
Advanced Glycation End Products (AGEs) and Chronic Kidney Disease: Does the Modern Diet AGE the Kidney?


Abstract
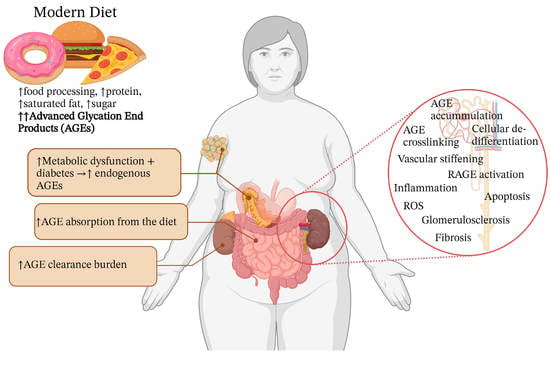
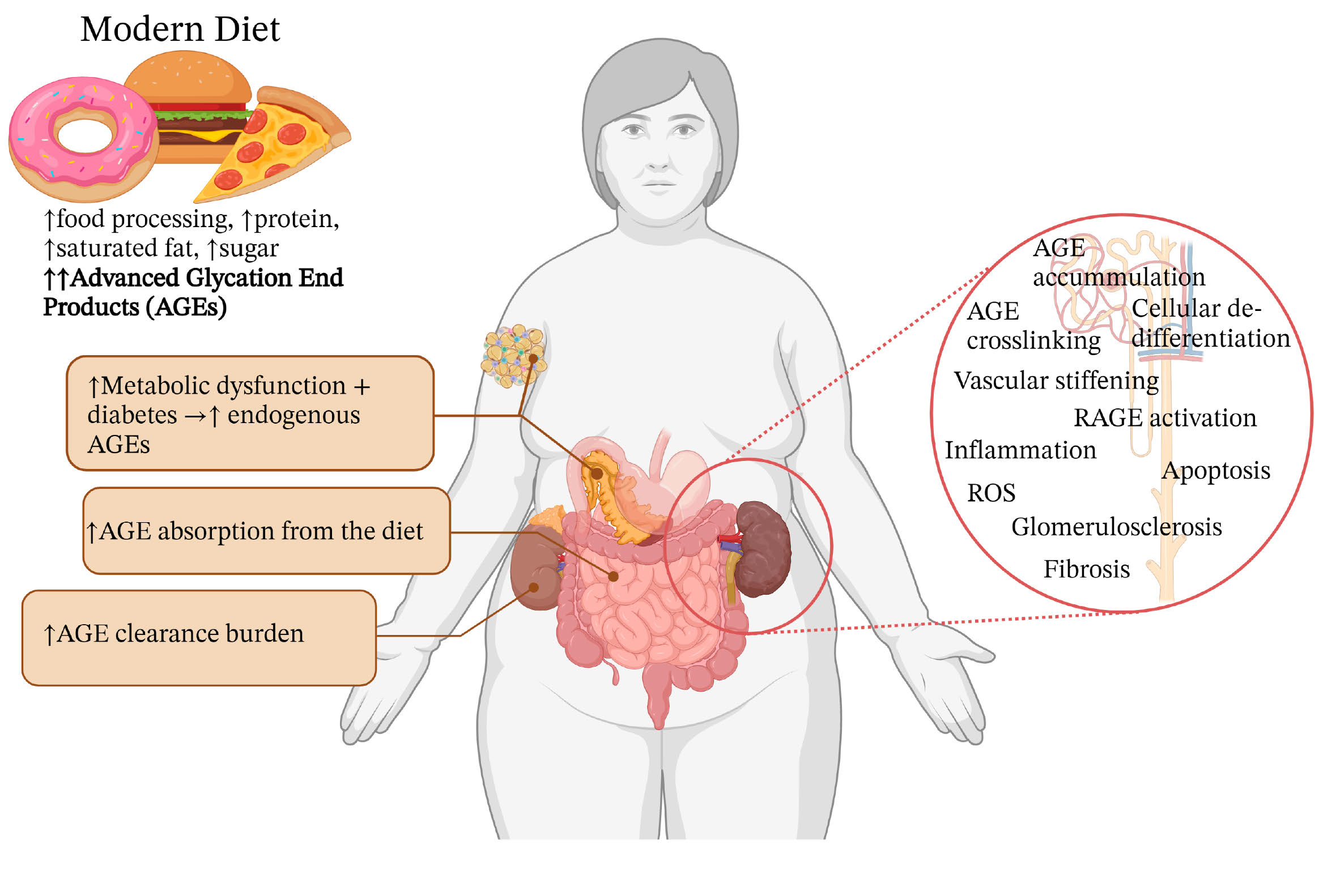
:
1. The Western Diet as a Risk Factor for Kidney Disease
2. AGE Chemistry
3. Factors Regulating AGE Accumulation and Turnover in the Body
3.1. Endogenous AGE Production—Beyond the Maillard Reaction
3.2. Exogenous AGE Sources
3.2.1. Quantifying AGEs in Commonly Consumed Foods
3.2.2. Absorption of Dietary AGEs
3.3. AGE Clearance
4. AGE Receptors—Facilitators of Clearance and Mediators of Pathology
4.1. RAGE
4.2. AGER1
5. AGE-Mediated Pathology
5.1. AGEs Can Induce Both Receptor Mediated and Non-Receptor Mediated Pathology
5.2. Dietary AGEs and AGE Pathology
5.3. Dietary AGEs and the Microbiome
6. AGEs and Kidney Disease
6.1. Mechanisms by Which AGEs Damage the Kidney
6.2. Renal Handling of AGEs
6.3. Exogenous AGEs and Kidney Function
6.4. Therapeutic Targeting of AGEs in Kidney Disease
6.4.1. Carbonyl Scavengers
6.4.2. Vitamin B and Its Derivatives
6.4.3. AGE Cross Link Breakers
6.4.4. RAGE Blockade
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Bikbov, B.; Purcell, C.A.; Levey, A.S.; Smith, M.; Abdoli, A.; Abebe, M.; Adebayo, O.M.; Afarideh, M.; Agarwal, S.K.; Agudelo-Botero, M.; et al. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef] [Green Version]
- Gur, E.; Levy, D.; Topaz, G.; Naser, R.; Wand, O.; Kitay-Cohen, Y.; Benchetrit, S.; Sarel, E.; Cohen-Hagai, K. Disease severity and renal outcomes of patients with chronic kidney disease infected with COVID-19. Clin. Exp. Nephrol. 2022, 26, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Williamson, E.J.; Walker, A.J.; Bhaskaran, K.; Bacon, S.; Bates, C.; Morton, C.E.; Curtis, H.J.; Mehrkar, A.; Evans, D.; Inglesby, P.; et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 2020, 584, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.-M.; Yang, C.-W. Chronic kidney disease: Global dimension and perspectives. Lancet 2013, 382, 260–272. [Google Scholar] [CrossRef]
- Cordain, L.; Eaton, S.B.; Sebastian, A.; Mann, N.; Lindeberg, S.; Watkins, B.A.; O’Keefe, J.H.; Brand-Miller, J. Origins and evolution of the Western diet: Health implications for the 21st century. Am. J. Clin. Nutr. 2005, 81, 341–354. [Google Scholar] [CrossRef]
- Odermatt, A. The Western-style diet: A major risk factor for impaired kidney function and chronic kidney disease. Am. J. Physiol.-Ren. Physiol. 2011, 301, F919–F931. [Google Scholar] [CrossRef] [Green Version]
- Hariharan, D.; Vellanki, K.; Kramer, H. The Western Diet and Chronic Kidney Disease. Curr. Hypertens. Rep. 2015, 17, 16. [Google Scholar] [CrossRef]
- Wang, L.; Martínez Steele, E.; Du, M.; Pomeranz, J.L.; O’Connor, L.E.; Herrick, K.A.; Luo, H.; Zhang, X.; Mozaffarian, D.; Zhang, F.F. Trends in Consumption of Ultraprocessed Foods Among US Youths Aged 2–19 Years, 1999–2018. JAMA 2021, 326, 519–530. [Google Scholar] [CrossRef]
- Fiolet, T.; Srour, B.; Sellem, L.; Kesse-Guyot, E.; Allès, B.; Méjean, C.; Deschasaux, M.; Fassier, P.; Latino-Martel, P.; Beslay, M.; et al. Consumption of ultra-processed foods and cancer risk: Results from NutriNet-Santé prospective cohort. BMJ 2018, 360, k322. [Google Scholar] [CrossRef] [Green Version]
- Srour, B.; Fezeu, L.K.; Kesse-Guyot, E.; Allès, B.; Debras, C.; Druesne-Pecollo, N.; Chazelas, E.; Deschasaux, M.; Hercberg, S.; Galan, P.; et al. Ultraprocessed Food Consumption and Risk of Type 2 Diabetes Among Participants of the NutriNet-Santé Prospective Cohort. JAMA Intern. Med. 2020, 180, 283–291. [Google Scholar] [CrossRef]
- Suksatan, W.; Moradi, S.; Naeini, F.; Bagheri, R.; Mohammadi, H.; Talebi, S.; Mehrabani, S.; Hojjati Kermani, M.A.; Suzuki, K. Ultra-Processed Food Consumption and Adult Mortality Risk: A Systematic Review and Dose–Response Meta-Analysis of 207,291 Participants. Nutrients 2022, 14, 174. [Google Scholar] [CrossRef]
- Srour, B.; Fezeu, L.K.; Kesse-Guyot, E.; Allès, B.; Méjean, C.; Andrianasolo, R.M.; Chazelas, E.; Deschasaux, M.; Hercberg, S.; Galan, P.; et al. Ultra-processed food intake and risk of cardiovascular disease: Prospective cohort study (NutriNet-Santé). BMJ 2019, 365, l1451. [Google Scholar] [CrossRef] [Green Version]
- Schnabel, L.; Kesse-Guyot, E.; Allès, B.; Touvier, M.; Srour, B.; Hercberg, S.; Buscail, C.; Julia, C. Association Between Ultraprocessed Food Consumption and Risk of Mortality Among Middle-aged Adults in France. JAMA Intern. Med. 2019, 179, 490–498. [Google Scholar] [CrossRef]
- Rico-Campà, A.; Martínez-González, M.A.; Alvarez-Alvarez, I.; Mendonça, R.D.D.; de la Fuente-Arrillaga, C.; Gómez-Donoso, C.; Bes-Rastrollo, M. Association between consumption of ultra-processed foods and all cause mortality: SUN prospective cohort study. BMJ 2019, 365, l1949. [Google Scholar] [CrossRef] [Green Version]
- Bonaccio, M.; Costanzo, S.; Ruggiero, E.; Persichillo, M.; Esposito, S.; Olivieri, M.; Di Castelnuovo, A.; Cerletti, C.; Donati, M.B.; de Gaetano, G.; et al. Changes in ultra-processed food consumption during the first Italian lockdown following the COVID-19 pandemic and major correlates: Results from two population-based cohorts. Public Health Nutr. 2021, 24, 3905–3915. [Google Scholar] [CrossRef]
- Ruíz-Roso, M.B.; de Carvalho Padilha, P.; Matilla-Escalante, D.C.; Brun, P.; Ulloa, N.; Acevedo-Correa, D.; Arantes Ferreira Peres, W.; Martorell, M.; Rangel Bousquet Carrilho, T.; de Oliveira Cardoso, L.; et al. Changes of Physical Activity and Ultra-Processed Food Consumption in Adolescents from Different Countries during COVID-19 Pandemic: An Observational Study. Nutrients 2020, 12, 2289. [Google Scholar] [CrossRef]
- Poulsen, M.W.; Hedegaard, R.V.; Andersen, J.M.; de Courten, B.; Bugel, S.; Nielsen, J.; Skibsted, L.H.; Dragsted, L.O. Advanced glycation endproducts in food and their effects on health. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2013, 60, 10–37. [Google Scholar] [CrossRef]
- Liang, Z.; Chen, X.; Yang, Z.; Liu, Y.; Qiu, X.; Zeng, Z.; Lu, S.; Liu, Y. Sodium Ions Affect Pyrraline Formation in the Maillard Reaction with Lys-Containing Dipeptides and Tripeptides. Front. Nutr. 2022, 9, 874650. [Google Scholar] [CrossRef]
- Forbes, J.M.; Cooper, M.E. Mechanisms of diabetic complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef]
- Koska, J.; Gerstein, H.C.; Beisswenger, P.J.; Reaven, P.D. Advanced Glycation End Products Predict Loss of Renal Function and High-Risk Chronic Kidney Disease in Type 2 Diabetes. Diabetes Care 2022, 45, 684–691. [Google Scholar] [CrossRef]
- Saulnier, P.-J.; Wheelock, K.M.; Howell, S.; Weil, E.J.; Tanamas, S.K.; Knowler, W.C.; Lemley, K.V.; Mauer, M.; Yee, B.; Nelson, R.G.; et al. Advanced Glycation End Products Predict Loss of Renal Function and Correlate with Lesions of Diabetic Kidney Disease in American Indians with Type 2 Diabetes. Diabetes 2016, 65, 3744–3753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semba, R.D.; Ferrucci, L.; Sun, K.; Beck, J.; Dalal, M.; Varadhan, R.; Walston, J.; Guralnik, J.M.; Fried, L.P. Advanced glycation end products and their circulating receptors predict cardiovascular disease mortality in older community-dwelling women. Aging Clin. Exp. Res. 2009, 21, 182–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semba, R.D.; Bandinelli, S.; Sun, K.; Guralnik, J.M.; Ferrucci, L. Plasma Carboxymethyl-Lysine, an Advanced Glycation End Product, and All-Cause and Cardiovascular Disease Mortality in Older Community-Dwelling Adults. J. Am. Geriatr. Soc. 2009, 57, 1874–1880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nin, J.W.; Jorsal, A.; Ferreira, I.; Schalkwijk, C.G.; Prins, M.H.; Parving, H.-H.; Tarnow, L.; Rossing, P.; Stehouwer, C.D. Higher Plasma Levels of Advanced Glycation End Products Are Associated with Incident Cardiovascular Disease and All-Cause Mortality in Type 1 Diabetes: A 12-year follow-up study. Diabetes Care 2011, 34, 442–447. [Google Scholar] [CrossRef] [Green Version]
- Machowska, A.; Sun, J.; Qureshi, A.R.; Isoyama, N.; Leurs, P.; Anderstam, B.; Heimburger, O.; Barany, P.; Stenvinkel, P.; Lindholm, B. Plasma Pentosidine and Its Association with Mortality in Patients with Chronic Kidney Disease. PLoS ONE 2016, 11, e0163826. [Google Scholar] [CrossRef] [Green Version]
- Ulrich, P.; Cerami, A. Protein glycation, diabetes, and aging. Recent Prog. Horm. Res. 2001, 56, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Thorpe, S.R.; Baynes, J.W. Maillard reaction products in tissue proteins: New products and new perspectives. Amino Acids 2003, 25, 275–281. [Google Scholar] [CrossRef]
- Thornalley, P.J. Dicarbonyl Intermediates in the Maillard Reaction. Ann. N. Y. Acad. Sci. 2005, 1043, 111–117. [Google Scholar] [CrossRef]
- Skovsted, I.C.; Christensen, M.; Breinholt, J.; Mortensen, S.B. Characterisation of a novel AGE-compound derived from lysine and 3-deoxyglucosone. Cell. Mol. Biol. 1998, 44, 1159–1163. [Google Scholar]
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced glycation end-products: A review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef] [Green Version]
- Wells-Knecht, K.J.; Brinkmann, E.; Wells-Knecht, M.C.; Litchfield, J.E.; Ahmed, M.U.; Reddy, S.; Zyzak, D.V.; Thorpe, S.R.; Baynes, J.W. New biomarkers of Maillard reaction damage to proteins. Nephrol. Dial. Transpl. 1996, 11 (Suppl. 5), 41–47. [Google Scholar] [CrossRef]
- Uribarri, J.; Tuttle, K.R. Advanced glycation end products and nephrotoxicity of high-protein diets. Clin. J. Am. Soc. Nephrol. 2006, 1, 1293–1299. [Google Scholar] [CrossRef]
- Lorenzi, M. The polyol pathway as a mechanism for diabetic retinopathy: Attractive, elusive, and resilient. J. Diabetes Res. 2007, 2007, 061038. [Google Scholar] [CrossRef]
- Thornalley, P.J. Pharmacology of methylglyoxal: Formation, modification of proteins and nucleic acids, and enzymatic detoxification--a role in pathogenesis and antiproliferative chemotherapy. Gen. Pharmacol. 1996, 27, 565–573. [Google Scholar] [CrossRef]
