranni, P.A. Hypoadiponectinemia in obesity and type 2 diabetes: Close association with insulin resistance and hyperinsulinemia. J. Clin. Endocrinol. Metab. 2001, 86, 1930–1935. [Google Scholar] [CrossRef]
- Lara-Castro, C.; Luo, N.; Wallace, P.; Klein, R.L.; Garvey, W.T. Adiponectin multimeric complexes and the metabolic syndrome trait cluster. Diabetes 2006, 55, 249–259. [Google Scholar] [CrossRef]
- Matsubara, M.; Maruoka, S.; Katayose, S. Inverse relationship between plasma adiponectin and leptin concentrations in normal-weight and obese women. Eur. J. Endocrinol. 2002, 147, 173–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Montagnani, M.; Funahashi, T.; Shimomura, I.; Quon, M.J. Adiponectin stimulates production of nitric oxide in vascular endothelial cells. J. Biol. Chem. 2003, 278, 45021–45026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motoshima, H.; Wu, X.; Mahadev, K.; Goldstein, B.J. Adiponectin suppresses proliferation and superoxide generation and enhances eNOS activity in endothelial cells treated with oxidized LDL. Biochem. Biophys. Res. Commun. 2004, 315, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Ahima, R.S.; Flier, J.S. Leptin. Annu. Rev. Physiol. 2000, 62, 413–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, S.S.; Qasim, A.; Reilly, M.P. Leptin resistance: A possible interface of inflammation and metabolism in obesity-related cardiovascular disease. J. Am. Coll. Cardiol. 2008, 52, 1201–1210. [Google Scholar] [CrossRef] [Green Version]
- Vecchione, C.; Maffei, A.; Colella, S.; Aretini, A.; Poulet, R.; Frati, G.; Gentile, M.T.; Fratta, L.; Trimarco, V.; Trimarco, B.; et al. Leptin effect on endothelial nitric oxide is mediated through Akt-endothelial nitric oxide synthase phosphorylation pathway. Diabetes 2002, 51, 168–173. [Google Scholar] [CrossRef] [Green Version]
- Korda, M.; Kubant, R.; Patton, S.; Malinski, T. Leptin-induced endothelial dysfunction in obesity. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1514–H1521. [Google Scholar] [CrossRef] [Green Version]
- Söderberg, S.; Ahrén, B.; Jansson, J.H.; Johnson, O.; Hallmans, G.; Asplund, K.; Olsson, T. Leptin is associated with increased risk of myocardial infarction. J. Intern. Med. 1999, 246, 409–418. [Google Scholar] [CrossRef]
- Hajer, G.R.; van Haeften, T.W.; Visseren, F.L. Adipose tissue dysfunction in obesity, diabetes, and vascular diseases. Eur. Heart J. 2008, 29, 2959–2971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piatti, P.M.; Monti, L.D.; Conti, M.; Baruffaldi, L.; Galli, L.; Phan, C.V.; Guazzini, B.; Pontiroli, A.E.; Pozza, G. Hypertriglyceridemia and hyperinsulinemia are potent inducers of endothelin-1 release in humans. Diabetes 1996, 45, 316–321. [Google Scholar] [CrossRef]
- Schinzari, F.; Tesauro, M.; Cardillo, C. Increased endothelin-1-mediated vasoconstrictor tone in human obesity: Effects of gut hormones. Physiol. Res. 2018, 67, S69–S81. [Google Scholar] [CrossRef]
- Stow, L.R.; Jacobs, M.E.; Wingo, C.S.; Cain, B.D. Endothelin-1 gene regulation. FASEB J. 2011, 25, 16–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hotamisligil, G.S. Obesity-induced inflammatory changes in adipose tissue. J. Clin. Investig. 2003, 112, 1785–1788. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Rask-Madsen, C.; Domínguez, H.; Ihlemann, N.; Hermann, T.; Køber, L.; Torp-Pedersen, C. Tumor necrosis factor-alpha inhibits insulin’s stimulating effect on glucose uptake and endothelium-dependent vasodilation in humans. Circulation 2003, 108, 1815–1821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pittas, A.G.; Joseph, N.A.; Greenberg, A.S. Adipocytokines and insulin resistance. J. Clin. Endocrinol. Metab. 2004, 89, 447–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesauro, M.; Schinzari, F.; Rovella, V.; Melina, D.; Mores, N.; Barini, A.; Mettimano, M.; Lauro, D.; Iantorno, M.; Quon, M.J.; et al. Tumor necrosis factor-alpha antagonism improves vasodilation during hyperinsulinemia in metabolic syndrome. Diabetes Care 2008, 31, 1439–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yudkin, J.S.; Eringa, E.; Stehouwer, C.D. “Vasocrine” signalling from perivascular fat: A mechanism linking insulin resistance to vascular disease. Lancet 2005, 365, 1817–1820. [Google Scholar] [CrossRef]
- Schinzari, F.; Tesauro, M.; Veneziani, A.; Mores, N.; Di Daniele, N.; Cardillo, C. Favorable Vascular Actions of Angiotensin-(1-7) in Human Obesity. Hypertension 2018, 71, 185–191. [Google Scholar] [CrossRef]
- Aghamohammadzadeh, R.; Withers, S.; Lynch, F.; Greenstein, A.; Malik, R.; Heagerty, A. Perivascular adipose tissue from human systemic and coronary vessels: The emergence of a new pharmacotherapeutic target. Br. J. Pharmacol. 2012, 165, 670–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenstein, A.S.; Khavandi, K.; Withers, S.B.; Sonoyama, K.; Clancy, O.; Jeziorska, M.; Laing, I.; Yates, A.P.; Pemberton, P.W.; Malik, R.A.; et al. Local inflammation and hypoxia abolish the protective anticontractile properties of perivascular fat in obese patients. Circulation 2009, 119, 1661–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schinzari, F.; Tesauro, M.; Cardillo, C. Endothelial and Perivascular Adipose Tissue Abnormalities in Obesity-Related Vascular Dysfunction: Novel Targets for Treatment. J. Cardiovasc. Pharmacol. 2017, 69, 360–368. [Google Scholar] [CrossRef]
- Chatterjee, T.K.; Stoll, L.L.; Denning, G.M.; Harrelson, A.; Blomkalns, A.L.; Idelman, G.; Rothenberg, F.G.; Neltner, B.; Romig-Martin, S.A.; Dickson, E.W.; et al. Proinflammatory phenotype of perivascular adipocytes: Influence of high-fat feeding. Circ. Res. 2009, 104, 541–549. [Google Scholar] [CrossRef] [Green Version]
- Xia, N.; Horke, S.; Habermeier, A.; Closs, E.I.; Reifenberg, G.; Gericke, A.; Mikhed, Y.; Munzel, T.; Daiber, A.; Forstermann, U.; et al. Uncoupling of Endothelial Nitric Oxide Synthase in Perivascular Adipose Tissue of Diet-Induced Obese Mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 78–85. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Barrett, E.J.; Barrett, M.O.; Cao, W.; Liu, Z. Tumor necrosis factor-alpha induces insulin resistance in endothelial cells via a p38 mitogen-activated protein kinase-dependent pathway. Endocrinology 2007, 148, 3356–3363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberg, G.R.; Michell, B.J.; van Denderen, B.J.; Watt, M.J.; Carey, A.L.; Fam, B.C.; Andrikopoulos, S.; Proietto, J.; Görgün, C.Z.; Carling, D.; et al. Tumor necrosis factor alpha-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab. 2006, 4, 465–474. [Google Scholar] [CrossRef] [Green Version]
- Shulman, G.I. Cellular mechanisms of insulin resistance. J. Clin. Investig. 2000, 106, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, H.O.; Baron, A.D. Vascular function, insulin resistance and fatty acids. Diabetologia 2002, 45, 623–634. [Google Scholar] [CrossRef]
- Rahmouni, K.; Morgan, D.A.; Morgan, G.M.; Liu, X.; Sigmund, C.D.; Mark, A.L.; Haynes, W.G. Hypothalamic PI3K and MAPK differentially mediate regional sympathetic activation to insulin. J. Clin. Investig. 2004, 114, 652–658. [Google Scholar] [CrossRef] [Green Version]
- Cassaglia, P.A.; Hermes, S.M.; Aicher, S.A.; Brooks, V.L. Insulin acts in the arcuate nucleus to increase lumbar sympathetic nerve activity and baroreflex function in rats. J. Physiol. 2011, 589, 1643–1662. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.A.; Hoffman, R.P.; Balon, T.W.; Sinkey, C.A.; Mark, A.L. Hyperinsulinemia produces both sympathetic neural activation and vasodilation in normal humans. J. Clin. Investig. 1991, 87, 2246–2252. [Google Scholar] [CrossRef] [PubMed]
