
SGLT-2 Inhibitors and the Inflammasome: What’s Next in the 21st Century?
 ,
,
Abstract
:1. Introduction
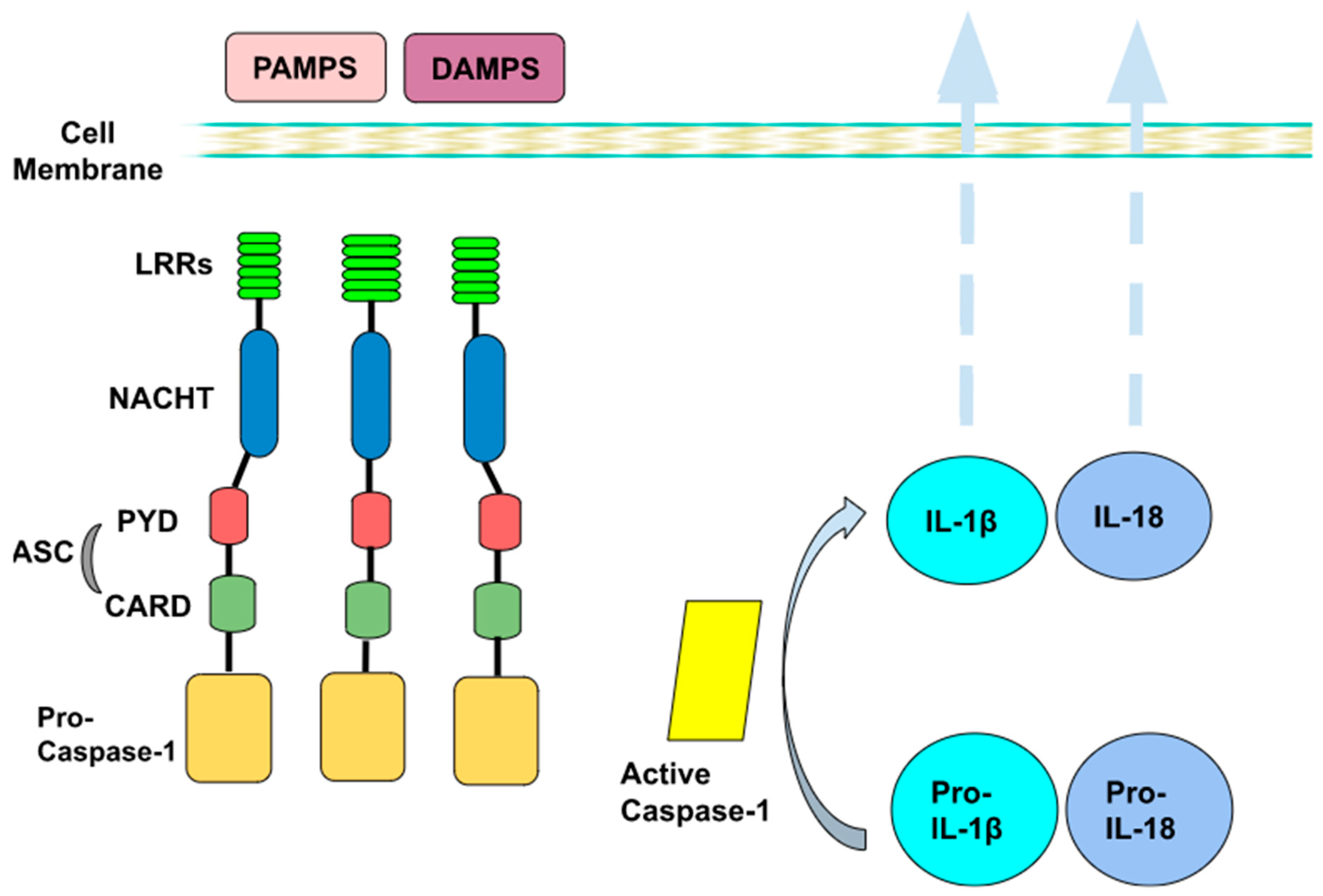
2. Inflammasome’s Mechanism of Action
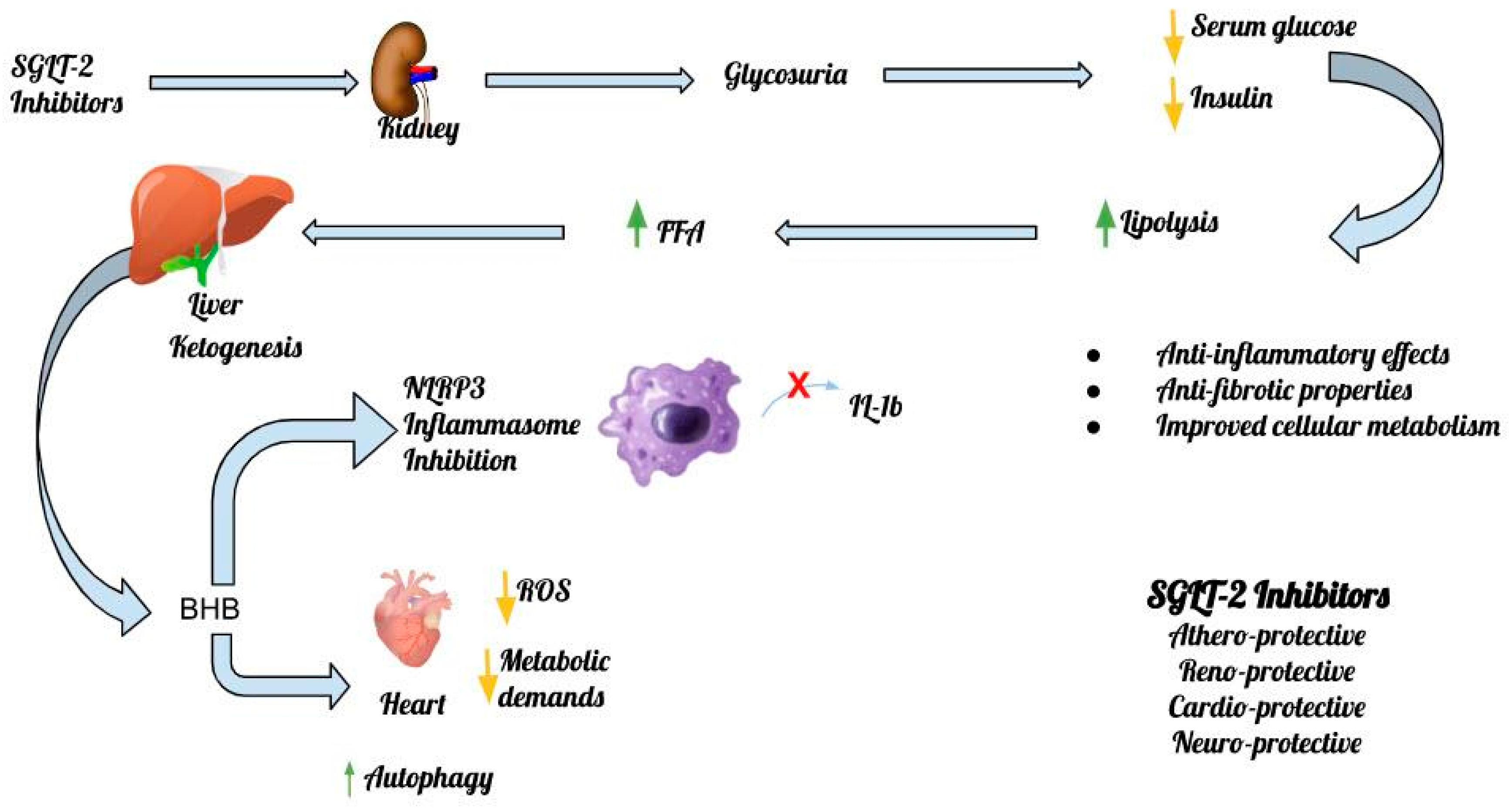
3. SGLT-2 Inhibitors’ Anti-Inflammatory Effects
4. SGLT-2 Inhibitors, the Inflammasome and the Heart
5. SGLT-2 Inhibitors, the Inflammasome and the Kidney
6. SGLT-2 Inhibitors, the Inflammasome and the CNS
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Caparrotta, T.M.; Greenhalgh, A.M.; Osinski, K.; Gifford, R.M.; Moser, S.; Wild, S.H.; Reynolds, R.M.; Webb, D.J.; Colhoun, H.M. Sodium–Glucose Co-Transporter 2 Inhibitors (SGLT2i) Exposure and Outcomes in Type 2 Diabetes: A Systematic Review of Population-Based Observational Studies. Diabetes Ther. 2021, 12, 991–1028. [Google Scholar] [CrossRef] [PubMed]
- Kang, A.; Jardine, M.J. SGLT2 inhibitors may offer benefit beyond diabetes. Nat. Rev. Nephrol. 2021, 17, 83–84. [Google Scholar] [CrossRef]
- Cefalu, W.T.; Riddle, M.C. SGLT2 Inhibitors: The Latest “New Kids on the Block”! Diabetes Care 2015, 38, 352–354. [Google Scholar] [CrossRef]
- Vallianou, N.G.; Christodoulatos, G.S.; Kounatidis, D.; Dalamaga, M. Sotagliflozin, a dual SGLT1 and SGLT2 inhibitor: In the heart of the problem. Metab. Open 2021, 10, 100089. [Google Scholar] [CrossRef] [PubMed]
- Petersen, C. Analyse des Phloridzins. Ann. Acad. Sci. Fenn. 1835, 15, 178. [Google Scholar] [CrossRef]
- White, J.R. Apple Trees to Sodium Glucose Co-Transporter Inhibitors: A Review of SGLT2 Inhibition. Clin. Diabetes 2010, 28, 5–10. [Google Scholar] [CrossRef]
- Vallianou, N.G.; Trigkidis, K.; Kazazis, C. Sodium glucose co-transporter 2 inhibitors and their nephroprotective potential. Clin. Nephrol. 2017, 87, 212–216. [Google Scholar] [CrossRef]
- Wolters Kluwer. Available online: https://www.uptodate.com (accessed on 15 April 2023).