- Thornalley, P.; Langborg, A.; Minhas, H. Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem. J. 1999, 344, 109–116. [Google Scholar] [CrossRef]
- Baynes, J.W.; Thorpe, S.R. Role of oxidative stress in diabetic complications: A new perspective on an old paradigm. Diabetes 1999, 48, 1–9. [Google Scholar] [CrossRef]
- Monnier, V.M.; Bautista, O.; Kenny, D.; Sell, D.R.; Fogarty, J.; Dahms, W.; Cleary, P.A.; Lachin, J.; Genuth, S. Skin collagen glycation, glycoxidation, and crosslinking are lower in subjects with long-term intensive versus conventional therapy of type 1 diabetes: Relevance of glycated collagen products versus HbA1c as markers of diabetic complications. DCCT Skin Collagen Ancillary Study Group. Diabetes Control and Complications Trial. Diabetes 1999, 48, 870–880. [Google Scholar] [CrossRef]
- Genuth, S.; Sun, W.; Cleary, P.; Gao, X.; Sell, D.R.; Lachin, J.; Monnier, V.M. Skin advanced glycation end products glucosepane and methylglyoxal hydroimidazolone are independently associated with long-term microvascular complication progression of type 1 diabetes. Diabetes 2015, 64, 266–278. [Google Scholar] [CrossRef] [Green Version]
- den Dekker, M.A.M.; Zwiers, M.; van den Heuvel, E.R.; de Vos, L.C.; Smit, A.J.; Zeebregts, C.J.; Oudkerk, M.; Vliegenthart, R.; Lefrandt, J.D.; Mulder, D.J. Skin autofluorescence, a non-invasive marker for AGE accumulation, is associated with the degree of atherosclerosis. PLoS ONE 2013, 8, e83084. [Google Scholar] [CrossRef]
- Atzeni, I.M.; van de Zande, S.C.; Westra, J.; Zwerver, J.; Smit, A.J.; Mulder, D.J. The AGE Reader: A non-invasive method to assess long-term tissue damage. Methods 2021, 203, 533–541. [Google Scholar] [CrossRef]
- Gugliucci, A.; Menini, T. Circulating advanced glycation peptides in streptozotocin-induced diabetic rats: Evidence for preferential modification of IgG light chains. Life Sci. 1998, 62, 2141–2150. [Google Scholar] [CrossRef]
- Miyata, T.; Wada, Y.; Cai, Z.; Iida, Y.; Horie, K.; Yasuda, Y.; Maeda, K.; Kurokawa, K.; van Ypersele de Strihou, C. Implication of an increased oxidative stress in the formation of advanced glycation end products in patients with end-stage renal failure. Kidney Int. 1997, 51, 1170–1181. [Google Scholar] [CrossRef] [Green Version]
- Wagner, Z.; Wittmann, I.; Mazak, I.; Schinzel, R.; Heidland, A.; Kientsch-Engel, R.; Nagy, J. N(epsilon)-(carboxymethyl)lysine levels in patients with type 2 diabetes: Role of renal function. Am. J. Kidney Dis. 2001, 38, 785–791. [Google Scholar] [CrossRef]
- Makita, Z.; Bucala, R.; Rayfield, E.J.; Friedman, E.A.; Kaufman, A.M.; Korbet, S.M.; Barth, R.H.; Winston, J.A.; Fuh, H.; Manogue, K.R.; et al. Reactive glycosylation endproducts in diabetic uraemia and treatment of renal failure. Lancet 1994, 343, 1519–1522. [Google Scholar] [CrossRef]
- Thomas, M.C.; Tsalamandris, C.; MacIsaac, R.; Medley, T.; Kingwell, B.; Cooper, M.E.; Jerums, G. Low-molecular-weight AGEs are associated with GFR and anemia in patients with type 2 diabetes. Kidney Int. 2004, 66, 1167–1172. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, J.; Morrissey, P.A. Nutritional and toxicological aspects of the Maillard browning reaction in foods. Crit. Rev. Food Sci. Nutr. 1989, 28, 211–248. [Google Scholar] [CrossRef]
- Goldberg, T.; Cai, W.; Peppa, M.; Dardaine, V.; Baliga, B.S.; Uribarri, J.; Vlassara, H. Advanced glycoxidation end products in commonly consumed foods. J. Am. Diet. Assoc. 2004, 104, 1287–1291. [Google Scholar] [CrossRef]
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced Glycation End Products in Foods and a Practical Guide to Their Reduction in the Diet. J. Am. Diet. Assoc. 2010, 110, 911–916.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, K.; Dhar, I.; Caspar-Bell, G. Role of Advanced Glycation End Products and Its Receptors in the Pathogenesis of Cigarette Smoke-Induced Cardiovascular Disease. Int. J. Angiol. 2015, 24, 75–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, M.; Takino, J.-I.; Furuno, S.; Shirai, H.; Kawakami, M.; Muramatsu, M.; Kobayashi, Y.; Yamagishi, S.-i. Assessment of the Concentrations of Various Advanced Glycation End-Products in Beverages and Foods That Are Commonly Consumed in Japan. PLoS ONE 2015, 10, e0118652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hull, G.L.J.; Woodside, J.V.; Ames, J.M.; Cuskelly, G.J. Nε-(carboxymethyl)lysine content of foods commonly consumed in a Western style diet. Food Chem. 2012, 131, 170–174. [Google Scholar] [CrossRef]
- Scheijen, J.L.J.M.; Clevers, E.; Engelen, L.; Dagnelie, P.C.; Brouns, F.; Stehouwer, C.D.A.; Schalkwijk, C.G. Analysis of advanced glycation endproducts in selected food items by ultra-performance liquid chromatography tandem mass spectrometry: Presentation of a dietary AGE database. Food Chem. 2016, 190, 1145–1150. [Google Scholar] [CrossRef]
- Snelson, M.; Coughlan, M.T. Dietary Advanced Glycation End Products: Digestion, Metabolism and Modulation of Gut Microbial Ecology. Nutrients 2019, 11, 215. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Sabol, J.; Mitsuhashi, T.; Vlassara, H. Dietary glycotoxins: Inhibition of reactive products by aminoguanidine facilitates renal clearance and reduces tissue sequestration. Diabetes 1999, 48, 1308–1315. [Google Scholar] [CrossRef]
- Koschinsky, T.; He, C.J.; Mitsuhashi, T.; Bucala, R.; Liu, C.; Buenting, C.; Heitmann, K.; Vlassara, H. Orally absorbed reactive glycation products (glycotoxins): An environmental risk factor in diabetic nephropathy. Proc. Natl. Acad. Sci. USA 1997, 94, 6474–6479. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Andrade, C.; Tessier, F.J.; Niquet-Leridon, C.; Seiquer, I.; Pilar Navarro, M. Study of the urinary and faecal excretion of Nepsilon-carboxymethyllysine in young human volunteers. Amino Acids 2012, 43, 595–602. [Google Scholar] [CrossRef]
- Sebekova, K.; Saavedra, G.; Zumpe, C.; Somoza, V.; Klenovicsova, K.; Birlouez-Aragon, I. Plasma concentration and urinary excretion of N epsilon-(carboxymethyl)lysine in breast milk- and formula-fed infants. Ann. N. Y. Acad. Sci. 2008, 1126, 177–180. [Google Scholar] [CrossRef]
- Faist, V.; Erbersdobler, H.F. Metabolic transit and in vivo effects of melanoidins and precursor compounds deriving from the Maillard reaction. Ann. Nutr. Metab. 2000, 45, 1–12. [Google Scholar] [CrossRef]
- Forster, A.; Kuhne, Y.; Henle, T. Studies on absorption and elimination of dietary maillard reaction products. Ann. N. Y. Acad. Sci. 2005, 1043, 474–481. [Google Scholar] [CrossRef]
- Hultsch, C.; Hellwig, M.; Pawelke, B.; Bergmann, R.; Rode, K.; Pietzsch, J.; Krause, R.; Henle, T. Biodistribution and catabolism of 18F-labeled N-ε-fructoselysine as a model of Amadori products. Nucl. Med. Biol. 2006, 33, 865–873. [Google Scholar] [CrossRef]
- Schwenger, V.; Morath, C.; Schönfelder, K.; Klein, W.; Weigel, K.; Deppisch, R.; Henle, T.; Ritz, E.; Zeier, M. An oral load of the early glycation compound lactuloselysine fails to accumulate in the serum of uraemic patients. Nephrol. Dial. Transpl. 2006, 21, 383–388. [Google Scholar] [CrossRef] [Green Version]
- Liardon, R.; De Weck-Gaudard, D.; Philippossian, G.; Finot, P.A. Identification of N.epsilon.-carboxymethyllysine: A new Maillard reaction product in rat urine. J. Agric. Food Chem. 1987, 35, 427–431. [Google Scholar] [CrossRef]
- Miyata, T.; Ueda, Y.; Horie, K.; Nangaku, M.; Tanaka, S.; de Strihou, C.V.Y.; Kurokawa, K. Renal catabolism of advanced glycation end products: The fate of pentosidine. Kidney Int. 1998, 53, 416–422. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, R.; Helling, R.; Heichert, C.; Scheunemann, M.; Mading, P.; Wittrisch, H.; Johannsen, B.; Henle, T. Radio fluorination and positron emission tomography (PET) as a new approach to study the in vivo distribution and elimination of the advanced glycation endproducts N epsilon-carboxymethyllysine (CML) and N epsilon-carboxyethyllysine (CEL). Nahrung 2001, 45, 182–188. [Google Scholar] [CrossRef]
- Somoza, V.; Wenzel, E.; Weiß, C.; Clawin-Rädecker, I.; Grübel, N.; Erbersdobler, H.F. Dose-dependent utilisation of casein-linked lysinoalanine, N (epsilon)-fructoselysine and N (epsilon)-carboxymethyllysine in rats. Mol. Nutr. Food Res. 2006, 50, 833–841. [Google Scholar] [CrossRef]
- Roncero-Ramos, I.; Delgado-Andrade, C.; Tessier, F.J.; Niquet-Leridon, C.; Strauch, C.; Monnier, V.M.; Navarro, M.P. Metabolic transit of N(epsilon)-carboxymethyl-lysine after consumption of AGEs from bread crust. Food Funct. 2013, 4, 1032–1039. [Google Scholar] [CrossRef]
- Alamir, I.; Niquet-Leridon, C.; Jacolot, P.; Rodriguez, C.; Orosco, M.; Anton, P.M.; Tessier, F.J. Digestibility of extruded proteins and metabolic transit of N epsilon -carboxymethyllysine in rats. Amino Acids 2013, 44, 1441–1449. [Google Scholar] [CrossRef]
- Xu, H.; Wang, Z.; Wang, Y.; Hu, S.; Liu, N. Biodistribution and elimination study of fluorine-18 labeled Nepsilon-carboxymethyl-lysine following intragastric and intravenous administration. PLoS ONE 2013, 8, e57897. [Google Scholar] [CrossRef] [Green Version]
- Tessier, F.J.; Niquet-Leridon, C.; Jacolot, P.; Jouquand, C.; Genin, M.; Schmidt, A.M.; Grossin, N.; Boulanger, E. Quantitative assessment of organ distribution of dietary protein-bound (13) C-labeled N(varepsilon) -carboxymethyllysine after a chronic oral exposure in mice. Mol. Nutr. Food Res. 2016, 60, 2446–2456. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, A.; Ogura, A.; Tahara, T.; Nozaki, S.; Urano, S.; Hara, M.; Kojima, S.; Kurbangalieva, A.; Onoe, H.; Watanabe, Y.; et al. In vivo imaging of advanced glycation end products (AGEs) of albumin: First observations of significantly reduced clearance and liver deposition properties in mice. Org. Biomol. Chem. 2016, 14, 5755–5760. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H. The AGE-receptor in the pathogenesis of diabetic complications. Diabetes/Metab. Res. Rev. 2001, 17, 436–443. [Google Scholar] [CrossRef]
