
Measurement of Serum Low Density Lipoprotein Cholesterol and Triglyceride-Rich Remnant Cholesterol as Independent Predictors of Atherosclerotic Cardiovascular Disease: Possibilities and Limitations

Abstract
:1. Introduction
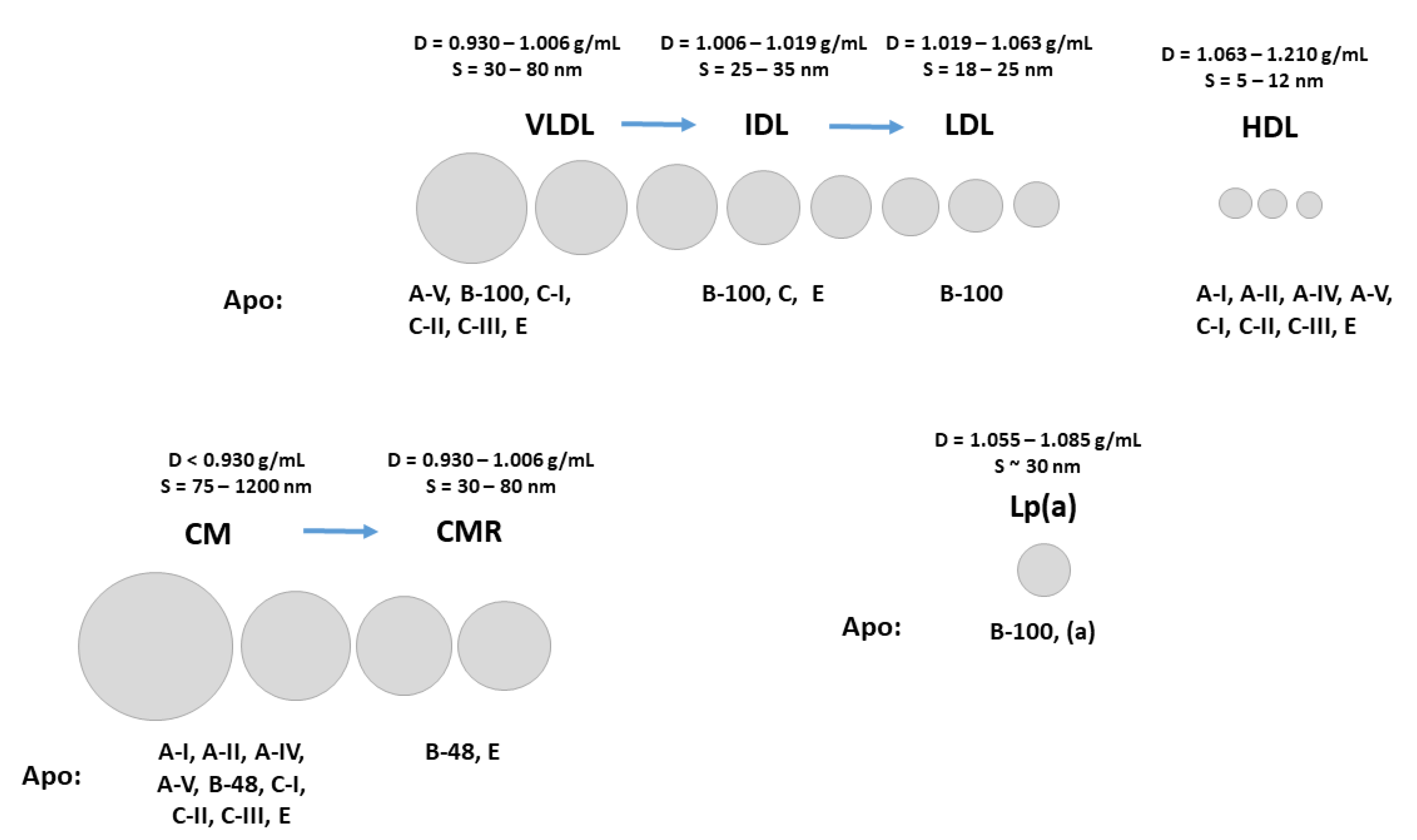
2. Atherogenic Lipoprotein C Concentrations as Indicators of Enhanced Risk for Atherosclerosis Development
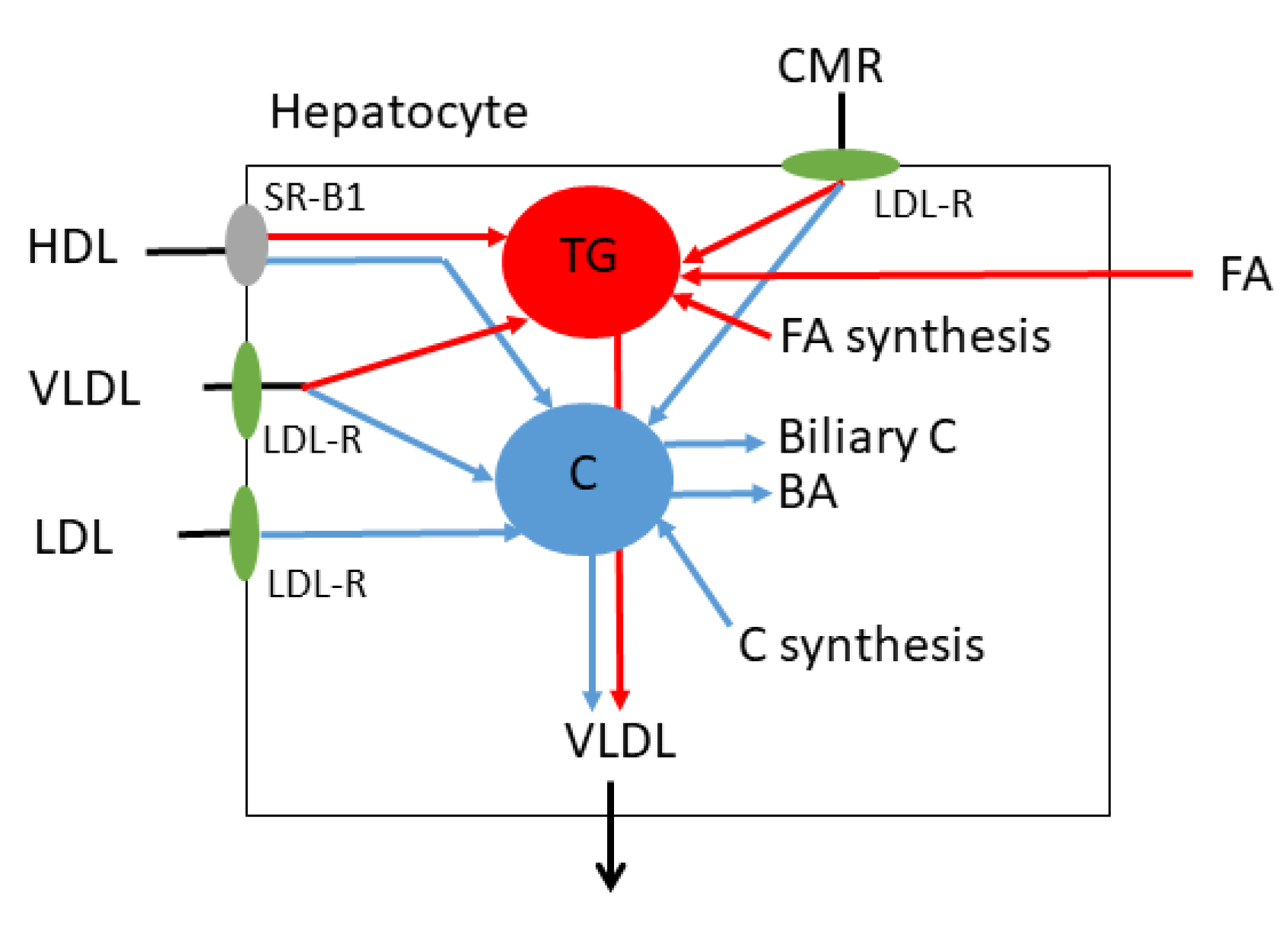
3. LDL-C
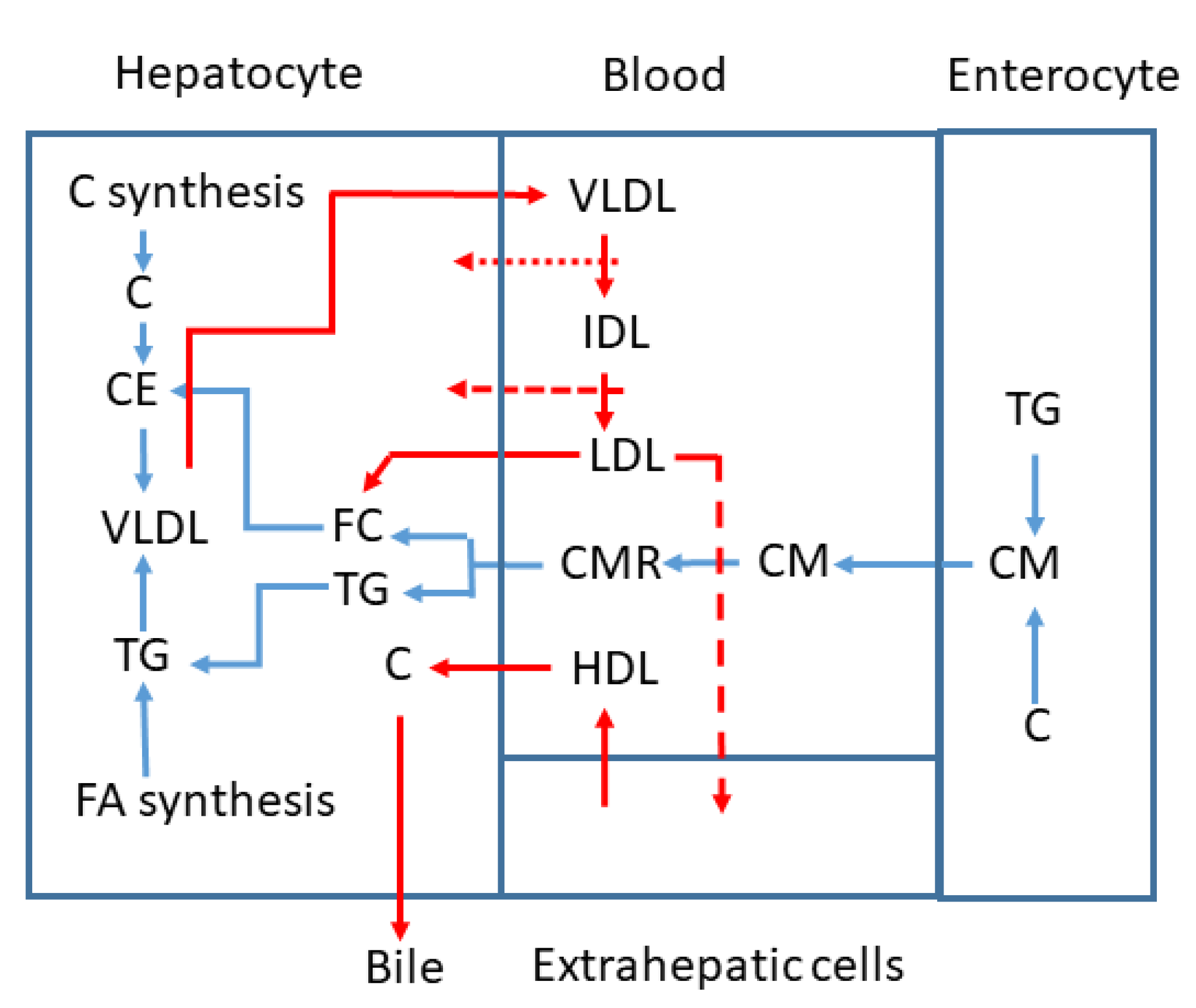
4. TG Rich Lipoprotein C (TRL-C) or Remnant C
5. Non HDL-C and ApoB
6. Personalized Diagnostics and Therapy
7. Limitations
8. Summary of Results
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gitt, A.K.; Parhofer, K.G.; Laufs, U.; Marz, W.; Paar, W.D.; Bramlage, P.; Marx, N. Hypercholesterolemia diagnosis, treatment patterns and target achievement in patients with acute coronary syndromes in Germany. Clin. Res. Cardiol. 