- Rowe, J.W.; Young, J.B.; Minaker, K.L.; Stevens, A.L.; Pallotta, J.; Landsberg, L. Effect of insulin and glucose infusions on sympathetic nervous system activity in normal man. Diabetes 1981, 30, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Young, C.N.; Deo, S.H.; Chaudhary, K.; Thyfault, J.P.; Fadel, P.J. Insulin enhances the gain of arterial baroreflex control of muscle sympathetic nerve activity in humans. J. Physiol. 2010, 588, 3593–3603. [Google Scholar] [CrossRef]
- Ardilouze, J.L.; Sotorník, R.; Dennis, L.A.; Fielding, B.A.; Frayn, K.N.; Karpe, F. Failure to increase postprandial blood flow in subcutaneous adipose tissue is associated with tissue resistance to adrenergic stimulation. Diabetes Metab. 2012, 38, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Emanuel, A.L.; Meijer, R.I.; Muskiet, M.H.; van Raalte, D.H.; Eringa, E.C.; Serné, E.H. Role of Insulin-Stimulated Adipose Tissue Perfusion in the Development of Whole-Body Insulin Resistance. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 411–418. [Google Scholar] [CrossRef] [Green Version]
- Gribble, F.M. The gut endocrine system as a coordinator of postprandial nutrient homoeostasis. Proc. Nutr. Soc. 2012, 71, 456–462. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.J.; Robinson, E.S.; Rivera, L.R.; McMillan, P.J.; Testro, A.; Nikfarjam, M.; Bravo, D.M.; Furness, J.B. Glucagon-like peptide 1 and peptide YY are in separate storage organelles in enteroendocrine cells. Cell Tissue Res. 2014, 357, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Egerod, K.L.; Engelstoft, M.S.; Grunddal, K.V.; Nøhr, M.K.; Secher, A.; Sakata, I.; Pedersen, J.; Windeløv, J.A.; Füchtbauer, E.M.; Olsen, J.; et al. A major lineage of enteroendocrine cells coexpress CCK, secretin, GIP, GLP-1, PYY, and neurotensin but not somatostatin. Endocrinology 2012, 153, 5782–5795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fothergill, L.J.; Callaghan, B.; Hunne, B.; Bravo, D.M.; Furness, J.B. Costorage of Enteroendocrine Hormones Evaluated at the Cell and Subcellular Levels in Male Mice. Endocrinology 2017, 158, 2113–2123. [Google Scholar] [CrossRef]
- Habib, A.M.; Richards, P.; Cairns, L.S.; Rogers, G.J.; Bannon, C.A.; Parker, H.E.; Morley, T.C.; Yeo, G.S.; Reimann, F.; Gribble, F.M. Overlap of endocrine hormone expression in the mouse intestine revealed by transcriptional profiling and flow cytometry. Endocrinology 2012, 153, 3054–3065. [Google Scholar] [CrossRef] [Green Version]
- Frias, J.P.; Nauck, M.A.; Van, J.; Kutner, M.E.; Cui, X.; Benson, C.; Urva, S.; Gimeno, R.E.; Milicevic, Z.; Robins, D.; et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: A randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet 2018, 392, 2180–2193. [Google Scholar] [CrossRef]
- Holman, R.R.; Paul, S.K.; Bethel, M.A.; Matthews, D.R.; Neil, H.A. 10-year follow-up of intensive glucose control in type 2 diabetes. N. Engl. J. Med. 2008, 359, 1577–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, S.S.; Kohl, B.A. Glycemic control and weight reduction without causing hypoglycemia: The case for continued safe aggressive care of patients with type 2 diabetes mellitus and avoidance of therapeutic inertia. Mayo Clin. Proc. 2010, 85, S15–S26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchwald, H.; Estok, R.; Fahrbach, K.; Banel, D.; Jensen, M.D.; Pories, W.J.; Bantle, J.P.; Sledge, I. Weight and type 2 diabetes after bariatric surgery: Systematic review and meta-analysis. Am. J. Med. 2009, 122, 248–256.e245. [Google Scholar] [CrossRef] [PubMed]
- Nauck, M.A.; Homberger, E.; Siegel, E.G.; Allen, R.C.; Eaton, R.P.; Ebert, R.; Creutzfeldt, W. Incretin effects of increasing glucose loads in man calculated from venous insulin and C-peptide responses. J. Clin. Endocrinol. Metab. 1986, 63, 492–498. [Google Scholar] [CrossRef]
- Drucker, D.J. Biologic actions and therapeutic potential of the proglucagon-derived peptides. Nat. Clin. Pract. Endocrinol. Metab. 2005, 1, 22–31. [Google Scholar] [CrossRef]
- Fehmann, H.C.; Göke, R.; Göke, B. Cell and molecular biology of the incretin hormones glucagon-like peptide-I and glucose-dependent insulin releasing polypeptide. Endocr. Rev. 1995, 16, 390–410. [Google Scholar] [CrossRef]
- Deacon, C.F.; Nauck, M.A.; Toft-Nielsen, M.; Pridal, L.; Willms, B.; Holst, J.J. Both subcutaneously and intravenously administered glucagon-like peptide I are rapidly degraded from the NH2-terminus in type II diabetic patients and in healthy subjects. Diabetes 1995, 44, 1126–1131. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Tamvakopoulos, C.; Xie, D.; Dragovic, J.; Shen, X.; Fenyk-Melody, J.E.; Schmidt, K.; Bagchi, A.; Griffin, P.R.; Thornberry, N.A.; et al. The role of dipeptidyl peptidase IV in the cleavage of glucagon family peptides: In vivo metabolism of pituitary adenylate cyclase activating polypeptide-(1–38). J. Biol. Chem. 2003, 278, 22418–22423. [Google Scholar] [CrossRef] [Green Version]
- Nauck, M.A.; Kleine, N.; Orskov, C.; Holst, J.J.; Willms, B.; Creutzfeldt, W. Normalization of fasting hyperglycaemia by exogenous glucagon-like peptide 1 (7–36 amide) in type 2 (non-insulin-dependent) diabetic patients. Diabetologia 1993, 36, 741–744. [Google Scholar] [CrossRef] [Green Version]
- Drucker, D.J. The Cardiovascular Biology of Glucagon-like Peptide-1. Cell Metab. 2016, 24, 15–30. [Google Scholar] [CrossRef]
- Widenmaier, S.B.; Ao, Z.; Kim, S.J.; Warnock, G.; McIntosh, C.H. Suppression of p38 MAPK and JNK via Akt-mediated inhibition of apoptosis signal-regulating kinase 1 constitutes a core component of the beta-cell pro-survival effects of glucose-dependent insulinotropic polypeptide. J. Biol. Chem. 2009, 284, 30372–30382. [Google Scholar] [CrossRef] [Green Version]
- Bataille, D.; Gespach, C.; Tatemoto, K.; Marie, J.C.; Coudray, A.M.; Rosselin, G.; Mutt, V. Bioactive enteroglucagon (oxyntomodulin): Present knowledge on its chemical structure and its biological activities. Peptides 1981, 2 (Suppl. 2), 41–44. [Google Scholar] [CrossRef]
- Baldissera, F.G.; Holst, J.J.; Knuhtsen, S.; Hilsted, L.; Nielsen, O.V. Oxyntomodulin (glicentin-(33–69)): Pharmacokinetics, binding to liver cell membranes, effects on isolated perfused pig pancreas, and secretion from isolated perfused lower small intestine of pigs. Regul. Pept. 1988, 21, 151–166. [Google Scholar] [CrossRef]
- Gros, L.; Thorens, B.; Bataille, D.; Kervran, A. Glucagon-like peptide-1-(7–36) amide, oxyntomodulin, and glucagon interact with a common receptor in a somatostatin-secreting cell line. Endocrinology 1993, 133, 631–638. [Google Scholar] [CrossRef]
- Chen, H.Y.; Trumbauer, M.E.; Chen, A.S.; Weingarth, D.T.; Adams, J.R.; Frazier, E.G.; Shen, Z.; Marsh, D.J.; Feighner, S.D.; Guan, X.M.; et al. Orexigenic action of peripheral ghrelin is mediated by neuropeptide Y and agouti-related protein. Endocrinology 2004, 145, 2607–2612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Liu, C.; Uchida, A.; Chuang, J.C.; Walker, A.; Liu, T.; Osborne-Lawrence, S.; Mason, B.L.; Mosher, C.; Berglund, E.D.; et al. Arcuate AgRP neurons mediate orexigenic and glucoregulatory actions of ghrelin. Mol. Metab. 2014, 3, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Tesauro, M.; Schinzari, F.; Iantorno, M.; Rizza, S.; Melina, D.; Lauro, D.; Cardillo, C. Ghrelin improves endothelial function in patients with metabolic syndrome. Circulation 2005, 112, 2986–2992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, T.D.; Nogueiras, R.; Andermann, M.L.; Andrews, Z.B.; Anker, S.D.; Argente, J.; Batterham, R.L.; Benoit, S.C.; Bowers, C.Y.; Broglio, F.; et al. Ghrelin. Mol. Metab. 2015, 4, 437–460. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.V.; Ren, P.G.; Avsian-Kretchmer, O.; Luo, C.W.; Rauch, R.; Klein, C.; Hsueh, A.J. Obestatin, a peptide encoded by the ghrelin gene, opposes ghrelin’s effects on food intake. Science 2005, 310, 996–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowan, E.; Burch, K.J.; Green, B.D.; Grieve, D.J. Obestatin as a key regulator of metabolism and cardiovascular function with emerging therapeutic potential for diabetes. Br. J. Pharmacol. 2016, 173, 2165–2181. [Google Scholar] [CrossRef] [Green Version]