- Smedsrød, B.; Melkko, J.; Araki, N.; Sano, H.; Horiuchi, S. Advanced glycation end products are eliminated by scavenger-receptor-mediated endocytosis in hepatic sinusoidal Kupffer and endothelial cells. Biochem. J. 1997, 322 Pt 2, 567–573. [Google Scholar] [CrossRef] [Green Version]
- Nagai, R.; Mera, K.; Nakajou, K.; Fujiwara, Y.; Iwao, Y.; Imai, H.; Murata, T.; Otagiri, M. The ligand activity of AGE-proteins to scavenger receptors is dependent on their rate of modification by AGEs. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2007, 1772, 1192–1198. [Google Scholar] [CrossRef] [Green Version]
- Gugliucci, A.; Bendayan, M. Renal fate of circulating advanced glycated end products (AGE): Evidence for reabsorption and catabolism of AGE-peptides by renal proximal tubular cells. Diabetologia 1996, 39, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Miyata, T.; Ueda, Y.; Shinzato, T.; Iida, Y.; Tanaka, S.; Kurokawa, K.; van Ypersele de Strihou, C.; Maeda, K. Accumulation of albumin-linked and free-form pentosidine in the circulation of uremic patients with end-stage renal failure: Renal implications in the pathophysiology of pentosidine. J. Am. Soc. Nephrol. 1996, 7, 1198–1206. [Google Scholar] [CrossRef]
- Makita, Z.; Radoff, S.; Rayfield, E.J.; Yang, Z.; Skolnik, E.; Delaney, V.; Friedman, E.A.; Cerami, A.; Vlassara, H. Advanced glycosylation end products in patients with diabetic nephropathy. N. Engl. J. Med. 1991, 325, 836–842. [Google Scholar] [CrossRef]
- Perkins, R.K.; Miranda, E.R.; Karstoft, K.; Beisswenger, P.J.; Solomon, T.P.J.; Haus, J.M. Experimental Hyperglycemia Alters Circulating Concentrations and Renal Clearance of Oxidative and Advanced Glycation End Products in Healthy Obese Humans. Nutrients 2019, 11, 532. [Google Scholar] [CrossRef] [Green Version]
- Forbes, J.M.; Cooper, M.E.; Oldfield, M.D.; Thomas, M.C. Role of Advanced Glycation End Products in Diabetic Nephropathy. J. Am. Soc. Nephrol. 2003, 14 (Suppl. 3), S254–S258. [Google Scholar] [CrossRef] [Green Version]
- Harcourt, B.E.; Sourris, K.C.; Coughlan, M.T.; Walker, K.Z.; Dougherty, S.L.; Andrikopoulos, S.; Morley, A.L.; Thallas-Bonke, V.; Chand, V.; Penfold, S.A.; et al. Targeted reduction of advanced glycation improves renal function in obesity. Kidney Int. 2011, 80, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Tan, A.L.; Forbes, J.M.; Cooper, M.E. AGE, RAGE, and ROS in diabetic nephropathy. Semin. Nephrol. 2007, 27, 130–143. [Google Scholar] [CrossRef]
- Zheng, F.; He, C.; Cai, W.; Hattori, M.; Steffes, M.; Vlassara, H. Prevention of diabetic nephropathy in mice by a diet low in glycoxidation products. Diabetes/Metab. Res. Rev. 2002, 18, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Stinghen, A.E.M.; Massy, Z.A.; Vlassara, H.; Striker, G.E.; Boullier, A. Uremic Toxicity of Advanced Glycation End Products in CKD. J. Am. Soc. Nephrol. 2015, 27, 354–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, R.; Baker, S.S.; Liu, W.; Desai, S.; Alkhouri, R.; Kozielski, R.; Mastrandrea, L.; Sarfraz, A.; Cai, W.; Vlassara, H.; et al. Effect of Dietary Advanced Glycation End Products on Mouse Liver. PLoS ONE 2012, 7, e35143. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.M.; Steffes, M.; Donnelly, T.; Liu, C.; Fuh, H.; Basgen, J.; Bucala, R.; Vlassara, H. Prevention of cardiovascular and renal pathology of aging by the advanced glycation inhibitor aminoguanidine. Proc. Natl. Acad. Sci. USA 1996, 93, 3902–3907. [Google Scholar] [CrossRef] [Green Version]
- Leung, C.; Herath, C.B.; Jia, Z.; Goodwin, M.; Mak, K.Y.; Watt, M.J.; Forbes, J.M.; Angus, P.W. Dietary glycotoxins exacerbate progression of experimental fatty liver disease. J. Hepatol. 2014, 60, 832–838. [Google Scholar] [CrossRef]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced Glycation End Products: Sparking the Development of Diabetic Vascular Injury. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef] [Green Version]
- Bucciarelli, L.G.; Wendt, T.; Rong, L.; Lalla, E.; Hofmann, M.A.; Goova, M.T.; Taguchi, A.; Yan, S.F.; Yan, S.D.; Stern, D.M.; et al. RAGE is a multiligand receptor of the immunoglobulin superfamily: Implications for homeostasis and chronic disease. Cell. Mol. Life Sci. 2002, 59, 1117–1128. [Google Scholar] [CrossRef]
- Neeper, M.; Schmidt, A.M.; Brett, J.; Yan, S.D.; Wang, F.; Pan, Y.C.; Elliston, K.; Stern, D.; Shaw, A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 1992, 267, 14998–15004. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Vianna, M.; Gerlach, M.; Brett, J.; Ryan, J.; Kao, J.; Esposito, C.; Hegarty, H.; Hurley, W.; Clauss, M.; et al. Isolation and characterization of two binding proteins for advanced glycosylation end products from bovine lung which are present on the endothelial cell surface. J. Biol. Chem. 1992, 267, 14987–14997. [Google Scholar] [CrossRef]
- Hofmann, M.A.; Drury, S.; Fu, C.; Qu, W.; Taguchi, A.; Lu, Y.; Avila, C.; Kambham, N.; Bierhaus, A.; Nawroth, P.; et al. RAGE mediates a novel proinflammatory axis: A central cell surface receptor for S100/calgranulin polypeptides. Cell 1999, 97, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Hori, O.; Brett, J.; Slattery, T.; Cao, R.; Zhang, J.; Chen, J.X.; Nagashima, M.; Lundh, E.R.; Vijay, S.; Nitecki, D.; et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J. Biol. Chem. 1995, 270, 25752–25761. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Adsit, F.G.; Boyington, J.C. The 1.5 A crystal structure of human receptor for advanced glycation endproducts (RAGE) ectodomains reveals unique features determining ligand binding. J. Biol. Chem. 2010, 285, 40762–40770. [Google Scholar] [CrossRef] [Green Version]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef]
- Sousa, M.M.; Yan, S.D.; Stern, D.; Saraiva, M.J. Interaction of the receptor for advanced glycation end products (RAGE) with transthyretin triggers nuclear transcription factor kB (NF-kB) activation. Lab. Investig. 2000, 80, 1101–1110. [Google Scholar] [CrossRef] [Green Version]
- Chavakis, T.; Bierhaus, A.; Al-Fakhri, N.; Schneider, D.; Witte, S.; Linn, T.; Nagashima, M.; Morser, J.; Arnold, B.; Preissner, K.T.; et al. The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: A novel pathway for inflammatory cell recruitment. J. Exp. Med. 2003, 198, 1507–1515. [Google Scholar] [CrossRef]
- Ma, W.; Rai, V.; Hudson, B.I.; Song, F.; Schmidt, A.M.; Barile, G.R. RAGE binds C1q and enhances C1q-mediated phagocytosis. Cell. Immunol. 2012, 274, 72–82. [Google Scholar] [CrossRef]
- Brett, J.; Schmidt, A.M.; Yan, S.D.; Zou, Y.S.; Weidman, E.; Pinsky, D.; Nowygrod, R.; Neeper, M.; Przysiecki, C.; Shaw, A.; et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am. J. Pathol. 1993, 143, 1699–1712. [Google Scholar]
- Soulis, T.; Thallas, V.; Youssef, S.; Gilbert, R.E.; McWilliam, B.G.; Murray-McIntosh, R.P.; Cooper, M.E. Advanced glycation end products and their receptors co-localise in rat organs susceptible to diabetic microvascular injury. Diabetologia 1997, 40, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Ritthaler, U.; Deng, Y.; Zhang, Y.; Greten, J.; Abel, M.; Sido, B.; Allenberg, J.; Otto, G.; Roth, H.; Bierhaus, A.; et al. Expression of receptors for advanced glycation end products in peripheral occlusive vascular disease. Am. J. Pathol. 1995, 146, 688–694. [Google Scholar]
- Tanji, N.; Markowitz, G.S.; Fu, C.; Kislinger, T.; Taguchi, A.; Pischetsrieder, M.; Stern, D.; Schmidt, A.M.; D’agati, V.D. Expression of Advanced Glycation End Products and Their Cellular Receptor RAGE in Diabetic Nephropathy and Nondiabetic Renal Disease. J. Am. Soc. Nephrol. 2000, 11, 1656–1666. [Google Scholar] [CrossRef]
- Wendt, T.M.; Tanji, N.; Guo, J.; Kislinger, T.R.; Qu, W.; Lu, Y.; Bucciarelli, L.G.; Rong, L.L.; Moser, B.; Markowitz, G.S.; et al. RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am. J. Pathol. 2003, 162, 1123–1137. [Google Scholar] [CrossRef] [Green Version]
- Sugaya, K.; Fukagawa, T.; Matsumoto, K.; Mita, K.; Takahashi, E.; Ando, A.; Inoko, H.; Ikemura, T. Three genes in the human MHC class III region near the junction with the class II: Gene for receptor of advanced glycosylation end products, PBX2 homeobox gene and a notch homolog, human counterpart of mouse mammary tumor gene int-3. Genomics 1994, 23, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Kalea, A.Z.; Reiniger, N.; Yang, H.; Arriero, M.; Schmidt, A.M.; Hudson, B.I. Alternative splicing of the murine receptor for advanced glycation end-products (RAGE) gene. FASEB J. 2009, 23, 1766–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bierhaus, A.; Humpert, P.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.; Nawroth, P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, N.; Dhawan, V. Receptor for advanced glycation end products (RAGE) in vascular and inflammatory diseases. Int. J. Cardiol. 2013, 168, 1788–1794. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.I.; Carter, A.M.; Harja, E.; Kalea, A.Z.; Arriero, M.; Yang, H.; Grant, P.J.; Schmidt, A.M. Identification, classification, and expression of RAGE gene splice variants. FASEB J. 2008, 22, 1572–1580. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Du Yan, S.; Yan, S.F.; Stern, D.M. The biology of the receptor for advanced glycation end products and its ligands. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2000, 1498, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, A.; Blood, D.C.; del Toro, G.; Canet, A.; Lee, D.C.; Qu, W.; Tanji, N.; Lu, Y.; Lalla, E.; Fu, C.; et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature 2000, 405, 354–360. [Google Scholar] [CrossRef]
- Lander, H.M.; Tauras, J.M.; Ogiste, J.S.; Hori, O.; Moss, R.A.; Schmidt, A.M. Activation of the receptor for advanced glycation end products triggers a p21(ras)-dependent mitogen-activated protein kinase pathway regulated by oxidant stress. J. Biol. Chem. 1997, 272, 17810–17814. [Google Scholar] [CrossRef] [Green Version]