- The Human Protein Atlas Single Cell Type—NLRP3. Available online: https://www.proteinatlas.org/ENSG00000162711-NLRP3/single+cell+type (accessed on 12 April 2023).
- Marcuzzi, A.; Melloni, E.; Zauli, G.; Romani, A.; Secchiero, P.; Maximova, N.; Rimondi, E. Autoinflammatory Diseases and Cytokine Storms—Imbalances of Innate and Adaptative Immunity. Int. J. Mol. Sci. 2021, 22, 11241. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Burdette, B.E.; Esparza, A.N.; Zhu, H.; Wang, S. Gasdermin D in pyroptosis. Acta Pharm. Sin. B 2021, 11, 2768–2782. [Google Scholar] [CrossRef]
- Lee, H.-M.; Kim, J.-J.; Kim, H.J.; Shong, M.; Ku, B.J.; Jo, E.-K. Upregulated NLRP3 Inflammasome Activation in Patients with Type 2 Diabetes. Diabetes 2013, 62, 194–204. [Google Scholar] [CrossRef]
- Abbate, A.; Toldo, S.; Marchetti, C.; Kron, J.; Van Tassell, B.W.; Dinarello, C.A. Interleukin-1 and the Inflammasome as Therapeutic Targets in Cardiovascular Disease. Circ. Res. 2020, 126, 1260–1280. [Google Scholar] [CrossRef]
- Xiong, W.; Meng, X.-F.; Zhang, C. NLRP3 Inflammasome in Metabolic-Associated Kidney Diseases: An Update. Front. Immunol. 2021, 12, 714340. [Google Scholar] [CrossRef] [PubMed]
- Feijóo-Bandín, S.; Aragón-Herrera, A.; Otero-Santiago, M.; Anido-Varela, L.; Moraña-Fernández, S.; Tarazón, E.; Roselló-Lletí, E.; Portolés, M.; Gualillo, O.; González-Juanatey, J.R.; et al. Role of Sodium-Glucose Co-Transporter 2 Inhibitors in the Regulation of Inflammatory Processes in Animal Models. Int. J. Mol. Sci. 2022, 23, 5634. [Google Scholar] [CrossRef] [PubMed]
- Al Mamun, A.; Akter, A.; Hossain, S.; Sarker, T.; Safa, S.A.; Mustafa, Q.G.; Muhammad, S.A.; Munir, F. Role of NLRP3 In-flammasome in Liver Disease. J. Dig. Dis. 2020, 21, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Lee, S.-G.; Kim, S.H.; Kim, J.H.; Choi, E.; Cho, W.; Rim, J.H.; Hwang, I.; Lee, C.J.; Lee, M.; et al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat. Commun. 2020, 11, 2127. [Google Scholar] [CrossRef]
- Elrakaybi, A.; Laubner, K.; Zhou, Q.; Hug, M.J.; Seufert, J. Cardiovascular protection by SGLT2 inhibitors—Do anti-inflammatory mechanisms play a role? Mol. Metab. 2022, 64, 101549. [Google Scholar] [CrossRef] [PubMed]
- Tsigalou, C.; Vallianou, N.; Dalamaga, M. Autoantibody Production in Obesity: Is There Evidence for a Link Between Obesity and Autoimmunity? Curr. Obes. Rep. 2020, 9, 245–254. [Google Scholar] [CrossRef]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Paulus, W.J.; Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 23, 263–271. [Google Scholar] [CrossRef]
- Masood, H.; Che, R.; Zhang, A. Inflammasomes in the Pathophysiology of Kidney Diseases. Kidney Dis. 2015, 1, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.L.; Davidson, M.T.; Kurishima, C.; Vega, R.B.; Powers, J.C.; Matsuura, T.R.; Petucci, C.; Lewandowski, E.D.; Crawford, P.A.; Muoio, D.M.; et al. The failing heart utilizes 3-hydroxybutyrate as a metabolic stress defense. J. Clin. Investig. 2019, 4, e124079. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.-H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.-D.; et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome–mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.R.; Kim, D.H.; Park, M.H.; Lee, B.; Kim, M.J.; Lee, E.K.; Chung, K.W.; Kim, S.M.; Im, D.S.; Chung, H.Y. β-Hydroxybutyrate suppresses inflammasome formation by ameliorating endoplasmic reticulum stress via AMPK activation. Oncotarget 2016, 7, 66444–66454. [Google Scholar] [CrossRef]