2023, 112, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Holme, I.; Solberg, L.A.; Weissfeld, L.; Helgeland, A.; Hjermann, I.; Leren, P.; Strong, J.P.; Williams, O.D. Coronary risk factors and their pathway of action through coronary raised lesions, coronary stenoses and coronary death. Multivariate statistical analysis of an autopsy series: The Oslo Study. Am. J. Cardiol. 1985, 55, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Kannel, W.B.; Castelli, W.P.; Gordon, T. Cholesterol in the prediction of atherosclerotic disease. New perspectives based on the Framingham study. Ann. Intern. Med. 1979, 90, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Newman, W.P., 3rd; Freedman, D.S.; Voors, A.W.; Gard, P.D.; Srinivasan, S.R.; Cresanta, J.L.; Williamson, G.D.; Webber, L.S.; Berenson, G.S. Relation of serum lipoprotein levels and systolic blood pressure to early atherosclerosis. The Bogalusa Heart Study. N. Engl. J. Med. 1986, 314, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Tertov, V.V.; Orekhov, A.N.; Martsenyuk, O.N.; Perova, N.V.; Smirnov, V.N. Low-density lipoproteins isolated from the blood of patients with coronary heart disease induce the accumulation of lipids in human aortic cells. Exp. Mol. Pathol. 