- Agnew, A.J.; Robinson, E.; McVicar, C.M.; Harvey, A.P.; Ali, I.H.; Lindsay, J.E.; McDonald, D.M.; Green, B.D.; Grieve, D.J. The gastrointestinal peptide obestatin induces vascular relaxation via specific activation of endothelium-dependent NO signalling. Br. J. Pharmacol. 2012, 166, 327–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schinzari, F.; Veneziani, A.; Mores, N.; Barini, A.; Di Daniele, N.; Cardillo, C.; Tesauro, M. Vascular Effects of Obestatin in Lean and Obese Subjects. Diabetes 2017, 66, 1214–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habib, A.M.; Richards, P.; Rogers, G.J.; Reimann, F.; Gribble, F.M. Co-localisation and secretion of glucagon-like peptide 1 and peptide YY from primary cultured human L cells. Diabetologia 2013, 56, 1413–1416. [Google Scholar] [CrossRef] [Green Version]
- Savage, A.P.; Adrian, T.E.; Carolan, G.; Chatterjee, V.K.; Bloom, S.R. Effects of peptide YY (PYY) on mouth to caecum intestinal transit time and on the rate of gastric emptying in healthy volunteers. Gut 1987, 28, 166–170. [Google Scholar] [CrossRef]
- Wu, T.; Rayner, C.K.; Young, R.L.; Horowitz, M. Gut motility and enteroendocrine secretion. Curr. Opin. Pharmacol. 2013, 13, 928–934. [Google Scholar] [CrossRef]
- Batterham, R.L.; Cohen, M.A.; Ellis, S.M.; Le Roux, C.W.; Withers, D.J.; Frost, G.S.; Ghatei, M.A.; Bloom, S.R. Inhibition of food intake in obese subjects by peptide YY3-36. N. Engl. J. Med. 2003, 349, 941–948. [Google Scholar] [CrossRef] [Green Version]
- Billing, L.J.; Smith, C.A.; Larraufie, P.; Goldspink, D.A.; Galvin, S.; Kay, R.G.; Howe, J.D.; Walker, R.; Pruna, M.; Glass, L.; et al. Co-storage and release of insulin-like peptide-5, glucagon-like peptide-1 and peptideYY from murine and human colonic enteroendocrine cells. Mol. Metab. 2018, 16, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Ang, S.Y.; Hutchinson, D.S.; Patil, N.; Evans, B.A.; Bathgate, R.A.D.; Halls, M.L.; Hossain, M.A.; Summers, R.J.; Kocan, M. Signal transduction pathways activated by insulin-like peptide 5 at the relaxin family peptide RXFP4 receptor. Br. J. Pharmacol. 2017, 174, 1077–1089. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, K.C.; Raybould, H.E.; Walsh, J.H. Cholecystokinin inhibits gastric acid secretion through type “A” cholecystokinin receptors and somatostatin in rats. Am. J. Physiol. 1992, 263, G287–G292. [Google Scholar] [CrossRef]
- Ahrén, B.; Hedner, P.; Lundquist, I. Interaction of gastric inhibitory polypeptide (GIP) and cholecystokinin (CCK-8) with basal and stimulated insulin secretion in mice. Acta Endocrinol. 1983, 102, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Rehfeld, J.F. Cholecystokinin as satiety signal. Int. J. Obes. 1981, 5, 465–469. [Google Scholar] [PubMed]
- Koop, I.; Schindler, M.; Bosshammer, A.; Scheibner, J.; Stange, E.; Koop, H. Physiological control of cholecystokinin release and pancreatic enzyme secretion by intraduodenal bile acids. Gut 1996, 39, 661–667. [Google Scholar] [CrossRef] [Green Version]
- Rogers, R.C.; Hermann, G.E. Mechanisms of action of CCK to activate central vagal afferent terminals. Peptides 2008, 29, 1716–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holst, J.J. The physiology of glucagon-like peptide 1. Physiol. Rev. 2007, 87, 1409–1439. [Google Scholar] [CrossRef] [PubMed]
- Tesauro, M.; Schinzari, F.; Adamo, A.; Rovella, V.; Martini, F.; Mores, N.; Barini, A.; Pitocco, D.; Ghirlanda, G.; Lauro, D.; et al. Effects of GLP-1 on forearm vasodilator function and glucose disposal during hyperinsulinemia in the metabolic syndrome. Diabetes Care 2013, 36, 683–689. [Google Scholar] [CrossRef] [Green Version]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jódar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef] [Green Version]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Alhadeff, A.L.; Grill, H.J. Hindbrain nucleus tractus solitarius glucagon-like peptide-1 receptor signaling reduces appetitive and motivational aspects of feeding. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R465–R470. [Google Scholar] [CrossRef] [Green Version]
- Dickson, S.L.; Shirazi, R.H.; Hansson, C.; Bergquist, F.; Nissbrandt, H.; Skibicka, K.P. The glucagon-like peptide 1 (GLP-1) analogue, exendin-4, decreases the rewarding value of food: A new role for mesolimbic GLP-1 receptors. J. Neurosci. 2012, 32, 4812–4820. [Google Scholar] [CrossRef] [PubMed]
- Kanoski, S.E.; Hayes, M.R.; Skibicka, K.P. GLP-1 and weight loss: Unraveling the diverse neural circuitry. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R885–R895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollag, R.J.; Zhong, Q.; Phillips, P.; Min, L.; Zhong, L.; Cameron, R.; Mulloy, A.L.; Rasmussen, H.; Qin, F.; Ding, K.H.; et al. Osteoblast-derived cells express functional glucose-dependent insulinotropic peptide receptors. Endocrinology 2000, 141, 1228–1235. [Google Scholar] [CrossRef]
- Faivre, E.; Gault, V.A.; Thorens, B.; Hölscher, C. Glucose-dependent insulinotropic polypeptide receptor knockout mice are impaired in learning, synaptic plasticity, and neurogenesis. J. Neurophysiol. 2011, 105, 1574–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yip, R.G.; Boylan, M.O.; Kieffer, T.J.; Wolfe, M.M. Functional GIP receptors are present on adipocytes. Endocrinology 1998, 139, 4004–4007. [Google Scholar] [CrossRef]
- Meier, J.J.; Hücking, K.; Holst, J.J.; Deacon, C.F.; Schmiegel, W.H.; Nauck, M.A. Reduced insulinotropic effect of gastric inhibitory polypeptide in first-degree relatives of patients with type 2 diabetes. Diabetes 2001, 50, 2497–2504. [Google Scholar] [CrossRef] [Green Version]
- Mentis, N.; Vardarli, I.; Köthe, L.D.; Holst, J.J.; Deacon, C.F.; Theodorakis, M.; Meier, J.J.; Nauck, M.A. GIP does not potentiate the antidiabetic effects of GLP-1 in hyperglycemic patients with type 2 diabetes. Diabetes 2011, 60, 1270–1276. [Google Scholar] [CrossRef] [Green Version]
- Lund, A.; Vilsbøll, T.; Bagger, J.I.; Holst, J.J.; Knop, F.K. The separate and combined impact of the intestinal hormones, GIP, GLP-1, and GLP-2, on glucagon secretion in type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E1038–E1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gögebakan, Ö.; Andres, J.; Biedasek, K.; Mai, K.; Kühnen, P.; Krude, H.; Isken, F.; Rudovich, N.; Osterhoff, M.A.; Kintscher, U.; et al. Glucose-dependent insulinotropic polypeptide reduces fat-specific expression and activity of 11β-hydroxysteroid dehydrogenase type 1 and inhibits release of free fatty acids. Diabetes 2012, 61, 292–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.J.; Nian, C.; Karunakaran, S.; Clee, S.M.; Isales, C.M.; McIntosh, C.H. GIP-overexpressing mice demonstrate reduced diet-induced obesity and steatosis, and improved glucose homeostasis. PLoS ONE 2012, 7, e40156. [Google Scholar] [CrossRef]
- Baggio, L.L.; Huang, Q.; Brown, T.J.; Drucker, D.J. Oxyntomodulin and glucagon-like peptide-1 differentially regulate murine food intake and energy expenditure. Gastroenterology 2004, 127, 546–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dakin, C.L.; Small, C.J.; Batterham, R.L.; Neary, N.M.; Cohen, M.A.; Patterson, M.; Ghatei, M.A.; Bloom, S.R. Peripheral oxyntomodulin reduces food intake and body weight gain in rats. Endocrinology 2004, 145, 2687–2695. [Google Scholar] [CrossRef] [Green Version]
- Scott, R.; Minnion, J.; Tan, T.; Bloom, S.R. Oxyntomodulin analogue increases energy expenditure via the glucagon receptor. Peptides 2018, 104, 70–77. [Google Scholar] [CrossRef]
- Wynne, K.; Park, A.J.; Small, C.J.; Meeran, K.; Ghatei, M.A.; Frost, G.S.; Bloom, S.R. Oxyntomodulin increases energy expenditure in addition to decreasing energy intake in overweight and obese humans: A randomised controlled trial. Int. J. Obes. 2006, 30, 1729–1736. [Google Scholar] [CrossRef] [Green Version]