- Yeh, C.-H.; Sturgis, L.; Haidacher, J.; Zhang, X.-N.; Sherwood, S.J.; Bjercke, R.J.; Juhasz, O.; Crow, M.T.; Tilton, R.G.; Denner, L. Requirement for p38 and p44/p42 Mitogen-Activated Protein Kinases in RAGE-Mediated Nuclear Factor-κB Transcriptional Activation and Cytokine Secretion. Diabetes 2001, 50, 1495–1504. [Google Scholar] [CrossRef] [Green Version]
- Grimm, S.; Ott, C.; Horlacher, M.; Weber, D.; Hohn, A.; Grune, T. Advanced-glycation-end-product-induced formation of immunoproteasomes: Involvement of RAGE and Jak2/STAT1. Biochem. J. 2012, 448, 127–139. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.S.; Guh, J.Y.; Chen, H.C.; Hung, W.C.; Lai, Y.H.; Chuang, L.Y. Role of receptor for advanced glycation end-product (RAGE) and the JAK/STAT-signaling pathway in AGE-induced collagen production in NRK-49F cells. J. Cell. Biochem. 2001, 81, 102–113. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Fages, C.; Rauvala, H. Receptor for Advanced Glycation End Products (RAGE)-mediated Neurite Outgrowth and Activation of NF-κB Require the Cytoplasmic Domain of the Receptor but Different Downstream Signaling Pathways. J. Biol. Chem. 1999, 274, 19919–19924. [Google Scholar] [CrossRef] [Green Version]
- Sorci, G.; Riuzzi, F.; Arcuri, C.; Giambanco, I.; Donato, R. Amphoterin stimulates myogenesis and counteracts the antimyogenic factors basic fibroblast growth factor and S100B via RAGE binding. Mol. Cell. Biol. 2004, 24, 4880–4894. [Google Scholar] [CrossRef] [Green Version]
- Hudson, B.I.; Kalea, A.Z.; Del Mar Arriero, M.; Harja, E.; Boulanger, E.; D’Agati, V.; Schmidt, A.M. Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J. Biol. Chem. 2008, 283, 34457–34468. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.W.; Vlassara, H.; Peten, E.P.; He, C.J.; Striker, G.E.; Striker, L.J. Advanced glycation end products up-regulate gene expression found in diabetic glomerular disease. Proc. Natl. Acad. Sci. USA 1994, 91, 9436–9440. [Google Scholar] [CrossRef] [Green Version]
- Zeng, S.; Dun, H.; Ippagunta, N.; Rosario, R.; Zhang, Q.Y.; Lefkowitch, J.; Yan, S.F.; Schmidt, A.M.; Emond, J.C. Receptor for advanced glycation end product (RAGE)-dependent modulation of early growth response-1 in hepatic ischemia/reperfusion injury. J. Hepatol. 2009, 50, 929–936. [Google Scholar] [CrossRef]
- Xie, J.; Mendez, J.D.; Mendez-Valenzuela, V.; Aguilar-Hernandez, M.M. Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell. Signal. 2013, 25, 2185–2197. [Google Scholar] [CrossRef]
- Cai, W.; He, J.C.; Zhu, L.; Chen, X.; Zheng, F.; Striker, G.E.; Vlassara, H. Oral glycotoxins determine the effects of calorie restriction on oxidant stress, age-related diseases, and lifespan. Am. J. Pathol. 2008, 173, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Buchs, A.E.; Kornberg, A.; Zahavi, M.; Aharoni, D.; Zarfati, C.; Rapoport, M.J. Increased expression of tissue factor and receptor for advanced glycation end products in peripheral blood mononuclear cells of patients with type 2 diabetes mellitus with vascular complications. Exp. Diabesity Res. 2004, 5, 163–169. [Google Scholar] [CrossRef]
- Koyama, H.; Shoji, T.; Yokoyama, H.; Motoyama, K.; Mori, K.; Fukumoto, S.; Emoto, M.; Shoji, T.; Tamei, H.; Matsuki, H.; et al. Plasma level of endogenous secretory RAGE is associated with components of the metabolic syndrome and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2587–2593. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.; van Ree, R.M.; Oterdoom, L.H.; de Vries, A.P.J.; van Son, W.J.; de Jong, P.E.; Navis, G.J.; Zuurman, M.W.; Bierhaus, A.; Gans, R.O.B.; et al. Low Levels of sRAGE Are Associated With Increased Risk for Mortality in Renal Transplant Recipients. Transplantation 2007, 84, 659–663. [Google Scholar] [CrossRef]
- Selvin, E.; Halushka, M.; Rawlings, A.; Hoogeveen, R.C.; Ballantyne, C.M.; Coresh, J.; Astor, B.C. sRAGE and risk of diabetes, cardiovascular disease and death. Diabetes 2013, 62, 2116–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loomis, S.J.; Chen, Y.; Sacks, D.B.; Christenson, E.S.; Christenson, R.H.; Rebholz, C.M.; Selvin, E. Cross-sectional Analysis of AGE-CML, sRAGE, and esRAGE with Diabetes and Cardiometabolic Risk Factors in a Community-Based Cohort. Clin. Chem. 2017, 63, 980–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basta, G.; Sironi, A.M.; Lazzerini, G.; Del Turco, S.; Buzzigoli, E.; Casolaro, A.; Natali, A.; Ferrannini, E.; Gastaldelli, A. Circulating soluble receptor for advanced glycation end products is inversely associated with glycemic control and S100A12 protein. J. Clin. Endocrinol. Metab. 2006, 91, 4628–4634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basta, G.; Leonardis, D.; Mallamaci, F.; Cutrupi, S.; Pizzini, P.; Gaetano, L.; Tripepi, R.; Tripepi, G.; De Caterina, R.; Zoccali, C. Circulating soluble receptor of advanced glycation end product inversely correlates with atherosclerosis in patients with chronic kidney disease. Kidney Int. 2010, 77, 225–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laudenslager, M.; Lazo, M.; Wang, D.; Selvin, E.; Chen, P.-H.; Pankow, J.S.; Clark, J.M. Association between the soluble receptor for advanced glycation end products (sRAGE) and NAFLD in participants in the Atherosclerosis Risk in Communities Study. Dig. Liver Dis. 2021, 53, 873–878. [Google Scholar] [CrossRef]
- Geroldi, D.; Falcone, C.; Minoretti, P.; Emanuele, E.; Arra, M.; D’Angelo, A. High levels of soluble receptor for advanced glycation end products may be a marker of extreme longevity in humans. J. Am. Geriatr. Soc. 2006, 54, 1149–1150. [Google Scholar] [CrossRef]
- Scavello, F.; Tedesco, C.C.; Castiglione, S.; Maciag, A.; Sangalli, E.; Veglia, F.; Spinetti, G.; Puca, A.A.; Raucci, A. Modulation of soluble receptor for advanced glycation end products isoforms and advanced glycation end products in long-living individuals. Biomark Med. 2021, 15, 785–796. [Google Scholar] [CrossRef]
- Park, L.; Raman, K.G.; Lee, K.J.; Lu, Y.; Ferran, L.J.; Chow, W.S.; Stern, D.; Schmidt, A.M. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat. Med. 1998, 4, 1025–1031. [Google Scholar] [CrossRef]
- Bierhaus, A.; Haslbeck, K.M.; Humpert, P.M.; Liliensiek, B.; Dehmer, T.; Morcos, M.; Sayed, A.A.; Andrassy, M.; Schiekofer, S.; Schneider, J.G.; et al. Loss of pain perception in diabetes is dependent on a receptor of the immunoglobulin superfamily. J. Clin. Investig. 2004, 114, 1741–1751. [Google Scholar] [CrossRef] [Green Version]
- Kislinger, T.; Tanji, N.; Wendt, T.; Qu, W.; Lu, Y.; Ferran, L.J., Jr.; Taguchi, A.; Olson, K.; Bucciarelli, L.; Goova, M.; et al. Receptor for advanced glycation end products mediates inflammation and enhanced expression of tissue factor in vasculature of diabetic apolipoprotein E-null mice. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 905–910. [Google Scholar] [CrossRef] [Green Version]
- Kalea, A.Z.; Schmidt, A.M.; Hudson, B.I. RAGE: A novel biological and genetic marker for vascular disease. Clin. Sci. 2009, 116, 621–637. [Google Scholar] [CrossRef] [Green Version]
- Yonekura, H.; Yamamoto, Y.; Sakurai, S.; Petrova, R.G.; Abedin, M.J.; Li, H.; Yasui, K.; Takeuchi, M.; Makita, Z.; Takasawa, S.; et al. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem. J. 2003, 370, 1097–1109. [Google Scholar] [CrossRef]
- Li, Y.M.; Mitsuhashi, T.; Wojciechowicz, D.; Shimizu, N.; Li, J.; Stitt, A.; He, C.; Banerjee, D.; Vlassara, H. Molecular identity and cellular distribution of advanced glycation endproduct receptors: Relationship of p60 to OST-48 and p90 to 80K-H membrane proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 11047–11052. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Makita, Z.; Horii, Y.; Brunelle, S.; Cerami, A.; Sehajpal, P.; Suthanthiran, M.; Vlassara, H. Two novel rat liver membrane proteins that bind advanced glycosylation endproducts: Relationship to macrophage receptor for glucose-modified proteins. J. Exp. Med. 1991, 174, 515–524. [Google Scholar] [CrossRef]
- Kelleher, D.J.; Kreibich, G.; Gilmore, R. Oligosaccharyltransferase activity is associated with a protein complex composed of ribophorins I and II and a 48 kd protein. Cell 1992, 69, 55–65. [Google Scholar] [CrossRef]
- Thul, P.J.; Åkesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Blal, H.A.; Alm, T.; Asplund, A.; Björk, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356, eaal3321. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Vlassara, H.; Cai, W.; Goodman, S.; Pyzik, R.; Yong, A.; Chen, X.; Zhu, L.; Neade, T.; Beeri, M.; Silverman, J.M.; et al. Protection against loss of innate defenses in adulthood by low advanced glycation end products (AGE) intake: Role of the antiinflammatory AGE receptor-1. J. Clin. Endocrinol. Metab. 2009, 94, 4483–4491. [Google Scholar] [CrossRef] [Green Version]
- Uribarri, J.; Cai, W.; Ramdas, M.; Goodman, S.; Pyzik, R.; Chen, X.; Zhu, L.; Striker, G.E.; Vlassara, H. Restriction of advanced glycation end products improves insulin resistance in human type 2 diabetes: Potential role of AGER1 and SIRT1. Diabetes Care 2011, 34, 1610–1616. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; He, J.C.; Cai, W.; Liu, H.; Zhu, L.; Vlassara, H. Advanced glycation endproduct (AGE) receptor 1 is a negative regulator of the inflammatory response to AGE in mesangial cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11767–11772. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; He, J.C.; Zhu, L.; Chen, X.; Wallenstein, S.; Striker, G.E.; Vlassara, H. Reduced Oxidant Stress and Extended Lifespan in Mice Exposed to a Low Glycotoxin Diet: Association with Increased AGER1 Expression. Am. J. Pathol. 2007, 170, 1893–1902. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Moreno, J.; Quintana-Navarro, G.M.; Delgado-Lista, J.; Garcia-Rios, A.; Alcala-Diaz, J.F.; Gomez-Delgado, F.; Camargo, A.; Perez-Martinez, P.; Tinahones, F.J.; Striker, G.E.; et al. Mediterranean Diet Supplemented with Coenzyme Q10 Modulates the Postprandial Metabolism of Advanced Glycation End Products in Elderly Men and Women. J. Gerontol. Ser. A 2018, 73, 340–346. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Mariscal, F.M.; Cardelo, M.P.; de la Cruz, S.; Alcala-Diaz, J.F.; Roncero-Ramos, I.; Guler, I.; Vals-Delgado, C.; López-Moreno, A.; Luque, R.M.; Delgado-Lista, J.; et al. Reduction in Circulating Advanced Glycation End Products by Mediterranean Diet Is Associated with Increased Likelihood of Type 2 Diabetes Remission in Patients with Coronary Heart Disease: From the Cordioprev Study. Mol. Nutr. Food Res. 2021, 65, 1901290. [Google Scholar] [CrossRef]