- Deng, Y.; Xie, M.; Li, Q.; Xu, X.; Ou, W.; Zhang, Y.; Xiao, H.; Yu, H.; Zheng, Y.; Liang, Y.; et al. Targeting Mitochondria-Inflammation Circuit by β-Hydroxybutyrate Mitigates HFpEF. Circ. Res. 2021, 128, 232–245. [Google Scholar] [CrossRef]
- Liakos, A.; Tsapas, A.; Bekiari, E. Some glucose-lowering drugs reduce risk for major adverse cardiac events. Ann. Intern. Med. 2020, 173, JC9. [Google Scholar] [CrossRef]
- Nasiri-Ansari, N.; Nikolopoulou, C.; Papoutsi, K.; Kyrou, I.; Mantzoros, C.S.; Kyriakopoulos, G.; Chatzigeorgiou, A.; Kalotychou, V.; Randeva, M.S.; Chatha, K.; et al. Empagliflozin Attenuates Non-Alcoholic Fatty Liver Disease (NAFLD) in High Fat Diet Fed ApoE(−/−) Mice by Activating Autophagy and Reducing ER Stress and Apoptosis. Int. J. Mol. Sci. 2021, 22, 818. [Google Scholar] [CrossRef]
- Gordon, M.; Meagher, P.; Connelly, K.A. Effect of Empagliflozin and Liraglutide on the Nucleotide-Binding and Oligomerization Domain-Like Receptor Family Pyrin Domain-Containing 3 Inflammasome in a Rodent Model of Type 2 Diabetes Mellitus. Can. J. Diabetes 2021, 45, 553–556. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Carbone, S.; Dixon, D.L. The CANVAS Program: Implications of canagliflozin on reducing cardiovascular risk in patients with type 2 diabetes mellitus. Cardiovasc. Diabetol. 2019, 18, 64. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [PubMed]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner–La Rocca, H.-P.; Choi, D.-J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Theofilis, P.; Antonopoulos, A.S.; Katsimichas, T.; Oikonomou, E.; Siasos, G.; Aggeli, C.; Tsioufis, K.; Tousoulis, D. The impact of SGLT2 inhibition on imaging markers of cardiac function: A systematic review and meta-analysis. Pharmacol. Res. 2022, 180, 6243. [Google Scholar] [CrossRef] [PubMed]
- Theofilis, P.; Sagris, M.; Oikonomou, E.; Antonopoulos, A.S.; Siasos, G.; Tsioufis, K.; Tousoulis, D. Pleiotropic effects of SGLT2 inhibitors and heart failure outcomes. Diabetes Res. Clin. Pract. 2022, 188, 109927. [Google Scholar] [CrossRef]
- Vallianou, N.G.; Tsilingiris, D.; Kounatidis, D.; Lempesis, I.G.; Karampela, I.; Dalamaga, M. Sodium glucose cotransporter 2 inhibitors in obesity and associated cardiometabolic disorders: Where do we stand? Pol. Arch. Intern. Med. 2022, 132, 6342. [Google Scholar] [CrossRef]
- Braunwald, E. Gliflozins in the Management of Cardiovascular Disease. N. Engl. J. Med. 2022, 386, 2024–2034. [Google Scholar] [CrossRef]
- Theofilis, P.; Oikonomou, E.; Tsioufis, K.; Tousoulis, D. Diabetes Mellitus and Heart Failure: Epidemiology, Pathophysiologic Mechanisms, and the Role of SGLT2 Inhibitors. Life 2023, 13, 497. [Google Scholar] [CrossRef]
- Masson, W.; Lavalle-Cobo, A.; Nogueira, J.P. Effect of SGLT2-Inhibitors on Epicardial Adipose Tissue: A Meta-Analysis. Cells 2021, 10, 2150. [Google Scholar] [CrossRef]
- Uzu, T.; Yokoyama, H.; Itoh, H.; Koya, D.; Nakagawa, A.; Nishizawa, M.; Maegawa, H.; Yokomaku, Y.; Araki, S.-I.; Abiko, A.; et al. Elevated serum levels of interleukin-18 in patients with overt diabetic nephropathy: Effects of miglitol. Clin. Exp. Nephrol. 2011, 15, 58–63. [Google Scholar] [CrossRef]