- Finan, B.; Yang, B.; Ottaway, N.; Smiley, D.L.; Ma, T.; Clemmensen, C.; Chabenne, J.; Zhang, L.; Habegger, K.M.; Fischer, K.; et al. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat. Med. 2015, 21, 27–36. [Google Scholar] [CrossRef]
- Sun, Y.; Garcia, J.M.; Smith, R.G. Ghrelin and growth hormone secretagogue receptor expression in mice during aging. Endocrinology 2007, 148, 1323–1329. [Google Scholar] [CrossRef]
- Kaneko, K.; Yoshikawa, M.; Ohinata, K. Novel orexigenic pathway prostaglandin D2-NPY system—Involvement in orally active orexigenic delta opioid peptide. Neuropeptides 2012, 46, 353–357. [Google Scholar] [CrossRef]
- Sohn, J.W. Network of hypothalamic neurons that control appetite. BMB Rep. 2015, 48, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Ghigo, E.; Broglio, F.; Arvat, E.; Maccario, M.; Papotti, M.; Muccioli, G. Ghrelin: More than a natural GH secretagogue and/or an orexigenic factor. Clin. Endocrinol. 2005, 62, 1–17. [Google Scholar] [CrossRef]
- Pöykkö, S.M.; Kellokoski, E.; Hörkkö, S.; Kauma, H.; Kesäniemi, Y.A.; Ukkola, O. Low plasma ghrelin is associated with insulin resistance, hypertension, and the prevalence of type 2 diabetes. Diabetes 2003, 52, 2546–2553. [Google Scholar] [CrossRef]
- Korbonits, M.; Goldstone, A.P.; Gueorguiev, M.; Grossman, A.B. Ghrelin—A hormone with multiple functions. Front. Neuroendocrinol. 2004, 25, 27–68. [Google Scholar] [CrossRef]
- Zhao, T.J.; Sakata, I.; Li, R.L.; Liang, G.; Richardson, J.A.; Brown, M.S.; Goldstein, J.L.; Zigman, J.M. Ghrelin secretion stimulated by {beta}1-adrenergic receptors in cultured ghrelinoma cells and in fasted mice. Proc. Natl. Acad. Sci. USA 2010, 107, 15868–15873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagnon, J.; Baggio, L.L.; Drucker, D.J.; Brubaker, P.L. Ghrelin Is a Novel Regulator of GLP-1 Secretion. Diabetes 2015, 64, 1513–1521. [Google Scholar] [CrossRef] [Green Version]
- Tschöp, M.; Smiley, D.L.; Heiman, M.L. Ghrelin induces adiposity in rodents. Nature 2000, 407, 908–913. [Google Scholar] [CrossRef] [PubMed]
- Dixit, V.D.; Schaffer, E.M.; Pyle, R.S.; Collins, G.D.; Sakthivel, S.K.; Palaniappan, R.; Lillard, J.W., Jr.; Taub, D.D. Ghrelin inhibits leptin- and activation-induced proinflammatory cytokine expression by human monocytes and T cells. J. Clin. Investig. 2004, 114, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Li, W.G.; Gavrila, D.; Liu, X.; Wang, L.; Gunnlaugsson, S.; Stoll, L.L.; McCormick, M.L.; Sigmund, C.D.; Tang, C.; Weintraub, N.L. Ghrelin inhibits proinflammatory responses and nuclear factor-kappaB activation in human endothelial cells. Circulation 2004, 109, 2221–2226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldanzi, G.; Filigheddu, N.; Cutrupi, S.; Catapano, F.; Bonissoni, S.; Fubini, A.; Malan, D.; Baj, G.; Granata, R.; Broglio, F.; et al. Ghrelin and des-acyl ghrelin inhibit cell death in cardiomyocytes and endothelial cells through ERK1/2 and PI 3-kinase/AKT. J. Cell Biol. 2002, 159, 1029–1037. [Google Scholar] [CrossRef] [Green Version]
- Isgaard, J.; Granata, R. Ghrelin in cardiovascular disease and atherogenesis. Mol. Cell. Endocrinol. 2011, 340, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Virdis, A.; Duranti, E.; Colucci, R.; Ippolito, C.; Tirotta, E.; Lorenzini, G.; Bernardini, N.; Blandizzi, C.; Taddei, S. Ghrelin restores nitric oxide availability in resistance circulation of essential hypertensive patients: Role of NAD(P)H oxidase. Eur. Heart J. 2015, 36, 3023–3030. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.J.; Kosari, S.; Hunne, B.; Callaghan, B.; Rivera, L.R.; Bravo, D.M.; Furness, J.B. Differences in hormone localisation patterns of K and L type enteroendocrine cells in the mouse and pig small intestine and colon. Cell Tissue Res. 2015, 359, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Holzer, P.; Reichmann, F.; Farzi, A. Neuropeptide Y, peptide YY and pancreatic polypeptide in the gut-brain axis. Neuropeptides 2012, 46, 261–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greiner, T.U.; Backhed, F. Microbial regulation of GLP-1 and L-cell biology. Mol. Metab. 2016, 5, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Gribble, F.M.; Reimann, F. Enteroendocrine Cells: Chemosensors in the Intestinal Epithelium. Annu. Rev. Physiol. 2016, 78, 277–299. [Google Scholar] [CrossRef] [PubMed]
- Mentlein, R.; Dahms, P.; Grandt, D.; Krüger, R. Proteolytic processing of neuropeptide Y and peptide YY by dipeptidyl peptidase IV. Regul. Pept. 1993, 49, 133–144. [Google Scholar] [CrossRef]
- Le Roux, C.W.; Borg, C.M.; Murphy, K.G.; Vincent, R.P.; Ghatei, M.A.; Bloom, S.R. Supraphysiological doses of intravenous PYY3-36 cause nausea, but no additional reduction in food intake. Ann. Clin. Biochem. 2008, 45, 93–95. [Google Scholar] [CrossRef] [Green Version]
- Dirksen, C.; Damgaard, M.; Bojsen-Møller, K.N.; Jørgensen, N.B.; Kielgast, U.; Jacobsen, S.H.; Naver, L.S.; Worm, D.; Holst, J.J.; Madsbad, S.; et al. Fast pouch emptying, delayed small intestinal transit, and exaggerated gut hormone responses after Roux-en-Y gastric bypass. Neurogastroenterol. Motil. 2013, 25, 346-e255. [Google Scholar] [CrossRef]
- Pournaras, D.J.; Aasheim, E.T.; Bueter, M.; Ahmed, A.R.; Welbourn, R.; Olbers, T.; le Roux, C.W. Effect of bypassing the proximal gut on gut hormones involved with glycemic control and weight loss. Surg. Obes. Relat. Dis. 2012, 8, 371–374. [Google Scholar] [CrossRef] [Green Version]
- Adrian, T.E.; Sagor, G.R.; Savage, A.P.; Bacarese-Hamilton, A.J.; Hall, G.M.; Bloom, S.R. Peptide YY kinetics and effects on blood pressure and circulating pancreatic and gastrointestinal hormones and metabolites in man. J. Clin. Endocrinol. Metab. 1986, 63, 803–807. [Google Scholar] [CrossRef] [PubMed]
- Ahren, B.; Larsson, H. Peptide YY does not inhibit glucose-stimulated insulin secretion in humans. Eur. J. Endocrinol. 1996, 134, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Grosse, J.; Heffron, H.; Burling, K.; Akhter Hossain, M.; Habib, A.M.; Rogers, G.J.; Richards, P.; Larder, R.; Rimmington, D.; Adriaenssens, A.A.; et al. Insulin-like peptide 5 is an orexigenic gastrointestinal hormone. Proc. Natl. Acad. Sci. USA 2014, 111, 11133–11138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.S.; De Vadder, F.; Tremaroli, V.; Wichmann, A.; Mithieux, G.; Bäckhed, F. Insulin-like peptide 5 is a microbially regulated peptide that promotes hepatic glucose production. Mol. Metab. 2016, 5, 263–270. [Google Scholar] [CrossRef]
- Meng, A.H.; Ling, Y.L.; Zhang, X.P.; Zhang, J.L. Anti-inflammatory effect of cholecystokinin and its signal transduction mechanism in endotoxic shock rat. World J. Gastroenterol. 2002, 8, 712–717. [Google Scholar] [CrossRef] [PubMed]
- Lovick, T.A. CCK as a modulator of cardiovascular function. J. Chem. Neuroanat. 2009, 38, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Spreckley, E.; Murphy, K.G. The L-Cell in Nutritional Sensing and the Regulation of Appetite. Front. Nutr. 2015, 2, 23. [Google Scholar] [CrossRef] [Green Version]
- Verhoeckx, K.; Cotter, P.; López-Expósito, I.; Kleiveland, C.; Lea, T.; Mackie, A.; Requena, T.; Swiatecka, D.; Wichers, H. (Eds.) The Impact of Food Bioactives on Health: In vitro and Ex Vivo Models; Springer: Cham, Switzerland, 2015. [Google Scholar] [CrossRef]
- Noble, F.; Wank, S.A.; Crawley, J.N.; Bradwejn, J.; Seroogy, K.B.; Hamon, M.; Roques, B.P. International Union of Pharmacology. XXI. Structure, distribution, and functions of cholecystokinin receptors. Pharmacol. Rev. 1999, 51, 745–781. [Google Scholar]
- Lavine, J.A.; Raess, P.W.; Stapleton, D.S.; Rabaglia, M.E.; Suhonen, J.I.; Schueler, K.L.; Koltes, J.E.; Dawson, J.A.; Yandell, B.S.; Samuelson, L.C.; et al. Cholecystokinin is up-regulated in obese mouse islets and expands beta-cell mass by increasing beta-cell survival. Endocrinology 2010, 151, 3577–3588. [Google Scholar] [CrossRef]