- Lopez-Moreno, J.; Quintana-Navarro, G.M.; Camargo, A.; Jimenez-Lucena, R.; Delgado-Lista, J.; Marin, C.; Tinahones, F.J.; Striker, G.E.; Roche, H.M.; Perez-Martinez, P.; et al. Dietary fat quantity and quality modifies advanced glycation end products metabolism in patients with metabolic syndrome. Mol. Nutr. Food Res. 2017, 61, 1601029. [Google Scholar] [CrossRef]
- Zhuang, A.; Yap, F.Y.T.; McCarthy, D.; Leung, C.; Sourris, K.C.; Penfold, S.A.; Thallas-Bonke, V.; Coughlan, M.T.; Schulz, B.L.; Forbes, J.M. Globally elevating the AGE clearance receptor, OST48, does not protect against the development of diabetic kidney disease, despite improving insulin secretion. Sci. Rep. 2019, 9, 13664. [Google Scholar] [CrossRef]
- Zhuang, A.; Yap, F.Y.T.; Borg, D.J.; McCarthy, D.; Fotheringham, A.; Leung, S.; Penfold, S.A.; Sourris, K.C.; Coughlan, M.T.; Schulz, B.L.; et al. The AGE receptor, OST48 drives podocyte foot process effacement and basement membrane expansion (alters structural composition). Endocrinol. Diabetes Metab. 2021, 4, e00278. [Google Scholar] [CrossRef]
- Bai, P.; Phua, K.; Hardt, T.; Cernadas, M.; Brodsky, B. Glycation alters collagen fibril organization. Connect. Tissue Res. 1992, 28, 1–12. [Google Scholar] [CrossRef]
- Hadley, J.C.; Meek, K.M.; Malik, N.S. Glycation changes the charge distribution of type I collagen fibrils. Glycoconj. J. 1998, 15, 835–840. [Google Scholar] [CrossRef]
- Barlovic, D.P.; Soro-Paavonen, A.; Jandeleit-Dahm, K.A. RAGE biology, atherosclerosis and diabetes. Clin. Sci. 2011, 121, 43–55. [Google Scholar] [CrossRef] [Green Version]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Luevano-Contreras, C.; Chapman-Novakofski, K. Dietary advanced glycation end products and aging. Nutrients 2010, 2, 1247–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bengmark, S. Impact of nutrition on ageing and disease. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Cai, W.; Peppa, M.; Goodman, S.; Ferrucci, L.; Striker, G.; Vlassara, H. Circulating Glycotoxins and Dietary Advanced Glycation Endproducts: Two Links to Inflammatory Response, Oxidative Stress, and Aging. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2007, 62, 427–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlassara, H.; Cai, W.; Crandall, J.; Goldberg, T.; Oberstein, R.; Dardaine, V.; Peppa, M.; Rayfield, E.J. Inflammatory mediators are induced by dietary glycotoxins, a major risk factor for diabetic angiopathy. Proc. Natl. Acad. Sci. USA 2002, 99, 15596–15601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linkens, A.M.A.; Houben, A.J.; Niessen, P.M.; Wijckmans, N.; de Goei, E.; Van den Eynde, M.D.; Scheijen, J.; Waarenburg, M.; Mari, A.; Berendschot, T.T.; et al. A 4-week high-AGE diet does not impair glucose metabolism and vascular function in obese individuals. JCI Insight 2022, 7, e156950. [Google Scholar] [CrossRef]
- Vlassara, H.; Striker, L.J.; Teichberg, S.; Fuh, H.; Li, Y.M.; Steffes, M. Advanced glycation end products induce glomerular sclerosis and albuminuria in normal rats. Proc. Natl. Acad. Sci. USA 1994, 91, 11704–11708. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Ramdas, M.; Zhu, L.; Chen, X.; Striker, G.E.; Vlassara, H. Oral advanced glycation endproducts (AGEs) promote insulin resistance and diabetes by depleting the antioxidant defenses AGE receptor-1 and sirtuin 1. Proc. Natl. Acad. Sci. USA 2012, 109, 15888–15893. [Google Scholar] [CrossRef] [Green Version]
- Forbes, J.M.; Cowan, S.P.; Andrikopoulos, S.; Morley, A.L.; Ward, L.C.; Walker, K.Z.; Cooper, M.E.; Coughlan, M.T. Glucose homeostasis can be differentially modulated by varying individual components of a western diet. J. Nutr. Biochem. 2013, 24, 1251–1257. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Zhao, C.; Zhang, X.H.; Zheng, F.; Cai, W.; Vlassara, H.; Ma, Z.A. Advanced Glycation End Products Inhibit Glucose-Stimulated Insulin Secretion through Nitric Oxide-Dependent Inhibition of Cytochrome c Oxidase and Adenosine Triphosphate Synthesis. Endocrinology 2009, 150, 2569–2576. [Google Scholar] [CrossRef]
- Coughlan, M.T.; Yap, F.Y.; Tong, D.C.; Andrikopoulos, S.; Gasser, A.; Thallas-Bonke, V.; Webster, D.E.; Miyazaki, J.; Kay, T.W.; Slattery, R.M.; et al. Advanced glycation end products are direct modulators of beta-cell function. Diabetes 2011, 60, 2523–2532. [Google Scholar] [CrossRef] [Green Version]
- Coughlan, M.T.; Forbes, J.M. Temporal increases in urinary carboxymethyllysine correlate with albuminuria development in diabetes. Am. J. Nephrol. 2011, 34, 9–17. [Google Scholar] [CrossRef]
- Sebekova, K.; Hofmann, T.; Boor, P.; SebekovaJr, K.; Ulicna, O.; Erbersdobler, H.F.; Baynes, J.W.; Thorpe, S.R.; Heidland, A.; Somoza, V. Renal effects of oral maillard reaction product load in the form of bread crusts in healthy and subtotally nephrectomized rats. Ann. N. Y. Acad. Sci. 2005, 1043, 482–491. [Google Scholar] [CrossRef]
- Snelson, M.; Tan, S.M.; Clarke, R.E.; Pasquale, C.D.; Thallas-Bonke, V.; Nguyen, T.-V.; Penfold, S.A.; Harcourt, B.E.; Sourris, K.C.; Lindblom, R.S.; et al. Processed foods drive intestinal barrier permeability and microvascular diseases. Sci. Adv. 2021, 7, eabe4841. [Google Scholar] [CrossRef]
- Peppa, M.; He, C.; Hattori, M.; McEvoy, R.; Zheng, F.; Vlassara, H. Fetal or neonatal low-glycotoxin environment prevents autoimmune diabetes in NOD mice. Diabetes 2003, 52, 1441–1448. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, S.M.; Dong, H.J.; Li, Z.; Cai, W.; Altomonte, J.; Thung, S.N.; Zeng, F.; Fisher, E.A.; Vlassara, H. Improved insulin sensitivity is associated with restricted intake of dietary glycoxidation products in the db/db mouse. Diabetes 2002, 51, 2082–2089. [Google Scholar] [CrossRef] [Green Version]
- Tan, A.L.; Sourris, K.C.; Harcourt, B.E.; Thallas-Bonke, V.; Penfold, S.; Andrikopoulos, S.; Thomas, M.C.; O’Brien, R.C.; Bierhaus, A.; Cooper, M.E.; et al. Disparate effects on renal and oxidative parameters following RAGE deletion, AGE accumulation inhibition, or dietary AGE control in experimental diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2010, 298, F763–F770. [Google Scholar] [CrossRef] [Green Version]
- Peppa, M.; Brem, H.; Ehrlich, P.; Zhang, J.G.; Cai, W.; Li, Z.; Croitoru, A.; Thung, S.; Vlassara, H. Adverse effects of dietary glycotoxins on wound healing in genetically diabetic mice. Diabetes 2003, 52, 2805–2813. [Google Scholar] [CrossRef] [Green Version]
- Uribarri, J.; Peppa, M.; Cai, W.; Goldberg, T.; Lu, M.; Baliga, S.; Vassalotti, J.A.; Vlassara, H. Dietary glycotoxins correlate with circulating advanced glycation end product levels in renal failure patients. Am. J. Kidney Dis. 2003, 42, 532–538. [Google Scholar] [CrossRef]
- Chao, P.C.; Huang, C.N.; Hsu, C.C.; Yin, M.C.; Guo, Y.R. Association of dietary AGEs with circulating AGEs, glycated LDL, IL-1alpha and MCP-1 levels in type 2 diabetic patients. Eur. J. Nutr. 2010, 49, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Lotan, R.; Ganmore, I.; Livny, A.; Itzhaki, N.; Waserman, M.; Shelly, S.; Zacharia, M.; Moshier, E.; Uribarri, J.; Beisswenger, P.; et al. Effect of Advanced Glycation End Products on Cognition in Older Adults with Type 2 Diabetes: Results from a Pilot Clinical Trial. J. Alzheimer’s Dis. 2021, 82, 1785–1795. [Google Scholar] [CrossRef] [PubMed]
- Birlouez-Aragon, I.; Saavedra, G.; Tessier, F.J.; Galinier, A.; Ait-Ameur, L.; Lacoste, F.; Niamba, C.N.; Alt, N.; Somoza, V.; Lecerf, J.M. A diet based on high-heat-treated foods promotes risk factors for diabetes mellitus and cardiovascular diseases. Am. J. Clin. Nutr. 2010, 91, 1220–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riboulet-Chavey, A.; Pierron, A.; Durand, I.; Murdaca, J.; Giudicelli, J.; Van Obberghen, E. Methylglyoxal impairs the insulin signaling pathways independently of the formation of intracellular reactive oxygen species. Diabetes 2006, 55, 1289–1299. [Google Scholar] [CrossRef] [Green Version]
- Hagiwara, S.; Gohda, T.; Tanimoto, M.; Ito, T.; Murakoshi, M.; Ohara, I.; Yamazaki, T.; Matsumoto, M.; Horikoshi, S.; Funabiki, K. Effects of pyridoxamine (K-163) on glucose intolerance and obesity in high-fat diet C57BL/6J mice. Metabolism 2009, 58, 934–945. [Google Scholar] [CrossRef]
- Sandu, O.; Song, K.; Cai, W.; Zheng, F.; Uribarri, J.; Vlassara, H. Insulin resistance and type 2 diabetes in high-fat-fed mice are linked to high glycotoxin intake. Diabetes 2005, 54, 2314–2319. [Google Scholar] [CrossRef] [Green Version]
- Mark, A.B.; Poulsen, M.W.; Andersen, S.; Andersen, J.M.; Bak, M.J.; Ritz, C.; Holst, J.J.; Nielsen, J.; de Courten, B.; Dragsted, L.O.; et al. Consumption of a Diet Low in Advanced Glycation End Products for 4 Weeks Improves Insulin Sensitivity in Overweight Women. Diabetes Care 2014, 37, 88–95. [Google Scholar] [CrossRef] [Green Version]
- Vaaler, S.; Hanssen, K.F.; Aagenaes, O. The effect of cooking upon the blood glucose response to ingested carrots and potatoes. Diabetes Care 1984, 7, 221–223. [Google Scholar] [CrossRef]
- Rizkalla, S.W.; Laromiguiere, M.; Champ, M.; Bruzzo, F.; Boillot, J.; Slama, G. Effect of baking process on postprandial metabolic consequences: Randomized trials in normal and type 2 diabetic subjects. Eur. J. Clin. Nutr. 2007, 61, 175–183. [Google Scholar] [CrossRef]