- Lei, Y.; Devarapu, S.K.; Motrapu, M.; Cohen, C.D.; Lindenmeyer, M.T.; Moll, S.; Kumar, S.V.; Anders, H.-J. Interleukin-1β Inhibition for Chronic Kidney Disease in Obese Mice with Type 2 Diabetes. Front. Immunol. 2019, 10, 1223. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, K.; Bock, F.; Dong, W.; Wang, H.; Kopf, S.; Kohli, S.; Al-Dabet, M.M.; Ranjan, S.; Wolter, J.; Wacker, C.; et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015, 87, 74–84. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, X.; Zhang, X.; Lu, D.; Guo, R. Electro-Acupuncture Protects Diabetic Nephropathy-Induced Inflammation Through Suppression of NLRP3 Inflammasome in Renal Macrophage Isolation. Endocr. Metab. Immune Disord. Drug Targets 2021, 21, 2075–2083. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Meng, X.-F.; Zhang, C. Inflammasome activation in podocytes: A new mechanism of glomerular diseases. Inflamm. Res. 2020, 69, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Yang, Z.; Zhang, C.; Shi, Y.; Han, W.; Song, S.; Mu, L.; Du, C.; Shi, Y. Inhibition of NLRP3 inflammasome ameliorates podocyte damage by suppressing lipid accumulation in diabetic nephropathy. Metabolism 2021, 118, 154748. [Google Scholar] [CrossRef]
- Xie, C.; Wu, W.; Tang, A.; Luo, N.; Tan, Y. lncRNA GAS5/miR-452-5p Reduces Oxidative Stress and Pyroptosis of High-Glucose-Stimulated Renal Tubular Cells. Diabetes Metabol. Syndr. Obes. 2019, 12, 2609–2617. [Google Scholar] [CrossRef]
- Yi, H.; Peng, R.; Zhang, L.-Y.; Sun, Y.; Peng, H.-M.; Liu, H.-D.; Yu, L.-J.; Li, A.-L.; Zhang, Y.-J.; Jiang, W.-H.; et al. LincRNA-Gm4419 knockdown ameliorates NF-κB/NLRP3 inflammasome-mediated inflammation in diabetic nephropathy. Cell Death Dis. 2017, 8, e2583. [Google Scholar] [CrossRef]
- Tung, C.-W.; Hsu, Y.-C.; Shih, Y.-H.; Chang, P.-J.; Lin, C.-L. Glomerular mesangial cell and podocyte injuries in diabetic nephropathy. Nephrology 2018, 23 (Suppl. 4), 32–37. [Google Scholar] [CrossRef]
- Zhang, C.; Zhu, X.; Li, L.; Ma, T.; Shi, M.; Yang, Y.; Fan, Q. A small molecule inhibitor MCC950 ameliorates kidney injury in diabetic nephropathy by inhibiting NLRP3 inflammasome activation. Diabetes Metab. Syndr. Obes. Targets Ther. 2019, 12, 1297–1309. [Google Scholar] [CrossRef]
- Wang, S.; Li, Y.; Fan, J.; Zhang, X.; Luan, J.; Bian, Q.; Ding, T.; Wang, Y.; Wang, Z.; Song, P.; et al. Interleukin-22 ameliorated renal injury and fibrosis in diabetic nephropathy through inhibition of NLRP3 inflammasome activation. Cell Death Dis. 2017, 8, e2937. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Han, W.; Song, S.; Du, Y.; Liu, C.; Chen, N.; Wu, H.; Shi, Y.; Duan, H. NLRP3 deficiency ameliorates renal inflammation and fibrosis in diabetic mice. Mol. Cell. Endocrinol. 2018, 478, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Soares, J.L.S.; Fernandes, F.P.; Patente, T.A.; Monteiro, M.B.; Parisi, M.C.; Giannella-Neto, D.; Corrêa-Giannella, M.L.; Pontillo, A. Gain-of-function Variants in NLRP1 Protect against the Development of Diabetic Kidney Disease: NLRP1 Inflammasome Role in Metabolic Stress Sensing? Clin. Immunol. 2018, 187, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Luan, P.; Zhuang, J.; Zou, J.; Li, H.; Shuai, P.; Xu, X.; Zhao, Y.; Kou, W.; Ji, S.; Peng, A.; et al. NLRC5 deficiency ameliorates diabetic nephropathy through alleviating inflammation. FASEB J. 2018, 32, 1070–1084. [Google Scholar] [CrossRef]