- Sanger, G.J.; Lee, K. Hormones of the gut-brain axis as targets for the treatment of upper gastrointestinal disorders. Nat. Rev. Drug Discov. 2008, 7, 241–254. [Google Scholar] [CrossRef]
- Pathak, V.; Flatt, P.R.; Irwin, N. Cholecystokinin (CCK) and related adjunct peptide therapies for the treatment of obesity and type 2 diabetes. Peptides 2018, 100, 229–235. [Google Scholar] [CrossRef]
- Bäckhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef] [Green Version]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Noce, A.; Marrone, G.; Di Daniele, F.; Ottaviani, E.; Wilson Jones, G.; Bernini, R.; Romani, A.; Rovella, V. Impact of Gut Microbiota Composition on Onset and Progression of Chronic Non-Communicable Diseases. Nutrients 2019, 11, 1073. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.H.; Hazen, S.L. The Gut Microbiome and Its Role in Cardiovascular Diseases. Circulation 2017, 135, 1008–1010. [Google Scholar] [CrossRef]
- Reinhardt, C.; Bergentall, M.; Greiner, T.U.; Schaffner, F.; Ostergren-Lunden, G.; Petersen, L.C.; Ruf, W.; Backhed, F. Tissue factor and PAR1 promote microbiota-induced intestinal vascular remodelling. Nature 2012, 483, 627–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Annalisa, N.; Alessio, T.; Claudette, T.D.; Erald, V.; Antonino de, L.; Nicola, D.D. Gut microbioma population: An indicator really sensible to any change in age, diet, metabolic syndrome, and life-style. Mediat. Inflamm. 2014, 2014, 901308. [Google Scholar] [CrossRef]
- Dumas, M.E.; Barton, R.H.; Toye, A.; Cloarec, O.; Blancher, C.; Rothwell, A.; Fearnside, J.; Tatoud, R.; Blanc, V.; Lindon, J.C.; et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc. Natl. Acad. Sci. USA 2006, 103, 12511–12516. [Google Scholar] [CrossRef] [Green Version]
- Belzer, C.; de Vos, W.M. Microbes inside—From diversity to function: The case of Akkermansia. ISME J. 2012, 6, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Kitai, T.; Hazen, S.L. Gut Microbiota in Cardiovascular Health and Disease. Circ. Res. 2017, 120, 1183–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.T.; Parajuli, N.; Sung, M.M.; Bairwa, S.C.; Levasseur, J.; Soltys, C.M.; Wishart, D.S.; Madsen, K.; Schertzer, J.D.; Dyck, J.R.B. Fecal transplant from resveratrol-fed donors improves glycaemia and cardiovascular features of the metabolic syndrome in mice. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E511–E519. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [Green Version]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Furet, J.P.; Kong, L.C.; Tap, J.; Poitou, C.; Basdevant, A.; Bouillot, J.L.; Mariat, D.; Corthier, G.; Doré, J.; Henegar, C.; et al. Differential adaptation of human gut microbiota to bariatric surgery-induced weight loss: Links with metabolic and low-grade inflammation markers. Diabetes 2010, 59, 3049–3057. [Google Scholar] [CrossRef] [Green Version]
- Kong, L.C.; Tap, J.; Aron-Wisnewsky, J.; Pelloux, V.; Basdevant, A.; Bouillot, J.L.; Zucker, J.D.; Doré, J.; Clément, K. Gut microbiota after gastric bypass in human obesity: Increased richness and associations of bacterial genera with adipose tissue genes. Am. J. Clin. Nutr. 2013, 98, 16–24. [Google Scholar] [CrossRef] [Green Version]
- Finucane, M.M.; Sharpton, T.J.; Laurent, T.J.; Pollard, K.S. A taxonomic signature of obesity in the microbiome? Getting to the guts of the matter. PLoS ONE 2014, 9, e84689. [Google Scholar] [CrossRef] [Green Version]
- Scott, K.P.; Gratz, S.W.; Sheridan, P.O.; Flint, H.J.; Duncan, S.H. The influence of diet on the gut microbiota. Pharmacol. Res. 2013, 69, 52–60. [Google Scholar] [CrossRef]
- Zhernakova, A.; Kurilshikov, A.; Bonder, M.J.; Tigchelaar, E.F.; Schirmer, M.; Vatanen, T.; Mujagic, Z.; Vila, A.V.; Falony, G.; Vieira-Silva, S.; et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 2016, 352, 565–569. [Google Scholar] [CrossRef] [Green Version]
- Mozaffarian, D.; Hao, T.; Rimm, E.B.; Willett, W.C.; Hu, F.B. Changes in diet and lifestyle and long-term weight gain in women and men. N. Engl. J. Med. 2011, 364, 2392–2404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Filippis, F.; Pellegrini, N.; Vannini, L.; Jeffery, I.B.; La Storia, A.; Laghi, L.; Serrazanetti, D.I.; Di Cagno, R.; Ferrocino, I.; Lazzi, C.; et al. High-level adherence to a Mediterranean diet beneficially impacts the gut microbiota and associated metabolome. Gut 2016, 65, 1812–1821. [Google Scholar] [CrossRef] [PubMed]
- Høverstad, T.; Midtvedt, T. Short-chain fatty acids in germfree mice and rats. J. Nutr. 1986, 116, 1772–1776. [Google Scholar] [CrossRef]
- Rabot, S.; Membrez, M.; Bruneau, A.; Gérard, P.; Harach, T.; Moser, M.; Raymond, F.; Mansourian, R.; Chou, C.J. Germ-free C57BL/6J mice are resistant to high-fat-diet-induced insulin resistance and have altered cholesterol metabolism. FASEB J. 2010, 24, 4948–4959. [Google Scholar] [CrossRef] [Green Version]
- Peng, L.; He, Z.; Chen, W.; Holzman, I.R.; Lin, J. Effects of butyrate on intestinal barrier function in a Caco-2 cell monolayer model of intestinal barrier. Pediatr. Res. 2007, 61, 37–41. [Google Scholar] [CrossRef] [Green Version]
- Holmes, E.; Li, J.V.; Marchesi, J.R.; Nicholson, J.K. Gut microbiota composition and activity in relation to host metabolic phenotype and disease risk. Cell Metab. 2012, 16, 559–564. [Google Scholar] [CrossRef] [Green Version]
- Den Besten, G.; Lange, K.; Havinga, R.; van Dijk, T.H.; Gerding, A.; van Eunen, K.; Müller, M.; Groen, A.K.; Hooiveld, G.J.; Bakker, B.M.; et al. Gut-derived short-chain fatty acids are vividly assimilated into host carbohydrates and lipids. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G900–G910. [Google Scholar] [CrossRef] [PubMed]
- De Vadder, F.; Kovatcheva-Datchary, P.; Goncalves, D.; Vinera, J.; Zitoun, C.; Duchampt, A.; Bäckhed, F.; Mithieux, G. Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell 2014, 156, 84–96. [Google Scholar] [CrossRef] [Green Version]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Houten, S.M.; Mataki, C.; Christoffolete, M.A.; Kim, B.W.; Sato, H.; Messaddeq, N.; Harney, J.W.; Ezaki, O.; Kodama, T.; et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 2006, 439, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Saha, P.K.; Chan, L.; Moore, D.D. Farnesoid X receptor is essential for normal glucose homeostasis. J. Clin. Investig. 2006, 116, 1102–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Lee, F.Y.; Barrera, G.; Lee, H.; Vales, C.; Gonzalez, F.J.; Willson, T.M.; Edwards, P.A. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc. Natl. Acad. Sci. USA 2006, 103, 1006–1011. [Google Scholar] [CrossRef] [Green Version]
- Duboc, H.; Taché, Y.; Hofmann, A.F. The bile acid TGR5 membrane receptor: From basic research to clinical application. Dig. Liver Dis. 2014, 46, 302–312. [Google Scholar] [CrossRef] [Green Version]
- Velazquez-Villegas, L.A.; Perino, A.; Lemos, V.; Zietak, M.; Nomura, M.; Pols, T.W.H.; Schoonjans, K. TGR5 signalling promotes mitochondrial fission and beige remodelling of white adipose tissue. Nat. Commun. 2018, 9, 245. [Google Scholar] [CrossRef] [Green Version]
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M.; et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009, 10, 167–177. [Google Scholar] [CrossRef] [Green Version]
- Ryan, K.K.; Tremaroli, V.; Clemmensen, C.; Kovatcheva-Datchary, P.; Myronovych, A.; Karns, R.; Wilson-Pérez, H.E.; Sandoval, D.A.; Kohli, R.; Bäckhed, F.; et al. FXR is a molecular target for the effects of vertical sleeve gastrectomy. Nature 2014, 509, 183–188. [Google Scholar] [CrossRef] [Green Version]
- Liou, A.P.; Paziuk, M.; Luevano, J.M., Jr.; Machineni, S.; Turnbaugh, P.J.; Kaplan, L.M. Conserved shifts in the gut microbiota due to gastric bypass reduce host weight and adiposity. Sci. Transl. Med. 2013, 5, 178ra141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchwald, H.; Oien, D.M. Metabolic/bariatric surgery Worldwide 2008. Obes. Surg. 2009, 19, 1605–1611. [Google Scholar] [CrossRef] [PubMed]