- de Courten, B.; de Courten, M.P.; Soldatos, G.; Dougherty, S.L.; Straznicky, N.; Schlaich, M.; Sourris, K.C.; Chand, V.; Scheijen, J.L.; Kingwell, B.A.; et al. Diet low in advanced glycation end products increases insulin sensitivity in healthy overweight individuals: A double-blind, randomized, crossover trial. Am. J. Clin. Nutr. 2016, 103, 1426–1433. [Google Scholar] [CrossRef]
- Seiquer, I.; Rubio, L.A.; Peinado, M.J.; Delgado-Andrade, C.; Navarro, M.P. Maillard reaction products modulate gut microbiota composition in adolescents. Mol. Nutr. Food Res. 2014, 58, 1552–1560. [Google Scholar] [CrossRef]
- Yacoub, R.; Nugent, M.; Cai, W.; Nadkarni, G.N.; Chaves, L.D.; Abyad, S.; Honan, A.M.; Thomas, S.A.; Zheng, W.; Valiyaparambil, S.A.; et al. Advanced glycation end products dietary restriction effects on bacterial gut microbiota in peritoneal dialysis patients; a randomized open label controlled trial. PLoS ONE 2017, 12, e0184789. [Google Scholar] [CrossRef] [Green Version]
- Mastrocola, R.; Collotta, D.; Gaudioso, G.; Le Berre, M.; Cento, A.S.; Ferreira Alves, G.; Chiazza, F.; Verta, R.; Bertocchi, I.; Manig, F.; et al. Effects of Exogenous Dietary Advanced Glycation End Products on the Cross-Talk Mechanisms Linking Microbiota to Metabolic Inflammation. Nutrients 2020, 12, 2497. [Google Scholar] [CrossRef]
- Bock, P.M.; Telo, G.H.; Ramalho, R.; Sbaraini, M.; Leivas, G.; Martins, A.F.; Schaan, B.D. The effect of probiotics, prebiotics or synbiotics on metabolic outcomes in individuals with diabetes: A systematic review and meta-analysis. Diabetologia 2021, 64, 26–41. [Google Scholar] [CrossRef]
- Farhangi, M.A.; Dehghan, P.; Namazi, N. Prebiotic supplementation modulates advanced glycation end-products (AGEs), soluble receptor for AGEs (sRAGE), and cardiometabolic risk factors through improving metabolic endotoxemia: A randomized-controlled clinical trial. Eur. J. Nutr. 2020, 59, 3009–3021. [Google Scholar] [CrossRef]
- Kellow, N.J.; Coughlan, M.T.; Savige, G.S.; Reid, C.M. Effect of dietary prebiotic supplementation on advanced glycation, insulin resistance and inflammatory biomarkers in adults with pre-diabetes: A study protocol for a double-blind placebo-controlled randomised crossover clinical trial. BMC Endocr. Disord. 2014, 14, 55. [Google Scholar] [CrossRef] [Green Version]
- Semba, R.D.; Fink, J.C.; Sun, K.; Bandinelli, S.; Guralnik, J.M.; Ferrucci, L. Carboxymethyl-lysine, an advanced glycation end product, and decline of renal function in older community-dwelling adults. Eur. J. Nutr. 2009, 48, 38–44. [Google Scholar] [CrossRef] [Green Version]
- Hou, F.F.; Ren, H.; OwenJr, W.F.; Guo, Z.J.; Chen, P.Y.; Schmidt, A.M.; Miyata, T.; Zhang, X. Enhanced expression of receptor for advanced glycation end products in chronic kidney disease. J. Am. Soc. Nephrol. 2004, 15, 1889–1896. [Google Scholar] [CrossRef] [Green Version]
- Semba, R.D.; Fink, J.C.; Sun, K.; Windham, B.G.; Ferrucci, L. Serum Carboxymethyl-Lysine, a Dominant Advanced Glycation End Product, Is Associated with Chronic Kidney Disease: The Baltimore Longitudinal Study of Aging. J. Ren. Nutr. 2010, 20, 74–81. [Google Scholar] [CrossRef] [Green Version]
- Gerdemann, A.; Lemke, H.D.; Nothdurft, A.; Heidland, A.; Münch, G.; Bahner, U.; Schinzel, R. Low-molecular but not high-molecular advanced glycation end products (AGEs) are removed by high-flux dialysis. Clin. Nephrol. 2000, 54, 276–283. [Google Scholar]
- Peppa, M.; Uribarri, J.; Cai, W.; Lu, M.; Vlassara, H. Glycoxidation and inflammation in renal failure patients. Am. J. Kidney Dis. 2004, 43, 690–695. [Google Scholar] [CrossRef] [PubMed]
- Agalou, S.; Ahmed, N.; Dawnay, A.; Thornalley, P.J. Removal of advanced glycation end products in clinical renal failure by peritoneal dialysis and haemodialysis. Biochem. Soc. Trans. 2003, 31, 1394–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witko-Sarsat, V.; Friedlander, M.; Capeillere-Blandin, C.; Nguyen-Khoa, T.; Nguyen, A.T.; Zingraff, J.; Jungers, P.; Descamps-Latscha, B. Advanced oxidation protein products as a novel marker of oxidative stress in uremia. Kidney Int. 1996, 49, 1304–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotomayor, C.G.; Gomes-Neto, A.W.; van Londen, M.; Gans, R.O.B.; Nolte, I.M.; Berger, S.P.; Navis, G.J.; Rodrigo, R.; Leuvenink, H.G.D.; Schalkwijk, C.G.; et al. Circulating Advanced Glycation Endproducts and Long-Term Risk of Cardiovascular Mortality in Kidney Transplant Recipients. Clin. J. Am. Soc. Nephrol. 2019, 14, 1512–1520. [Google Scholar] [CrossRef]
- Klein, R.; Horak, K.; Lee, K.E.; Danforth, L.; Cruickshanks, K.J.; Tsai, M.Y.; Gangnon, R.E.; Klein, B.E.K. The Relationship of Serum Soluble Receptor for Advanced Glycation End Products (sRAGE) and Carboxymethyl Lysine (CML) to the Incidence of Diabetic Nephropathy in Persons with Type 1 Diabetes. Diabetes Care 2017, 40, e117–e119. [Google Scholar] [CrossRef] [Green Version]
- Dalal, M.; Semba, R.D.; Sun, K.; Crasto, C.; Varadhan, R.; Bandinelli, S.; Fink, J.C.; Guralnik, J.M.; Ferrucci, L. Endogenous secretory receptor for advanced glycation end products and chronic kidney disease in the elderly population. Am. J. Nephrol. 2011, 33, 313–318. [Google Scholar] [CrossRef] [Green Version]
- Rebholz, C.M.; Astor, B.C.; Grams, M.E.; Halushka, M.K.; Lazo, M.; Hoogeveen, R.C.; Ballantyne, C.M.; Coresh, J.; Selvin, E. Association of plasma levels of soluble receptor for advanced glycation end products and risk of kidney disease: The Atherosclerosis Risk in Communities study. Nephrol. Dial. Transpl. 2015, 30, 77–83. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.C.; Woodward, M.; Neal, B.; Li, Q.; Pickering, R.; Marre, M.; Williams, B.; Perkovic, V.; Cooper, M.E.; Zoungas, S.; et al. The Relationship Between Levels of Advanced Glycation End-Products and Their Soluble Receptor and Adverse Outcomes in Adults with Type 2 Diabetes. Diabetes Care 2015, 38, 1891–1897. [Google Scholar] [CrossRef] [Green Version]
- McIntyre, N.J.; Fluck, R.J.; McIntyre, C.W.; Taal, M.W. Skin Autofluorescence and the Association with Renal and Cardiovascular Risk Factors in Chronic Kidney Disease Stage 3. Clin. J. Am. Soc. Nephrol. 2011, 6, 2356–2363. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Tani, Y.; Asai, J.; Nemoto, F.; Kusano, Y.; Suzuki, H.; Hayashi, Y.; Asahi, K.; Katoh, T.; Miyata, T.; et al. Skin autofluorescence is associated with renal function and cardiovascular diseases in pre-dialysis chronic kidney disease patients. Nephrol. Dial. Transpl. 2011, 26, 214–220. [Google Scholar] [CrossRef] [Green Version]
- Smit, A.J.; Gerrits, E.G. Skin autofluorescence as a measure of advanced glycation endproduct deposition: A novel risk marker in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2010, 19, 527–533. [Google Scholar] [CrossRef]
- Tanaka, K.; Nakayama, M.; Kanno, M.; Kimura, H.; Watanabe, K.; Tani, Y.; Kusano, Y.; Suzuki, H.; Hayashi, Y.; Asahi, K.; et al. Skin autofluorescence is associated with the progression of chronic kidney disease: A prospective observational study. PLoS ONE 2013, 8, e83799. [Google Scholar] [CrossRef] [Green Version]
- Genevieve, M.; Vivot, A.; Gonzalez, C.; Raffaitin, C.; Barberger-Gateau, P.; Gin, H.; Rigalleau, V. Skin autofluorescence is associated with past glycaemic control and complications in type 1 diabetes mellitus. Diabetes Metab. 2013, 39, 349–354. [Google Scholar] [CrossRef]
- Sugisawa, E.; Miura, J.; Iwamoto, Y.; Uchigata, Y. Skin autofluorescence reflects integration of past long-term glycemic control in patients with type 1 diabetes. Diabetes Care 2013, 36, 2339–2345. [Google Scholar] [CrossRef] [Green Version]
- Meerwaldt, R.; Hartog, J.W.L.; Graaff, R.; Huisman, R.J.; Links, T.P.; den Hollander, N.C.; Thorpe, S.R.; Baynes, J.W.; Navis, G.; Gans, R.O.B.; et al. Skin Autofluorescence, a Measure of Cumulative Metabolic Stress and Advanced Glycation End Products, Predicts Mortality in Hemodialysis Patients. J. Am. Soc. Nephrol. 2005, 16, 3687–3693. [Google Scholar] [CrossRef]
- Forbes, J.M.; Le Bagge, S.; Righi, S.; Fotheringham, A.K.; Gallo, L.A.; McCarthy, D.A.; Leung, S.; Baskerville, T.; Nisbett, J.; Morton, A.; et al. Advanced glycation end products as predictors of renal function in youth with type 1 diabetes. Sci. Rep. 2021, 11, 9422. [Google Scholar] [CrossRef]
- Yu, Y.; Thorpe, S.R.; Jenkins, A.J.; Shaw, J.N.; Sochaski, M.A.; McGee, D.; Aston, C.E.; Orchard, T.J.; Silvers, N.; Peng, Y.G.; et al. Advanced glycation end-products and methionine sulphoxide in skin collagen of patients with type 1 diabetes. Diabetologia 2006, 49, 2488–2498. [Google Scholar] [CrossRef] [Green Version]
- Orchard, T.J.; Lyons, T.J.; Cleary, P.A.; Braffett, B.H.; Maynard, J.; Cowie, C.; Gubitosi-Klug, R.A.; Way, J.; Anderson, K.; Barnie, A.; et al. The Association of Skin Intrinsic Fluorescence with Type 1 Diabetes Complications in the DCCT/EDIC Study. Diabetes Care 2013, 36, 3146–3153. [Google Scholar] [CrossRef] [Green Version]
- Weiss, M.F.; Erhard, P.; Kader-Attia, F.A.; Wu, Y.C.; Deoreo, P.B.; Araki, A.; Glomb, M.A.; Monnier, V.M. Mechanisms for the formation of glycoxidation products in end-stage renal disease. Kidney Int. 2000, 57, 2571–2585. [Google Scholar] [CrossRef] [Green Version]
- Coughlan, M.T.; Patel, S.K.; Jerums, G.; Penfold, S.A.; Nguyen, T.-V.; Sourris, K.C.; Panagiotopoulos, S.; Srivastava, P.M.; Cooper, M.E.; Burrell, L.M.; et al. Advanced glycation urinary protein-bound biomarkers and severity of diabetic nephropathy in man. Am. J. Nephrol. 2011, 34, 347–355. [Google Scholar] [CrossRef] [Green Version]
- Beisswenger, P.J.; Howell, S.K.; Russell, G.B.; Miller, M.E.; Rich, S.S.; Mauer, M. Early Progression of Diabetic Nephropathy Correlates with Methylglyoxal-Derived Advanced Glycation End Products. Diabetes Care 2013, 36, 3234–3239. [Google Scholar] [CrossRef] [Green Version]