- Yuan, F.; Kolb, R.; Pandey, G.; Li, W.; Sun, L.; Liu, F.; Sutterwala, F.S.; Liu, Y.; Zhang, W. Involvement of the NLRC4-Inflammasome in Diabetic Nephropathy. PLoS ONE 2016, 11, e0164135. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Huang, L.; Shi, X.; Yang, L.; Hua, F.; Ma, J.; Zhu, W.; Liu, X.; Xuan, R.; Shen, Y.; et al. Metformin protects against myocardial ischemia-reperfusion injury and cell pyroptosis via AMPK/NLRP3 inflammasome pathway. Aging 2020, 12, 24270–24287. [Google Scholar] [CrossRef]
- Lech, M.; Avila-Ferrufino, A.; Skuginna, V.; Susanti, H.E.; Anders, H.-J. Quantitative expression of RIG-like helicase, NOD-like receptor and inflammasome-related mRNAs in humans and mice. Int. Immunol. 2010, 22, 717–728. [Google Scholar] [CrossRef]
- Komada, T.; Chung, H.; Lau, A.; Platnich, J.M.; Beck, P.L.; Benediktsson, H.; Duff, H.J.; Jenne, C.N.; Muruve, D.A. Macrophage Uptake of Necrotic Cell DNA Activates the AIM2 Inflammasome to Regulate a Proinflammatory Phenotype in CKD. J. Am. Soc. Nephrol. 2018, 29, 1165–1181. [Google Scholar] [CrossRef]
- Komada, T.; Muruve, D.A. The role of inflammasomes in kidney disease. Nat. Rev. Nephrol. 2019, 15, 501–520. [Google Scholar] [CrossRef]
- Valiño-Rivas, L.; Cuarental, L.; Nuñez, G.; Sanz, A.B.; Ortiz, A.; Sanchez-Niño, M.D. Loss of NLRP6 expression increases the severity of acute kidney injury. Nephrol. Dial. Transplant. 2020, 35, 587–598. [Google Scholar] [CrossRef]
- Zhang, W.; Cai, Y.; Xu, W.; Yin, Z.; Gao, X.; Xiong, S. AIM2 Facilitates the Apoptotic DNA-Induced Systemic Lupus Erythematosus via Arbitrating Macrophage Functional Maturation. J. Clin. Immunol. 2013, 33, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Zhen, J.; Zhang, L.; Pan, J.; Ma, S.; Yu, X.; Li, X.; Chen, S.; Du, W. AIM2 Mediates Inflammation-Associated Renal Damage in Hepatitis B Virus-Associated Glomerulonephritis by Regulating Caspase-1, IL-1β, and IL-18. Mediat. Inflamm. 2014, 2014, 190860. [Google Scholar] [CrossRef] [PubMed]
- Ye, T.; Zhang, J.; Wu, D.; Shi, J.; Kuang, Z.; Ma, Y.; Xu, Q.; Chen, B.; Kan, C.; Sun, X.; et al. Empagliflozin Attenuates Obesity-Related Kidney Dysfunction and NLRP3 Inflammasome Activity Through the HO-1–Adiponectin Axis. Front. Endocrinol. 2022, 13, 907984. [Google Scholar] [CrossRef] [PubMed]
- Benetti, E.; Mastrocola, R.; Vitarelli, G.; Cutrin, J.C.; Nigro, D.; Chiazza, F.; Mayoux, E.; Collino, M.; Fantozzi, R. Empagliflozin Protects against Diet-Induced NLRP-3 Inflammasome Activation and Lipid Accumulation. J. Pharmacol. Exp. Ther. 2016, 359, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, Y.; Bajaj, M.; Yang, H.-C.; Ye, Y. Combined SGLT2 and DPP4 Inhibition Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Nephropathy in Mice with Type 2 Diabetes. Cardiovasc. Drugs Ther. 2018, 32, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; Von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B.; et al. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef]
- Ke, Q.; Shi, C.; Lv, Y.; Wang, L.; Luo, J.; Jiang, L.; Yang, J.; Zhou, Y. SGLT2 inhibitor counteracts NLRP3 inflammasome via tubular metabolite itaconate in fibrosis kidney. FASEB J. 2022, 36, e22078. [Google Scholar] [CrossRef]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Ye, Y.; Bajaj, M.; Yang, H.-C.; Perez-Polo, J.R.; Birnbaum, Y. SGLT-2 Inhibition with Dapagliflozin Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Cardiomyopathy in Mice with Type 2 Diabetes. Further Augmentation of the Effects with Saxagliptin, a DPP4 Inhibitor. Cardiovasc. Drugs Ther. 2017, 31, 119–132. [Google Scholar] [CrossRef]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.