- Robciuc, M.R.; Naukkarinen, J.; Ortega-Alonso, A.; Tyynismaa, H.; Raivio, T.; Rissanen, A.; Kaprio, J.; Ehnholm, C.; Jauhiainen, M.; Pietiläinen, K.H. Serum angiopoietin-like 4 protein levels and expression in adipose tissue are inversely correlated with obesity in monozygotic twins. J. Lipid Res. 2011, 52, 1575–1582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bäckhed, F.; Manchester, J.K.; Semenkovich, C.F.; Gordon, J.I. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. USA 2007, 104, 979–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, G.X.; Chen, G.R.; Xu, H.; Ge, R.S.; Lin, J. Activation of the AMP activated protein kinase by short-chain fatty acids is the main mechanism underlying the beneficial effect of a high fiber diet on the metabolic syndrome. Med. Hypotheses 2010, 74, 123–126. [Google Scholar] [CrossRef]
- Carvalho, B.M.; Guadagnini, D.; Tsukumo, D.M.L.; Schenka, A.A.; Latuf-Filho, P.; Vassallo, J.; Dias, J.C.; Kubota, L.T.; Carvalheira, J.B.C.; Saad, M.J.A. Modulation of gut microbiota by antibiotics improves insulin signalling in high-fat fed mice. Diabetologia 2012, 55, 2823–2834. [Google Scholar] [CrossRef]
- Hardie, D.G.; Carling, D. The AMP-activated protein kinase—Fuel gauge of the mammalian cell? Eur. J. Biochem. 1997, 246, 259–273. [Google Scholar] [CrossRef]
- Cryan, J.F.; Dinan, T.G. Mind-altering microorganisms: The impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci 2012, 13, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Breton, J.; Tennoune, N.; Lucas, N.; Francois, M.; Legrand, R.; Jacquemot, J.; Goichon, A.; Guérin, C.; Peltier, J.; Pestel-Caron, M.; et al. Gut Commensal, E. coli Proteins Activate Host Satiety Pathways following Nutrient-Induced Bacterial Growth. Cell Metab 2016, 23, 324–334. [Google Scholar] [CrossRef] [Green Version]
- Fetissov, S.O. Role of the gut microbiota in host appetite control: Bacterial growth to animal feeding behaviour. Nat. Rev. Endocrinol 2017, 13, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Martchenko, S.E.; Martchenko, A.; Cox, B.J.; Naismith, K.; Waller, A.; Gurges, P.; Sweeney, M.E.; Philpott, D.J.; Brubaker, P.L. Circadian GLP-1 Secretion in Mice Is Dependent on the Intestinal Microbiome for Maintenance of Diurnal Metabolic Homeostasis. Diabetes 2020, 69, 2589–2602. [Google Scholar] [CrossRef] [PubMed]
- Wick, L.M.; Quadroni, M.; Egli, T. Short- and long-term changes in proteome composition and kinetic properties in a culture of Escherichia coli during transition from glucose-excess to glucose-limited growth conditions in continuous culture and vice versa. Environ. Microbiol. 2001, 3, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Breton, J.; Legrand, R.; Akkermann, K.; Järv, A.; Harro, J.; Déchelotte, P.; Fetissov, S.O. Elevated plasma concentrations of bacterial ClpB protein in patients with eating disorders. Int. J. Eat. Disord. 2016, 49, 805–808. [Google Scholar] [CrossRef]
- Wichmann, A.; Allahyar, A.; Greiner, T.U.; Plovier, H.; Lunden, G.O.; Larsson, T.; Drucker, D.J.; Delzenne, N.M.; Cani, P.D.; Backhed, F. Microbial modulation of energy availability in the colon regulates intestinal transit. Cell Host Microbe 2013, 14, 582–590. [Google Scholar] [CrossRef] [Green Version]
- Gribble, F.M.; Reimann, F. Function and mechanisms of enteroendocrine cells and gut hormones in metabolism. Nat. Rev. Endocrinol 2019, 15, 226–237. [Google Scholar] [CrossRef]
- Liu, X.; Xu, X.; Shang, R.; Chen, Y. Asymmetric dimethylarginine (ADMA) as an important risk factor for the increased cardiovascular diseases and heart failure in chronic kidney disease. Nitric Oxide 2018, 78, 113–120. [Google Scholar] [CrossRef]
- Ohira, H.; Tsutsui, W.; Fujioka, Y. Are Short Chain Fatty Acids in Gut Microbiota Defensive Players for Inflammation and Atherosclerosis? J. Atheroscler. Thromb. 2017, 24, 660–672. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, N.; Hori, D.; Flavahan, S.; Steppan, J.; Flavahan, N.A.; Berkowitz, D.E.; Pluznick, J.L. Microbial short chain fatty acid metabolites lower blood pressure via endothelial G protein-coupled receptor 41. Physiol. Genom. 2016, 48, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Pluznick, J.L. Renal and cardiovascular sensory receptors and blood pressure regulation. Am. J. Physiol. Ren. Physiol. 2013, 305, F439–F444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluznick, J.L.; Protzko, R.J.; Gevorgyan, H.; Peterlin, Z.; Sipos, A.; Han, J.; Brunet, I.; Wan, L.X.; Rey, F.; Wang, T.; et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc. Natl. Acad. Sci. USA 2013, 110, 4410–4415. [Google Scholar] [CrossRef] [Green Version]
- Palazzo, M.; Balsari, A.; Rossini, A.; Selleri, S.; Calcaterra, C.; Gariboldi, S.; Zanobbio, L.; Arnaboldi, F.; Shirai, Y.F.; Serrao, G.; et al. Activation of enteroendocrine cells via TLRs induces hormone, chemokine, and defensin secretion. J. Immunol. 2007, 178, 4296–4303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.M.; Hazen, S.L. The gut microbial endocrine organ: Bacterially derived signals driving cardiometabolic diseases. Annu. Rev. Med. 2015, 66, 343–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [Green Version]
- Caesar, R.; Reigstad, C.S.; Backhed, H.K.; Reinhardt, C.; Ketonen, M.; Lunden, G.O.; Cani, P.D.; Backhed, F. Gut-derived lipopolysaccharide augments adipose macrophage accumulation but is not essential for impaired glucose or insulin tolerance in mice. Gut 2012, 61, 1701–1707. [Google Scholar] [CrossRef] [Green Version]
- Lackey, D.E.; Olefsky, J.M. Regulation of metabolism by the innate immune system. Nat. Rev. Endocrinol. 2016, 12, 15–28. [Google Scholar] [CrossRef]
- Liang, H.; Hussey, S.E.; Sanchez-Avila, A.; Tantiwong, P.; Musi, N. Effect of lipopolysaccharide on inflammation and insulin action in human muscle. PLoS ONE 2013, 8, e63983. [Google Scholar] [CrossRef]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Neal, M.D.; Leaphart, C.; Levy, R.; Prince, J.; Billiar, T.R.; Watkins, S.; Li, J.; Cetin, S.; Ford, H.; Schreiber, A.; et al. Enterocyte TLR4 mediates phagocytosis and translocation of bacteria across the intestinal barrier. J. Immunol. 2006, 176, 3070–3079. [Google Scholar] [CrossRef]
- Amar, J.; Chabo, C.; Waget, A.; Klopp, P.; Vachoux, C.; Bermúdez-Humarán, L.G.; Smirnova, N.; Bergé, M.; Sulpice, T.; Lahtinen, S.; et al. Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: Molecular mechanisms and probiotic treatment. EMBO Mol. Med. 2011, 3, 559–572. [Google Scholar] [CrossRef]
- Cani, P.D.; Possemiers, S.; Van de Wiele, T.; Guiot, Y.; Everard, A.; Rottier, O.; Geurts, L.; Naslain, D.; Neyrinck, A.; Lambert, D.M.; et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 2009, 58, 1091–1103. [Google Scholar] [CrossRef] [Green Version]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.H.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karbach, S.H.; Schonfelder, T.; Brandao, I.; Wilms, E.; Hormann, N.; Jackel, S.; Schuler, R.; Finger, S.; Knorr, M.; Lagrange, J.; et al. Gut Microbiota Promote Angiotensin II-Induced Arterial Hypertension and Vascular Dysfunction. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Nallu, A.; Sharma, S.; Ramezani, A.; Muralidharan, J.; Raj, D. Gut microbiome in chronic kidney disease: Challenges and opportunities. Transl. Res. 2017, 179, 24–37. [Google Scholar] [CrossRef] [Green Version]
- Pereira-Fantini, P.M.; Byars, S.G.; Pitt, J.; Lapthorne, S.; Fouhy, F.; Cotter, P.D.; Bines, J.E. Unravelling the metabolic impact of SBS-associated microbial dysbiosis: Insights from the piglet short bowel syndrome model. Sci. Rep. 2017, 7, 43326. [Google Scholar] [CrossRef] [PubMed]