- Perkins, B.A. Abstract #0097 Advanced glycation end products and other markers of protein damage in early diabetic nephropathy in type 1 diabetes. In Proceedings of the International Diabetes Federation, World Diabetes Congress, Melbourne, Australia, 3 December 2013. [Google Scholar]
- Thomas, M.C.; Forbes, J.M.; MacIsaac, R.; Jerums, G.; Cooper, M.E. Low-Molecular Weight Advanced Glycation End Products: Markers of Tissue AGE Accumulation and More? Ann. N. Y. Acad. Sci. 2005, 1043, 644–654. [Google Scholar] [CrossRef]
- Kern, E.F.O.; Erhard, P.; Sun, W.; Genuth, S.; Weiss, M.F. Early Urinary Markers of Diabetic Kidney Disease: A Nested Case-Control Study From the Diabetes Control and Complications Trial (DCCT). Am. J. Kidney Dis. 2010, 55, 824–834. [Google Scholar] [CrossRef] [Green Version]
- Nishad, R.; Tahaseen, V.; Kavvuri, R.; Motrapu, M.; Singh, A.K.; Peddi, K.; Pasupulati, A.K. Advanced-Glycation End-Products Induce Podocyte Injury and Contribute to Proteinuria. Front. Med. 2021, 8, 685447. [Google Scholar] [CrossRef]
- Kumar, P.A.; Welsh, G.I.; Raghu, G.; Menon, R.K.; Saleem, M.A.; Reddy, G.B. Carboxymethyl lysine induces EMT in podocytes through transcription factor ZEB2: Implications for podocyte depletion and proteinuria in diabetes mellitus. Arch. Biochem. Biophys. 2016, 590, 10–19. [Google Scholar] [CrossRef] [Green Version]
- Oldfield, M.D.; Bach, L.A.; Forbes, J.M.; Nikolic-Paterson, D.; McRobert, A.; Thallas, V.; Atkins, R.C.; Osicka, T.; Jerums, G.; Cooper, M.E. Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE). J. Clin. Investig. 2001, 108, 1853–1863. [Google Scholar] [CrossRef]
- Li, J.H.; Wang, W.; Huang, X.R.; Oldfield, M.; Schmidt, A.M.; Cooper, M.E.; Lan, H.Y. Advanced glycation end products induce tubular epithelial-myofibroblast transition through the RAGE-ERK1/2 MAP kinase signaling pathway. Am. J. Pathol. 2004, 164, 1389–1397. [Google Scholar] [CrossRef] [Green Version]
- Burns, W.C.; Twigg, S.M.; Forbes, J.M.; Pete, J.; Tikellis, C.; Thallas-Bonke, V.; Thomas, M.C.; Cooper, M.E.; Kantharidis, P. Connective Tissue Growth Factor Plays an Important Role in Advanced Glycation End Product–Induced Tubular Epithelial-to-Mesenchymal Transition: Implications for Diabetic Renal Disease. J. Am. Soc. Nephrol. 2006, 17, 2484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, F.; Sano, Y.; Haruki, H.; Kanda, T. Advanced glycation end-products induce basement membrane hypertrophy in endoneurial microvessels and disrupt the blood–nerve barrier by stimulating the release of TGF-β and vascular endothelial growth factor (VEGF) by pericytes. Diabetologia 2011, 54, 1517–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.H.; Huang, X.R.; Zhu, H.J.; Oldfield, M.; Cooper, M.; Truong, L.D.; Johnson, R.J.; Lan, H.Y. Advanced glycation end products activate Smad signaling via TGF-beta-dependent and independent mechanisms: Implications for diabetic renal and vascular disease. FASEB J. 2004, 18, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.E. Pathogenesis, prevention, and treatment of diabetic nephropathy. Lancet 1998, 352, 213–219. [Google Scholar] [CrossRef]
- Sourris, K.C.; Harcourt, B.E.; Forbes, J.M. A new perspective on therapeutic inhibition of advanced glycation in diabetic microvascular complications: Common downstream endpoints achieved through disparate therapeutic approaches? Am. J. Nephrol. 2009, 30, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Twigg, S.M.; Chen, M.M.; Joly, A.H.; Chakrapani, S.D.; Tsubaki, J.; Kim, H.S.; Oh, Y.; Rosenfeld, R.G. Advanced glycosylation end products up-regulate connective tissue growth factor (insulin-like growth factor-binding protein-related protein 2) in human fibroblasts: A potential mechanism for expansion of extracellular matrix in diabetes mellitus. Endocrinology 2001, 142, 1760–1769. [Google Scholar] [CrossRef] [PubMed]
- Isoda, K.; Folco, E.; Marwali, M.R.; Ohsuzu, F.; Libby, P. Glycated LDL increases monocyte CC chemokine receptor 2 expression and monocyte chemoattractant protein-1-mediated chemotaxis. Atherosclerosis 2008, 198, 307–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bierhaus, A.; Schiekofer, S.; Schwaninger, M.; Andrassy, M.; Humpert, P.M.; Chen, J.; Hong, M.; Luther, T.; Henle, T.; Kloting, I.; et al. Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes 2001, 50, 2792–2808. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, A.M.; Hori, O.; Chen, J.X.; Li, J.F.; Crandall, J.; Zhang, J.; Cao, R.; Yan, S.D.; Brett, J.; Stern, D. Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice. A potential mechanism for the accelerated vasculopathy of diabetes. J. Clin. Investig. 1995, 96, 1395–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coughlan, M.T.; Thorburn, D.R.; Penfold, S.A.; Laskowski, A.; Harcourt, B.E.; Sourris, K.C.; Tan, A.L.; Fukami, K.; Thallas-Bonke, V.; Nawroth, P.P. RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J. Am. Soc. Nephrol. 2009, 20, 742–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myint, K.-M.; Yamamoto, Y.; Doi, T.; Kato, I.; Harashima, A.; Yonekura, H.; Watanabe, T.; Shinohara, H.; Takeuchi, M.; Tsuneyama, K. RAGE control of diabetic nephropathy in a mouse model: Effects of RAGE gene disruption and administration of low-molecular weight heparin. Diabetes 2006, 55, 2510–2522. [Google Scholar] [CrossRef] [Green Version]
- Sourris, K.C.; Morley, A.L.; Koitka, A.; Samuel, P.; Coughlan, M.T.; Penfold, S.A.; Thomas, M.C.; Bierhaus, A.; Nawroth, P.P.; Yamamoto, H.; et al. Receptor for AGEs (RAGE) blockade may exert its renoprotective effects in patients with diabetic nephropathy via induction of the angiotensin II type 2 (AT2) receptor. Diabetologia 2010, 53, 2442–2451. [Google Scholar] [CrossRef] [Green Version]
- Ozdemir, A.M.; Hopfer, U.; Rosca, M.V.; Fan, X.J.; Monnier, V.M.; Weiss, M.F. Effects of Advanced Glycation End Product Modification on Proximal Tubule Epithelial Cell Processing of Albumin. Am. J. Nephrol. 2008, 28, 14–24. [Google Scholar] [CrossRef]
- Gallicchio, M.A.; Bach, L.A. Uptake of advanced glycation end products by proximal tubule epithelial cells via macropinocytosis. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2013, 1833, 2922–2932. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, A.; Takabatake, Y.; Kimura, T.; Maejima, I.; Namba, T.; Yamamoto, T.; Matsuda, J.; Minami, S.; Kaimori, J.-Y.; Matsui, I.; et al. Autophagy Inhibits the Accumulation of Advanced Glycation End Products by Promoting Lysosomal Biogenesis and Function in the Kidney Proximal Tubules. Diabetes 2017, 66, 1359–1372. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.J.; Shen, T.T.; Chen, R.H.; Wu, H.-L.; Wang, Y.J.; Deng, J.K.; Chen, Q.H.; Pan, Q.; Huang Fu, C.-M.; Tao, J.-L.; et al. Autophagy-Lysosome Pathway in Renal Tubular Epithelial Cells Is Disrupted by Advanced Glycation End Products in Diabetic Nephropathy. J. Biol. Chem. 2015, 290, 20499–20510. [Google Scholar] [CrossRef] [Green Version]
- Ejtahed, H.-S.; Angoorani, P.; Asghari, G.; Mirmiran, P.; Azizi, F. Dietary Advanced Glycation End Products and Risk of Chronic Kidney Disease. J. Ren. Nutr. 2016, 26, 308–314. [Google Scholar] [CrossRef]
- Clarke, R.; Dordevic, A.; Tan, S.; Ryan, L.; Coughlan, M. Dietary Advanced Glycation End Products and Risk Factors for Chronic Disease: A Systematic Review of Randomised Controlled Trials. Nutrients 2016, 8, 125. [Google Scholar] [CrossRef] [Green Version]
- Normand, G.; Lemoine, S.; Villien, M.; Le Bars, D.; Merida, I.; Irace, Z.; Troalen, T.; Costes, N.; Juillard, L. AGE Content of a Protein Load Is Responsible for Renal Performances: A Pilot Study. Diabetes Care 2018, 41, 1292–1294. [Google Scholar] [CrossRef] [Green Version]
- Uribarri, J.; Stirban, A.; Sander, D.; Cai, W.; Negrean, M.; Buenting, C.E.; Koschinsky, T.; Vlassara, H. Single oral challenge by advanced glycation end products acutely impairs endothelial function in diabetic and nondiabetic subjects. Diabetes Care 2007, 30, 2579–2582. [Google Scholar] [CrossRef] [Green Version]
- Semba, R.D.; Gebauer, S.K.; Baer, D.J.; Sun, K.; Turner, R.; Silber, H.A.; Talegawkar, S.; Ferrucci, L.; Novotny, J.A. Dietary intake of advanced glycation end products did not affect endothelial function and inflammation in healthy adults in a randomized controlled trial. J. Nutr. 2014, 144, 1037–1042. [Google Scholar] [CrossRef] [Green Version]
- Baye, E.; de Courten, M.P.; Walker, K.; Ranasinha, S.; Earnest, A.; Forbes, J.M.; de Courten, B. Effect of dietary advanced glycation end products on inflammation and cardiovascular risks in healthy overweight adults: A randomised crossover trial. Sci. Rep. 2017, 7, 4123. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Zeng, M.; He, Z.; Zheng, Z.; Qin, F.; Tao, G.; Zhang, S.; Chen, J. Increased Accumulation of Protein-bound N-(carboxymethyl)lysine in Tissues of Healthy Rats after Chronic Oral N-(carboxymethyl)lysine. J. Agric. Food. Chem. 2015, 63, 1658–1663. [Google Scholar] [CrossRef]
- Sebekova, K.; Faist, V.; Hofmann, T.; Schinzel, R.; Heidland, A. Effects of a diet rich in advanced glycation end products in the rat remnant kidney model. Am. J. Kidney Dis. 2003, 41, S48–S51. [Google Scholar] [CrossRef]
- Feng, J.X.; Hou, F.F.; Liang, M.; Wang, G.B.; Zhang, X.; Li, H.Y.; Xie, D.; Tian, J.W.; Liu, Z.Q. Restricted intake of dietary advanced glycation end products retards renal progression in the remnant kidney model. Kidney Int. 2007, 71, 901–911. [Google Scholar] [CrossRef] [Green Version]
- de Assis, A.M.; Rech, A.; Longoni, A.; da Silva Morrone, M.; de Bittencourt Pasquali, M.A.; Perry, M.L.; Souza, D.O.; Moreira, J.C. Dietary n-3 polyunsaturated fatty acids revert renal responses induced by a combination of 2 protocols that increase the amounts of advanced glycation end product in rats. Nutr. Res. 2015, 35, 512–522. [Google Scholar] [CrossRef] [Green Version]
- Teissier, T.; Quersin, V.; Gnemmi, V.; Daroux, M.; Howsam, M.; Delguste, F.; Lemoine, C.; Fradin, C.; Schmidt, A.-M.; Cauffiez, C.; et al. Knockout of receptor for advanced glycation end-products attenuates age-related renal lesions. Aging Cell 2019, 18, e12850. [Google Scholar] [CrossRef]