-F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Herrington, W.G.; Staplin, N.; Wanner, C.; Green, J.B.; Hauske, S.J.; Emberson, J.R.; Preiss, D.; Judge, P.; Mayne, K.J.; Ng, S.Y.A.; et al. Empagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2023, 388, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Tahara, A.; Takasu, T.; Yokono, M.; Imamura, M.; Kurosaki, E. Characterization and comparison of sodium-glucose cotransporter 2 inhibitors in pharmacokinetics, pharmacodynamics, and pharmacologic effects. J. Pharmacol. Sci. 2016, 130, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.K.; DeSilva, S.; Abbruscato, T.J. The Role of Glucose Transporters in Brain Disease: Diabetes and Alzheimer’s Disease. Int. J. Mol. Sci. 2012, 13, 12629–12655. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, H. Glucose transporters in brain in health and disease. Pflugers Arch. Eur. J. Physiol. 2020, 472, 1299–1343. [Google Scholar] [CrossRef]
- Enerson, B.E.; Drewes, L.R. The Rat Blood—Brain Barrier Transcriptome. J. Cereb. Blood Flow Metab. 2006, 26, 959–973. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Wen, S.; Gong, M.; Yuan, X.; Xu, D.; Wang, C.; Jin, J.; Zhou, L. Dapagliflozin Activates Neurons in the Central Nervous System and Regulates Cardiovascular Activity by Inhibiting SGLT-2 in Mice. Diabetes Metab. Syndr. Obes. Targets Ther. 2020, 13, 2781–2799. [Google Scholar] [CrossRef]
- Gaur, A.; Pal, G.K.; Ananthanarayanan, P.H.; Pal, P. Role of Ventromedial hypothalamus in high fat diet induced obesity in male rats: Association with lipid profile, thyroid profile and insulin resistance. Ann. Neurosci. 2014, 21, 104–107. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Jin, Y.; Fu, J. Novel Insights into the NLRP3 Inflammasome in Atherosclerosis. J. Am. Heart Assoc. 2019, 8, e012219. [Google Scholar] [CrossRef]
- Van der Heijden, T.; Kritikou, E.; Venema, W.; van Duijn, J.; van Santbrink, P.J.; Slütter, B.; Foks, A.C.; Bot, I.; Kuiper, J. NLRP3 Inflammasome Inhibition by MCC950 Reduces Atherosclerotic Lesion Development in Apolipoprotein E–Deficient Mice—Brief Report. Arter. Thromb. Vasc. Biol. 2017, 37, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Hierro-Bujalance, C.; Infante-Garcia, C.; del Marco, A.; Herrera, M.; Carranza-Naval, M.J.; Suarez, J.; Alves-Martinez, P.; Lubian-Lopez, S.; Garcia-Alloza, M. Empagliflozin reduces vascular damage and cognitive impairment in a mixed murine model of Alzheimer’s disease and type 2 diabetes. Alzheimer’s Res. Ther. 2020, 12, 40. [Google Scholar] [CrossRef] [PubMed]
- Tejera, D.; Mercan, D.; Sanchez-Caro, J.M.; Hanan, M.; Greenberg, D.; Soreq, H.; Latz, E.; Golenbock, D.; Heneka, M.T. Systemic inflammation impairs microglial Aβ clearance throughNLRP3 inflammasome. EMBO J. 2019, 38, e101064. [Google Scholar] [CrossRef] [PubMed]
- Lonnemann, N.; Hosseini, S.; Marchetti, C.; Skouras, D.B.; Stefanoni, D.; D’alessandro, A.; Dinarello, C.A.; Korte, M. The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2020, 117, 32145–32154. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-Y.; Iskander, C.; Wang, C.; Xiong, L.Y.; Shah, B.R.; Edwards, J.D.; Kapral, M.K.; Herrmann, N.; Lanctôt, K.L.; Masellis, M.; et al. Association of Sodium–Glucose Cotransporter 2 Inhibitors with Time to Dementia: A Population-Based Cohort Study. Diabetes Care 2023, 46, 297–304. [Google Scholar] [CrossRef]