- Webster, L.T.; Siddiqui, U.A.; Lucas, S.V.; Strong, J.M.; Mieyal, J.J. Identification of separate acyl-CoA:glycine and acyl-CoA:L-glutamine N-acyltransferase activities in mitochondrial fractions from liver of rhesus monkey and man. J. Biol. Chem. 1976, 251, 3352–3358. [Google Scholar] [CrossRef]
- Amedei, A.; Morbidelli, L. Circulating Metabolites Originating from Gut Microbiota Control Endothelial Cell Function. Molecules 2019, 24, 3992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pålsson, J.; Ricksten, S.E.; Delle, M.; Lundin, S. Changes in renal sympathetic nerve activity during experimental septic and endotoxin shock in conscious rats. Circ. Shock 1988, 24, 133–141. [Google Scholar] [PubMed]
- Santisteban, M.M.; Qi, Y.; Zubcevic, J.; Kim, S.; Yang, T.; Shenoy, V.; Cole-Jeffrey, C.T.; Lobaton, G.O.; Stewart, D.C.; Rubiano, A.; et al. Hypertension-Linked Pathophysiological Alterations in the Gut. Circ. Res. 2017, 120, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Kiouptsi, K.; Pontarollo, G.; Todorov, H.; Braun, J.; Jackel, S.; Koeck, T.; Bayer, F.; Karwot, C.; Karpi, A.; Gerber, S.; et al. Germ-free housing conditions do not affect aortic root and aortic arch lesion size of late atherosclerotic low-density lipoprotein receptor-deficient mice. Gut Microbes 2020, 11, 1809–1823. [Google Scholar] [CrossRef] [PubMed]
- Kiouptsi, K.; Jackel, S.; Pontarollo, G.; Grill, A.; Kuijpers, M.J.E.; Wilms, E.; Weber, C.; Sommer, F.; Nagy, M.; Neideck, C.; et al. The Microbiota Promotes Arterial Thrombosis in Low-Density Lipoprotein Receptor-Deficient Mice. mBio 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Niebauer, J.; Volk, H.D.; Kemp, M.; Dominguez, M.; Schumann, R.R.; Rauchhaus, M.; Poole-Wilson, P.A.; Coats, A.J.; Anker, S.D. Endotoxin and immune activation in chronic heart failure: A prospective cohort study. Lancet 1999, 353, 1838–1842. [Google Scholar] [CrossRef]
- Sandek, A.; Swidsinski, A.; Schroedl, W.; Watson, A.; Valentova, M.; Herrmann, R.; Scherbakov, N.; Cramer, L.; Rauchhaus, M.; Grosse-Herrenthey, A.; et al. Intestinal blood flow in patients with chronic heart failure: A link with bacterial growth, gastrointestinal symptoms, and cachexia. J. Am. Coll. Cardiol. 2014, 64, 1092–1102. [Google Scholar] [CrossRef] [Green Version]
- Dockray, G.J. Gastrointestinal hormones and the dialogue between gut and brain. J. Physiol. 2014, 592, 2927–2941. [Google Scholar] [CrossRef]
- Guarino, D.; Nannipieri, M.; Iervasi, G.; Taddei, S.; Bruno, R.M. The Role of the Autonomic Nervous System in the Pathophysiology of Obesity. Front. Physiol. 2017, 8, 665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthoud, H.R. The vagus nerve, food intake and obesity. Regul. Pept. 2008, 149, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Owyang, C. Endogenous cholecystokinin stimulates pancreatic enzyme secretion via vagal afferent pathway in rats. Gastroenterology 1994, 107, 525–531. [Google Scholar] [CrossRef]
- Alvarez, G.E.; Ballard, T.P.; Beske, S.D.; Davy, K.P. Subcutaneous obesity is not associated with sympathetic neural activation. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H414–H418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esler, M.; Straznicky, N.; Eikelis, N.; Masuo, K.; Lambert, G.; Lambert, E. Mechanisms of sympathetic activation in obesity-related hypertension. Hypertension 2006, 48, 787–796. [Google Scholar] [CrossRef] [Green Version]
- Grassi, G.; Seravalle, G.; Cattaneo, B.M.; Bolla, G.B.; Lanfranchi, A.; Colombo, M.; Giannattasio, C.; Brunani, A.; Cavagnini, F.; Mancia, G. Sympathetic activation in obese normotensive subjects. Hypertension 1995, 25, 560–563. [Google Scholar] [CrossRef] [PubMed]
- Landsberg, L. Diet, obesity and hypertension: An hypothesis involving insulin, the sympathetic nervous system, and adaptive thermogenesis. Q. J. Med. 1986, 61, 1081–1090. [Google Scholar]
- Kaelberer, M.M.; Buchanan, K.L.; Klein, M.E.; Barth, B.B.; Montoya, M.M.; Shen, X.; Bohórquez, D.V. A gut-brain neural circuit for nutrient sensory transduction. Science 2018, 361. [Google Scholar] [CrossRef] [Green Version]
- Grasset, E.; Burcelin, R. The gut microbiota to the brain axis in the metabolic control. Rev. Endocr. Metab. Disord. 2019, 20, 427–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, C.R.; Osadchiy, V.; Kalani, A.; Mayer, E.A. The Brain-Gut-Microbiome Axis. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 133–148. [Google Scholar] [CrossRef] [Green Version]
- Bravo, J.A.; Forsythe, P.; Chew, M.V.; Escaravage, E.; Savignac, H.M.; Dinan, T.G.; Bienenstock, J.; Cryan, J.F. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 16050–16055. [Google Scholar] [CrossRef] [Green Version]
- Samuel, B.S.; Shaito, A.; Motoike, T.; Rey, F.E.; Backhed, F.; Manchester, J.K.; Hammer, R.E.; Williams, S.C.; Crowley, J.; Yanagisawa, M.; et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc. Natl. Acad. Sci. USA 2008, 105, 16767–16772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolhurst, G.; Heffron, H.; Lam, Y.S.; Parker, H.E.; Habib, A.M.; Diakogiannaki, E.; Cameron, J.; Grosse, J.; Reimann, F.; Gribble, F.M. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 2012, 61, 364–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef] [Green Version]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell 2015, 161, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Wahlström, A.; Sayin, S.I.; Marschall, H.U.; Bäckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab 2016, 24, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Hsuchou, H.; Pan, W.; Kastin, A.J. Fibroblast growth factor 19 entry into brain. Fluids Barriers CNS 2013, 10, 32. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; John, L.M.; Adams, S.H.; Yu, X.X.; Tomlinson, E.; Renz, M.; Williams, P.M.; Soriano, R.; Corpuz, R.; Moffat, B.; et al. Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology 2004, 145, 2594–2603. [Google Scholar] [CrossRef] [Green Version]
- Marcelin, G.; Jo, Y.H.; Li, X.; Schwartz, G.J.; Zhang, Y.; Dun, N.J.; Lyu, R.M.; Blouet, C.; Chang, J.K.; Chua, S., Jr. Central action of FGF19 reduces hypothalamic AGRP/NPY neuron activity and improves glucose metabolism. Mol. Metab. 2014, 3, 19–28. [Google Scholar] [CrossRef]
- Morton, G.J.; Matsen, M.E.; Bracy, D.P.; Meek, T.H.; Nguyen, H.T.; Stefanovski, D.; Bergman, R.N.; Wasserman, D.H.; Schwartz, M.W. FGF19 action in the brain induces insulin-independent glucose lowering. J. Clin. Investig. 2013, 123, 4799–4808. [Google Scholar] [CrossRef] [Green Version]
- Ryan, K.K.; Kohli, R.; Gutierrez-Aguilar, R.; Gaitonde, S.G.; Woods, S.C.; Seeley, R.J. Fibroblast growth factor-19 action in the brain reduces food intake and body weight and improves glucose tolerance in male rats. Endocrinology 2013, 154, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Bohórquez, D.V.; Liddle, R.A. The gut connectome: Making sense of what you eat. J. Clin. Investig. 2015, 125, 888–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barajon, I.; Serrao, G.; Arnaboldi, F.; Opizzi, E.; Ripamonti, G.; Balsari, A.; Rumio, C. Toll-like receptors 3, 4, and 7 are expressed in the enteric nervous system and dorsal root ganglia. J. Histochem. Cytochem. 2009, 57, 1013–1023. [Google Scholar] [CrossRef]
- Haghikia, A.; Jörg, S.; Duscha, A.; Berg, J.; Manzel, A.; Waschbisch, A.; Hammer, A.; Lee, D.H.; May, C.; Wilck, N.; et al. Dietary Fatty Acids Directly Impact Central Nervous System Autoimmunity via the Small Intestine. Immunity 2015, 43, 817–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Wang, J.; Hu, W.; Wang, C.; Lu, X.; Tong, L.; Wu, F.; Zhang, W. Identification of functional farnesoid X receptors in brain neurons. FEBS Lett. 2016, 590, 3233–3242. [Google Scholar] [CrossRef]
- Keitel, V.; Görg, B.; Bidmon, H.J.; Zemtsova, I.; Spomer, L.; Zilles, K.; Häussinger, D. The bile acid receptor TGR5 (Gpbar-1) acts as a neurosteroid receptor in brain. Glia 2010, 58, 1794–1805. [Google Scholar] [CrossRef]