- Borg, D.J.; Forbes, J.M. Targeting advanced glycation with pharmaceutical agents: Where are we now? Glycoconj. J. 2016, 33, 653–670. [Google Scholar] [CrossRef]
- Soulis-Liparota, T.; Cooper, M.; Papazoglou, D.; Clarke, B.; Jerums, G. Retardation by Aminoguanidine of Development of Albuminuria, Mesangial Expansion, and Tissue Fluorescence in Streptozocin-Induced Diabetic Rat. Diabetes 1991, 40, 1328–1334. [Google Scholar] [CrossRef]
- Thornalley, P.J. Use of aminoguanidine (Pimagedine) to prevent the formation of advanced glycation endproducts. Arch. Biochem. Biophys. 2003, 419, 31–40. [Google Scholar] [CrossRef]
- Degenhardt, T.P.; Alderson, N.L.; Arrington, D.D.; Beattie, R.J.; Basgen, J.M.; Steffes, M.W.; Thorpe, S.R.; Baynes, J.W. Pyridoxamine inhibits early renal disease and dyslipidemia in the streptozotocin-diabetic rat. Kidney Int. 2002, 61, 939–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alderson, N.L.; Chachich, M.E.; Youssef, N.N.; Beattie, R.J.; Nachtigal, M.; Thorpe, S.R.; Baynes, J.W. The AGE inhibitor pyridoxamine inhibits lipemia and development of renal and vascular disease in Zucker obese rats. Kidney Int. 2003, 63, 2123–2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, F.; Zeng, Y.j.; Plati, A.R.; Elliot, S.J.; Berho, M.; Potier, M.; Striker, L.J.; Striker, G.E. Combined AGE inhibition and ACEi decreases the progression of established diabetic nephropathy in B6 db/db mice. Kidney Int. 2006, 70, 507–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, M.E.; Bolton, W.K.; Khalifah, R.G.; Degenhardt, T.P.; Schotzinger, R.J.; McGill, J.B. Effects of pyridoxamine in combined phase 2 studies of patients with type 1 and type 2 diabetes and overt nephropathy. Am. J. Nephrol. 2007, 27, 605–614. [Google Scholar] [CrossRef]
- Vasan, S.; Zhang, X.; Zhang, X.; Kapurniotu, A.; Bernhagen, J.; Teichberg, S.; Basgen, J.; Wagle, D.; Shih, D.; Terlecky, I.; et al. An agent cleaving glucose-derived protein crosslinks in vitro and in vivo. Nature 1996, 382, 275–278. [Google Scholar] [CrossRef]
- Cooper, M.E.; Thallas, V.; Forbes, J.; Scalbert, E.; Sastra, S.; Darby, I.; Soulis, T. The cross-link breaker, N-phenacylthiazolium bromide prevents vascular advanced glycation end-product accumulation. Diabetologia 2000, 43, 660–664. [Google Scholar] [CrossRef] [Green Version]
- Schwedler, S.B.; Verbeke, P.; Bakala, H.; Weiss, M.F.; Vilar, J.; Depreux, P.; Fourmaintraux, E.; Striker, L.J.; Striker, G.E. N-phenacylthiazolium bromide decreases renal and increases urinary advanced glycation end products excretion without ameliorating diabetic nephropathy in C57BL/6 mice. Diabetes Obes. Metab. 2001, 3, 230–239. [Google Scholar] [CrossRef]
- Oturai, P.S.; Christensen, M.; Rolin, B.; Pedersen, K.E.; Mortensen, S.B.; Boel, E. Effects of advanced glycation end-product inhibition and cross-link breakage in diabetic rats. Metab.-Clin. Exp. 2000, 49, 996–1000. [Google Scholar] [CrossRef]
- Thallas-Bonke, V.; Lindschau, C.; Rizkalla, B.; Bach, L.A.; Boner, G.; Meier, M.; Haller, H.; Cooper, M.E.; Forbes, J.M. Attenuation of Extracellular Matrix Accumulation in Diabetic Nephropathy by the Advanced Glycation End Product Cross-Link Breaker ALT-711 via a Protein Kinase C-α−Dependent Pathway. Diabetes 2004, 53, 2921–2930. [Google Scholar] [CrossRef] [Green Version]
- Forbes, J.M.; Thallas, V.; Thomas, M.C.; Founds, H.W.; Burns, W.C.; Jerums, G.; Cooper, M.E. The breakdown of preexisting advanced glycation end products is associated with reduced renal fibrosis in experimental diabetes. FASEB J. 2003, 17, 1762–1764. [Google Scholar] [CrossRef]
- Shapiro, B.P.; Owan, T.E.; Mohammed, S.F.; Meyer, D.M.; Mills, L.D.; Schalkwijk, C.G.; Redfield, M.M. Advanced glycation end products accumulate in vascular smooth muscle and modify vascular but not ventricular properties in elderly hypertensive canines. Circulation 2008, 118, 1002–1010. [Google Scholar] [CrossRef] [Green Version]
- Reiniger, N.; Lau, K.; McCalla, D.; Eby, B.; Cheng, B.; Lu, Y.; Qu, W.; Quadri, N.; Ananthakrishnan, R.; Furmansky, M.; et al. Deletion of the Receptor for Advanced Glycation End Products Reduces Glomerulosclerosis and Preserves Renal Function in the Diabetic OVE26 Mouse. Diabetes 2010, 59, 2043–2054. [Google Scholar] [CrossRef] [Green Version]
- Tesch, G.; Sourris, K.C.; Summers, S.A.; McCarthy, D.; Ward, M.S.; Borg, D.J.; Gallo, L.A.; Fotheringham, A.K.; Pettit, A.R.; Yap, F.Y.T.; et al. Deletion of bone-marrow-derived receptor for AGEs (RAGE) improves renal function in an experimental mouse model of diabetes. Diabetologia 2014, 57, 1977–1985. [Google Scholar] [CrossRef] [Green Version]
- Jensen, L.J.N.; Denner, L.; Schrijvers, B.F.; Tilton, R.G.; Rasch, R.; Flyvbjerg, A. Renal effects of a neutralising RAGE-antibody in long-term streptozotocin-diabetic mice. J. Endocrinol. 2006, 188, 493–501. [Google Scholar] [CrossRef]
- Flyvbjerg, A.; Denner, L.; Schrijvers, B.F.; Tilton, R.G.; Mogensen, T.H.; Paludan, S.R.; Rasch, R. Long-Term Renal Effects of a Neutralizing RAGE Antibody in Obese Type 2 Diabetic Mice. Diabetes 2004, 53, 166–172. [Google Scholar] [CrossRef] [Green Version]
- Kaida, Y.; Fukami, K.; Matsui, T.; Higashimoto, Y.; Nishino, Y.; Obara, N.; Nakayama, Y.; Ando, R.; Toyonaga, M.; Ueda, S.; et al. DNA Aptamer Raised Against AGEs Blocks the Progression of Experimental Diabetic Nephropathy. Diabetes 2013, 62, 3241–3250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, T.; Higashimoto, Y.; Nishino, Y.; Nakamura, N.; Fukami, K.; Yamagishi, S.-I. RAGE-Aptamer Blocks the Development and Progression of Experimental Diabetic Nephropathy. Diabetes 2017, 66, 1683–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taguchi, K.; Yamagishi, S.-I.; Yokoro, M.; Ito, S.; Kodama, G.; Kaida, Y.; Nakayama, Y.; Ando, R.; Yamada-Obara, N.; Asanuma, K.; et al. RAGE-aptamer attenuates deoxycorticosterone acetate/salt-induced renal injury in mice. Sci. Rep. 2018, 8, 2686. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author | Amadori Product (AGE Precursor) | Study Population | Methodology | Key Findings | Measurement Technique |
| [58] Erbesdobler and Faist, 2001 | Fructoselysine, Fructoseglycine | Rats and Humans | In vitro, everted gut sack, in vivo ligated jejunal segments |
| 14C Radioactivity |
| [59] Forster et al. 2005 | Pyralline, fructoselysine, pentosidine | Humans | Exclusion diet, followed by a high AGE test meal |
| Reversed Phase HPLC with UV detection |
| [60] Hultsch et al. 2006 | Fructoselysine | Rats | Injected and gavaged 18F flourobenzoylated fructoselysine. Biodistribution and catabolism study performed using PET scanning |
| PET scanning/Radioactivity counting |
| [61] Schwenger et al. 2006 | Lactuloselysine | Humans | Diet administered to healthy, diabetic and renal failure patients. Plasma concentrations and cumulative urinary excretion examined |
| Reverse Phase HPLC |
| Author | AGEs | Study Population | Methodology | Key Findings | Measurement Technique |
| [62] Liardon et al. 1987 | CML | Rats | Diets varying in quantity AGEs- timed urine collection and analysis for CML |
| Mass Spec |
| [55] Koschinsky et al. 1997 | Protein bound AGEs | Humans (incl. diabetes with/without DKD) | Single meal challenge, AGE egg white or fructose + egg white |
| ELISA |
| [63] Miyata et al.1998 | Pentosidine | Rats | IV injection with radiolabelled pentosidine, urine collected over 72 h |
|
Radioactivity/ ELISA |
| [54] He et al. 1999 | AGE-Ovalbumin | Rats | Fed a single dose of 14C or 125I labelled AGE-ovalbumin. Collected tissues, plasma and 72 h urine collection. |
|
Radioactivity/ ELISA |
| [64] Bergman et al. 2001 | CML and CEL (free) | Rats | Biodistribution study of intravenously administered 18F labelled AGEs using PET scanning and radioactive counting |
| PET scanning |
| [65] Somoza et al. 2006 | CML, LAL, FL (Casein linked) | Rats | Casein linked AGE feeding (2 dosages) to metabolic caged rodents |
| HP-LC-UV fluorescence |
| [57] Sebekova et al. 2008 | CML | Human infants | Comparison of circulating AGEs between breast and formula fed infants |
| LC-MS/MS |
| [66] Roncero-Ramos et al. 2013 | CML | Rats | 88 days on high or low AGE diet—Bread crust or it’s insoluble (HMW) or soluble fractions (LMW) |
| HPLC-MS/MS |
| [67] Alamir et al. 2013 | Extruded or non-extruded protein diet CML | Rats | 6 weeks feeding on extruded or non-extruded protein diet or single oral free CML challenge |
| LC-ESI-MS/MS |
| [68] Xu et al. 2013 | CML (free) | Mice | Biodistribution and elimination study 18F labelled CML in mice. Tracer labelled CML was administered either IV or intra-gastrically |
| PET scanning |
| [69] Tessier et al. 2016 | CML (protein bound) | Mice | 30 days feeding with a diet enriched with 13C-CML that could be differentiated from native CML in C57BL/6J and RAGE knock out mice |
| Stable isotope dilution analysis LC-MS/MS |
| [70] Tsutsui et al. 2016 | AGE-Albumin | Mice | Single IV injection of Cy 7.5 labelled AGE-BSA. Fluorescence kinetics assay performed |
| IVIS whole animal in vivo imaging system |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fotheringham, A.K.; Gallo, L.A.; Borg, D.J.; Forbes, J.M. Advanced Glycation End Products (AGEs) and Chronic Kidney Disease: Does the Modern Diet AGE the Kidney? Nutrients 2022, 14, 2675. https://0-doi-org.brum.beds.ac.uk/10.3390/nu14132675
Fotheringham AK, Gallo LA, Borg DJ, Forbes JM. Advanced Glycation End Products (AGEs) and Chronic Kidney Disease: Does the Modern Diet AGE the Kidney? Nutrients. 2022; 14(13):2675. https://0-doi-org.brum.beds.ac.uk/10.3390/nu14132675
Chicago/Turabian StyleFotheringham, Amelia K., Linda A. Gallo, Danielle J. Borg, and Josephine M. Forbes. 2022. "Advanced Glycation End Products (AGEs) and Chronic Kidney Disease: Does the Modern Diet AGE the Kidney?" Nutrients 14, no. 13: 2675. https://0-doi-org.brum.beds.ac.uk/10.3390/nu14132675