- Vallianou, N.G.; Geladari, E.; Kazazis, C.E. SGLT-2 inhibitors: Their pleiotropic properties. Diabetes Metab. Syndr. Clin. Res. Rev. 2017, 11, 311–315. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Title of Study/Reference | Animal Model | Findings of the Study | Remarks |
|---|---|---|---|
| List of Studies with SGLT-2 Inhibitors | |||
| SGLT inhibitor counteracts NLRP3 inflammasome via tubular metabolite itaconate in fibrosis kidney. Ke et al., 2022 [68,69]. | Animal model regarding the anti-fibrotic and anti-inflammatory effects of DAPA on renal parameters using the ischemia/reperfusion-induced fibrosis model. |
|
|
| Empagliflozin attenuates obesity related kidney dysfunction and NLRP3 inflammasome activity through the HO-1 Adiponectin axis. Ye T et al., 2022 [65,68]. | C57BL/6J mice were assigned to control, HFD groups and EMPA (10 mg/kg) groups. |
|
|
| Empagliflozin protects against diet-induced NLRP3 inflammasome activation and lipid accumulation. Benetti et al., 2016 [66,67,68]. | Male C57BL/6 mice were assigned to a control or HFHS diet for 4 months. Over the last 2 months, subsets of male mice were also treated with EMPA (1–10 mg/kg). |
|
|
| List of Studies with a Combination of SGLT-2 Inhibitors and GLP-1 Analogs and/or DPP4 Inhibitors | |||
| SGLT-2 inhibition with dapagliflozin reduced the activation of the NLRP3/ASC inflammasome and attenuates development of diabetic cardiomyopathy in mice with type 2 diabetes. Further augmentation of the effects with saxagliptin, a DPP4 inhibitor. Ye et al., 2017 [70,71]. | Type 2 diabetic (BTBR ob/ob) and WT mice were assigned to vehicle, DAPA or DAPA + SAXA groups for 8 weeks. Cardiac fibroblasts from WT and BTBR hearts were incubated with DAPA and exposed to LPS. |
|
|
| Combined SGLT2 and DPPA inhibition reduces the activation of the NLRP3/ASC inflammasome and attenuates the development of diabetic nephropathy in mice with type 2 diabetes. Birnbaum et al., 2018 [67]. | Male BTBR ob/ob and wild-type (WT) mice received a vehicle, DAPA or DAPA + SAXA for 8 weeks. |
| mRNA levels of NALP3, ASC, IL-1β, IL-6, caspase-1, TNF-α, collagen-1 and collagen-3 were significantly ↑ in the kidneys of the BTBR when compared with the WT mice. |
|
DAPA alone and, to a greater extent, DAPA + SAXA resulted in ↓↓ activation of the inflammasome. However, the combination of DAPA + SAXA did not result in greater attenuation of the collagen-1 and collagen-3 mRNA levels. | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kounatidis, D.; Vallianou, N.; Evangelopoulos, A.; Vlahodimitris, I.; Grivakou, E.; Kotsi, E.; Dimitriou, K.; Skourtis, A.; Mourouzis, I. SGLT-2 Inhibitors and the Inflammasome: What’s Next in the 21st Century? Nutrients 2023, 15, 2294. https://0-doi-org.brum.beds.ac.uk/10.3390/nu15102294
Kounatidis D, Vallianou N, Evangelopoulos A, Vlahodimitris I, Grivakou E, Kotsi E, Dimitriou K, Skourtis A, Mourouzis I. SGLT-2 Inhibitors and the Inflammasome: What’s Next in the 21st Century? Nutrients. 2023; 15(10):2294. https://0-doi-org.brum.beds.ac.uk/10.3390/nu15102294
Chicago/Turabian StyleKounatidis, Dimitris, Natalia Vallianou, Angelos Evangelopoulos, Ioannis Vlahodimitris, Eugenia Grivakou, Evangelia Kotsi, Krystalia Dimitriou, Alexandros Skourtis, and Iordanis Mourouzis. 2023. "SGLT-2 Inhibitors and the Inflammasome: What’s Next in the 21st Century?" Nutrients 15, no. 10: 2294. https://0-doi-org.brum.beds.ac.uk/10.3390/nu15102294