- Kimura, I.; Inoue, D.; Maeda, T.; Hara, T.; Ichimura, A.; Miyauchi, S.; Kobayashi, M.; Hirasawa, A.; Tsujimoto, G. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41). Proc. Natl. Acad. Sci. USA 2011, 108, 8030–8035. [Google Scholar] [CrossRef] [Green Version]
- Asano, Y.; Hiramoto, T.; Nishino, R.; Aiba, Y.; Kimura, T.; Yoshihara, K.; Koga, Y.; Sudo, N. Critical role of gut microbiota in the production of biologically active, free catecholamines in the gut lumen of mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G1288–G1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, E.; Ross, R.P.; O’Toole, P.W.; Fitzgerald, G.F.; Stanton, C. γ-Aminobutyric acid production by culturable bacteria from the human intestine. J. Appl. Microbiol. 2012, 113, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Özogul, F. Effects of specific lactic acid bacteria species on biogenic amine production by foodborne pathogen. Int. J. Food Sci. Technol. 2011, 46, 478–484. [Google Scholar] [CrossRef]
- Shishov, V.A.; Kirovskaia, T.A.; Kudrin, V.S.; Oleskin, A.V. Amine neuromediators, their precursors, and oxidation products in the culture of Escherichia coli K-12. Prikl. Biokhim. Mikrobiol. 2009, 45, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Tóth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 6, 263ra158. [Google Scholar] [CrossRef] [Green Version]
- Van Felius, I.D.; Akkermans, L.M.; Bosscha, K.; Verheem, A.; Harmsen, W.; Visser, M.R.; Gooszen, H.G. Interdigestive small bowel motility and duodenal bacterial overgrowth in experimental acute pancreatitis. Neurogastroenterol. Motil. 2003, 15, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Vandeputte, D.; Falony, G.; Vieira-Silva, S.; Tito, R.Y.; Joossens, M.; Raes, J. Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut 2016, 65, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Meddings, J.B.; Swain, M.G. Environmental stress-induced gastrointestinal permeability is mediated by endogenous glucocorticoids in the rat. Gastroenterology 2000, 119, 1019–1028. [Google Scholar] [CrossRef]
- Saunders, P.R.; Santos, J.; Hanssen, N.P.; Yates, D.; Groot, J.A.; Perdue, M.H. Physical and psychological stress in rats enhances colonic epithelial permeability via peripheral CRH. Dig. Dis Sci. 2002, 47, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Ho, S.B. Intestinal goblet cells and mucins in health and disease: Recent insights and progress. Curr. Gastroenterol. Rep. 2010, 12, 319–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubio, C.A.; Huang, C.B. Quantification of the sulphomucin-producing cell population of the colonic mucosa during protracted stress in rats. In vivo 1992, 6, 81–84. [Google Scholar]
- Mayer, E.A. Gut feelings: The emerging biology of gut-brain communication. Nat. Rev. Neurosci. 2011, 12, 453–466. [Google Scholar] [CrossRef]
- Lyte, M. The role of microbial endocrinology in infectious disease. J. Endocrinol. 1993, 137, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Mayer, E.A.; Savidge, T.; Shulman, R.J. Brain-gut microbiome interactions and functional bowel disorders. Gastroenterology 2014, 146, 1500–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alverdy, J.; Holbrook, C.; Rocha, F.; Seiden, L.; Wu, R.L.; Musch, M.; Chang, E.; Ohman, D.; Suh, S. Gut-derived sepsis occurs when the right pathogen with the right virulence genes meets the right host: Evidence for in vivo virulence expression in Pseudomonas aeruginosa. Ann. Surg. 2000, 232, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.B.; Hughes, D.T.; Zhu, C.; Boedeker, E.C.; Sperandio, V. The QseC sensor kinase: A bacterial adrenergic receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 10420–10425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cogan, T.A.; Thomas, A.O.; Rees, L.E.; Taylor, A.H.; Jepson, M.A.; Williams, P.H.; Ketley, J.; Humphrey, T.J. Norepinephrine increases the pathogenic potential of Campylobacter jejuni. Gut 2007, 56, 1060–1065. [Google Scholar] [CrossRef] [Green Version]
- Hughes, D.T.; Sperandio, V. Inter-kingdom signalling: Communication between bacteria and their hosts. Nat. Rev. Microbiol. 2008, 6, 111–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyte, M. The role of catecholamines in gram-negative sepsis. Med. Hypotheses 1992, 37, 255–258. [Google Scholar] [CrossRef]
- Booth, A.; Magnuson, A.; Fouts, J.; Foster, M.T. Adipose tissue: An endocrine organ playing a role in metabolic regulation. Horm. Mol. Biol. Clin. Investig. 2016, 26, 25–42. [Google Scholar] [CrossRef]




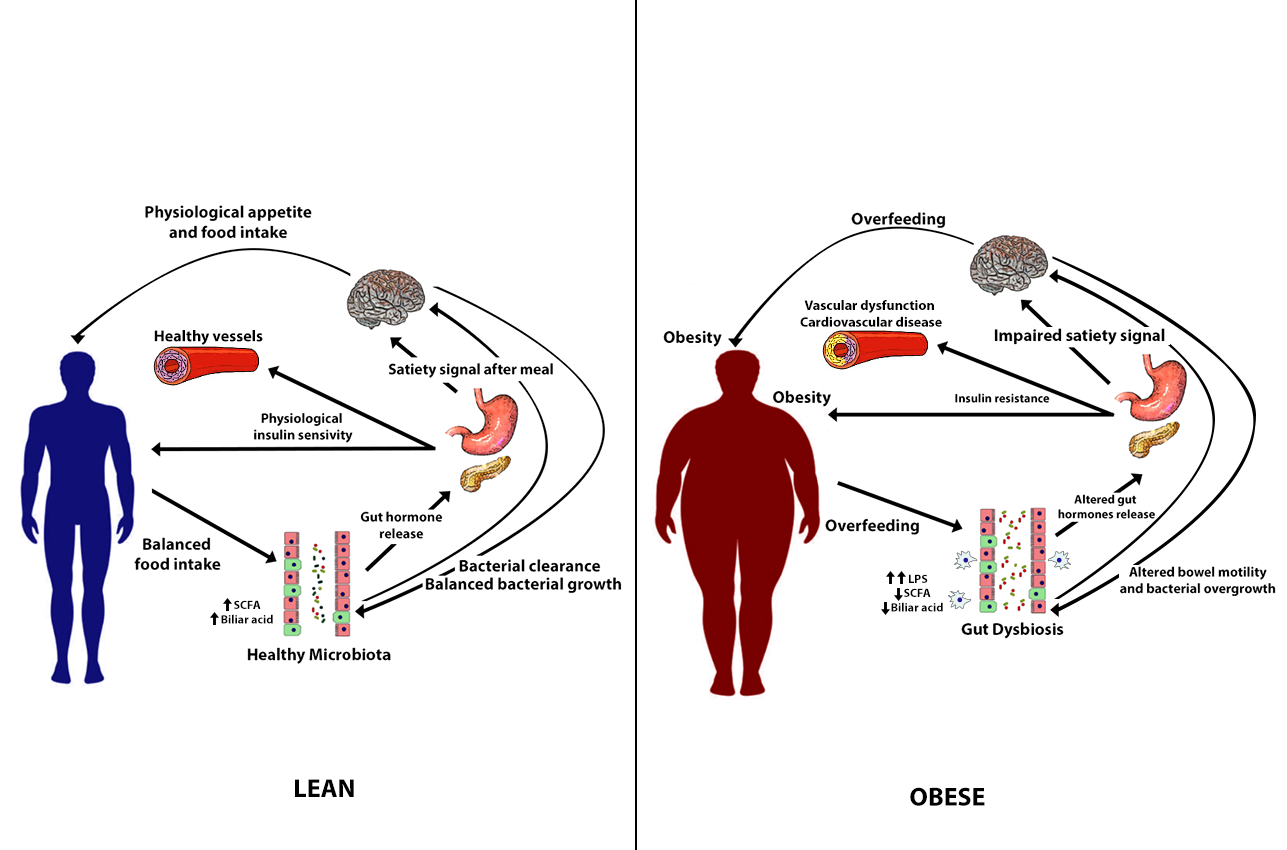{kind=link}
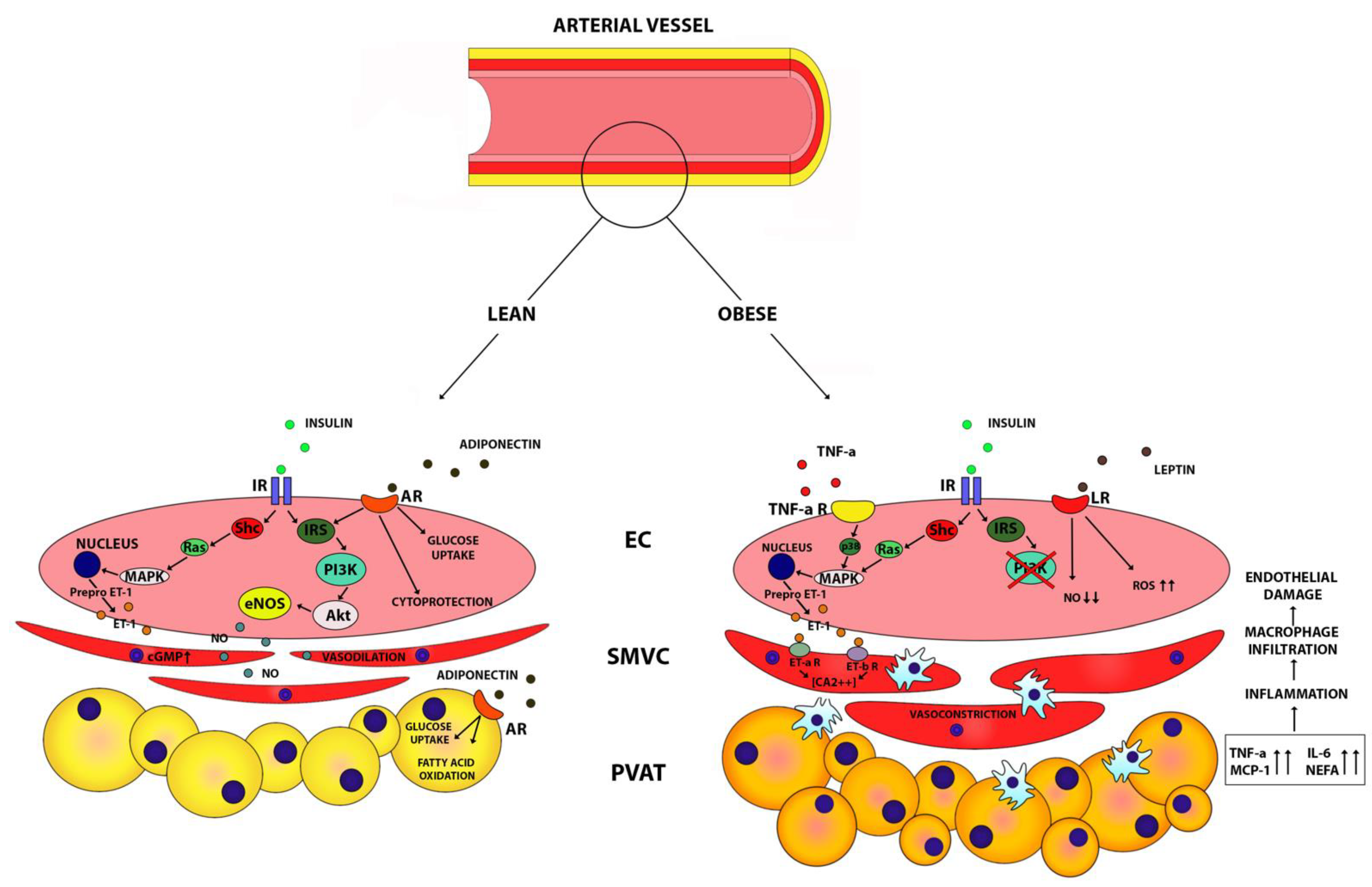{kind=link}
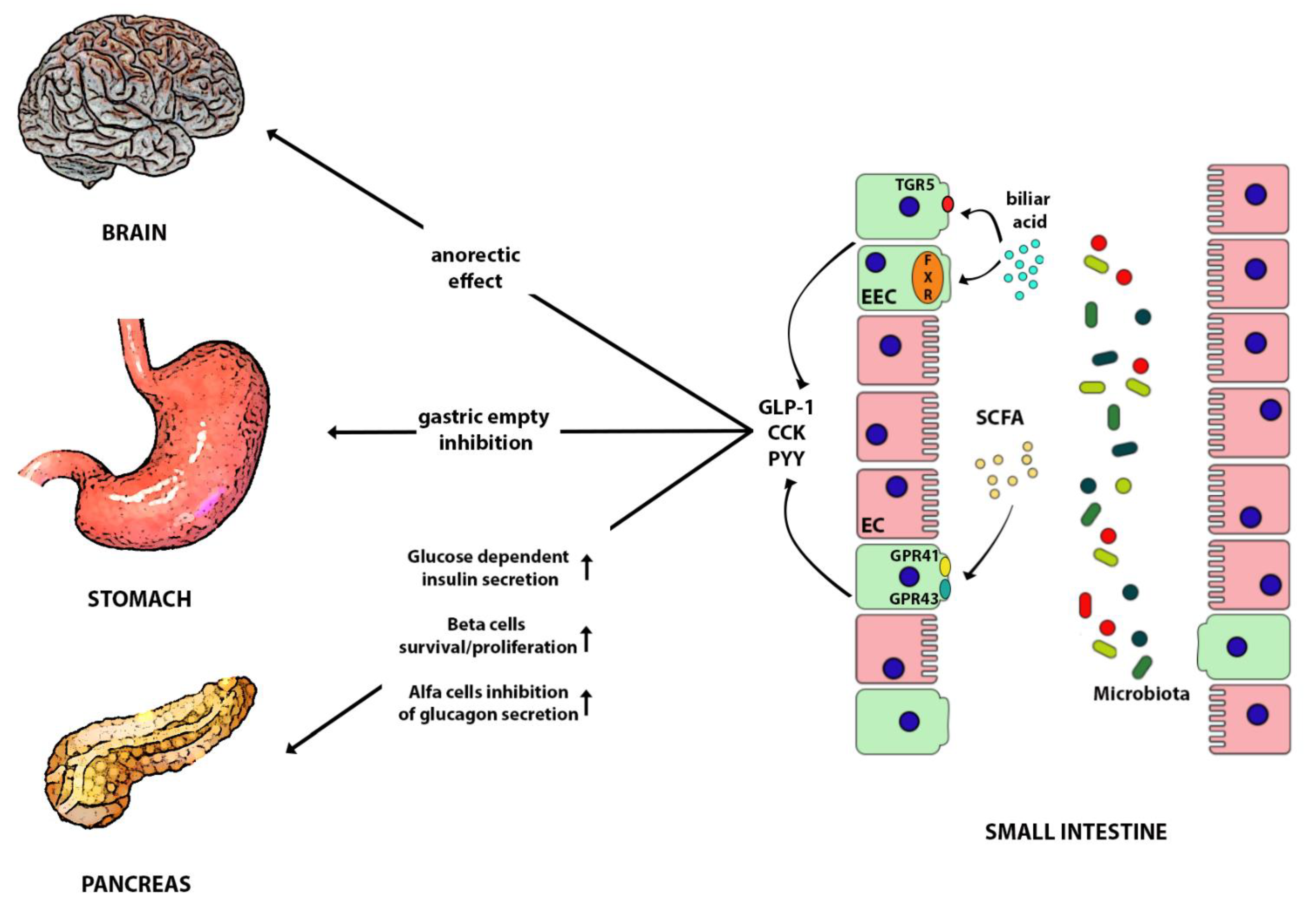{kind=link}
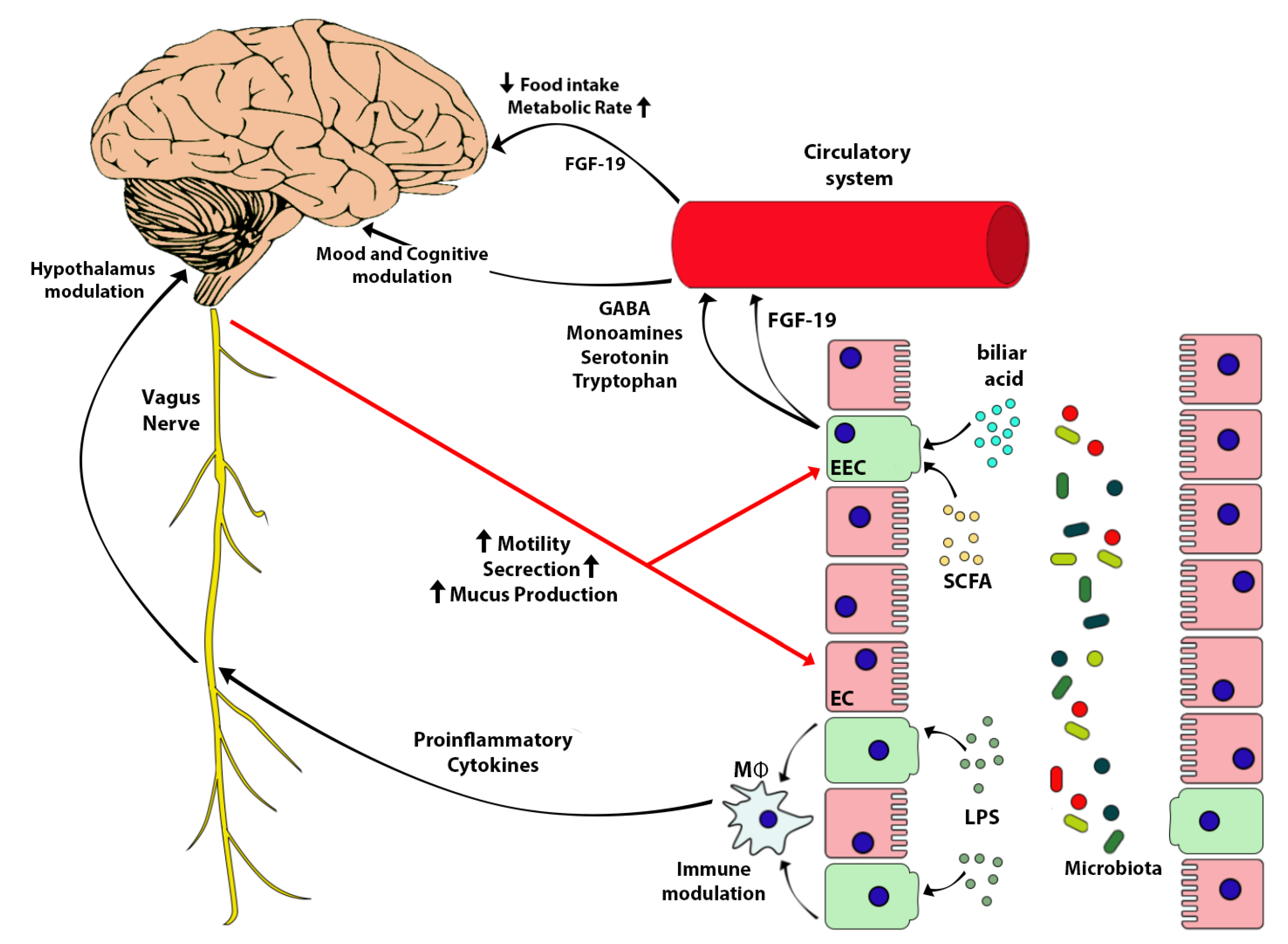{kind=link}
| Adipokines | Site of Release | Stimulated by | Function | Reference |
|---|---|---|---|---|
| Adiponectin | PVAT | Plasma concentration is inversely correlated with BMI and visceral fat | ↑ insulin-mediated glucose uptake in skeletal muscle ↑ liver insulin sensitivity ↑ endothelium vasoactivity ↑ glucose production by gluconeogenesis | [52,53,54,55] |
| Leptin | PVAT | Plasma levels ↑ with fat stores | Regulator of energy intake Adaptation to fasting ↑ oxidative cellular stress by long exposure ↓ NO endothelial availability by long exposure | [58,59,60,61] |
| Gut Hormones | Site of release | Stimulated by | Function | Reference |
|---|---|---|---|---|
| GLP-1 | Intestine L cells | Secreted after meal | ↑ insulin secretion ↑ pancreatic beta cells survival and proliferation ↑ pancreatic alfa cells inhibition of glucagon secretion ↓ lipid secretion ↓ glucose production by liver ↓ gastric motility ↑ sense of satiety ↓ food ingestion ↓ oxidative stress/platelet aggregation ↑ insulin stimulated vasodilator reactivity | [106,107] |
| GIP | Intestine K cells | Secreted after meal | ↑ insulin secretion ↑ beta cells proliferation ↓ beta cell apoptosis | [107] |
| OMX | Intestine L cells | Secreted after meal | ↑ insulin secretion ↓ food ingestion | [108,109,110] |
| Ghrelin | Gastric P/D1 cells | Secreted during fasting | ↑ food ingestion ↑ GLP-1 secretory ↑ NO endothelial availability | [111,112,113] |
| Obestatin | Gastric P/D1 cells | Secreted during fasting | ↑ food ingestion ↑ NO endothelial availability ↑ pancreatic beta cells survival and proliferation ↓ lipolysis in adipose tissue | [114,115,116,117,118] |
| PYY | Intestine L cells | Secreted after meal | ↓ gastric motility/↓ intestinal motility ↑ sense of satiety ↑ pancreatic secretion | [119,120,121,122] |
| INSL-5 | Intestine L cells | Secreted during fasting | ↑ food ingestion | [123,124] |
| CKK | Intestine I cells | Secreted after meal | ↑ gallbladder contraction inhibition of gastric acid secretion ↑ insulin secretion ↑ pancreatic beta cells survival and proliferation ↑ sense of satiety | [95,125,126,127,128,129] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rovella, V.; Rodia, G.; Di Daniele, F.; Cardillo, C.; Campia, U.; Noce, A.; Candi, E.; Della-Morte, D.; Tesauro, M. Association of Gut Hormones and Microbiota with Vascular Dysfunction in Obesity. Nutrients 2021, 13, 613. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13020613
Rovella V, Rodia G, Di Daniele F, Cardillo C, Campia U, Noce A, Candi E, Della-Morte D, Tesauro M. Association of Gut Hormones and Microbiota with Vascular Dysfunction in Obesity. Nutrients. 2021; 13(2):613. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13020613
Chicago/Turabian StyleRovella, Valentina, Giuseppe Rodia, Francesca Di Daniele, Carmine Cardillo, Umberto Campia, Annalisa Noce, Eleonora Candi, David Della-Morte, and Manfredi Tesauro. 2021. "Association of Gut Hormones and Microbiota with Vascular Dysfunction in Obesity" Nutrients 13, no. 2: 613. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13020613