1989, 50, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Werner, C.; Laufs, U. Moving beyond the “LDL hypothesis”. Vasa 2015, 44, 333–340. [Google Scholar] [CrossRef]
- Bruckert, E. New advances in lipid-modifying therapies for reducing cardiovascular risk. Cardiology 2002, 97, 59–66. [Google Scholar] [CrossRef]
- Gaine, S.P.; Quispe, R.; Patel, J.; Michos, E.D. New Strategies for Lowering Low Density Lipoprotein Cholesterol for Cardiovascular Disease Prevention. Curr. Cardiovasc. Risk Rep. 2022, 16, 69–78. [Google Scholar] [CrossRef]
- Makhmudova, U.; Samadifar, B.; Maloku, A.; Haxhikadrija, P.; Geiling, J.A.; Romer, R.; Lauer, B.; Mobius-Winkler, S.; Otto, S.; Schulze, P.C.; et al. Intensive lipid-lowering therapy for early achievement of guideline-recommended LDL-cholesterol levels in patients with ST-elevation myocardial infarction (“Jena auf Ziel”). Clin. Res. Cardiol. 2023. Online ahead of print. [Google Scholar] [CrossRef]
- Patnaik, S.; Pollevick, M.E.; Lara-Breitinger, K.M.; Stone, N.J. Inter-Individual Variability in Lipid Response: A Narrative Review. Am. J. Med. 2022, 135, 1427–1433.e7. [Google Scholar] [CrossRef]
- Sun, L.; Wolska, A.; Amar, M.; Zubiran, R.; Remaley, A.T. Approach to the Patient with a Suboptimal Statin Response: Causes and Algorithm for Clinical Management. J. Clin. Endocrinol. Metab. 2023. Online ahead of print. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; Giugliano, R.P.; Wiviott, S.D.; Atar, D.; Keech, A.; Kuder, J.F.; Im, K.; Murphy, S.A.; Flores-Arredondo, J.H.; Lopez, J.A.G.; et al. Long-Term Evolocumab in Patients With Established Atherosclerotic Cardiovascular Disease. Circulation 2022, 146, 1109–1119. [Google Scholar] [CrossRef]
- Laufs, U.; Birkenfeld, A.L.; Fraass, U.; Hohenstein, B.; Siegert, C.; Klotsche, J.; Steinhagen-Thiessen, E.; Pittrow, D.; Dexl, S.; Salmen, S.; et al. Novel Insights into the Management of Patients with Very High Cardiovascular Risk Eligible for PCSK9 Inhibitor Treatment: Baseline Findings from the PERI-DYS Study. Cardiovasc. Drugs Ther. 2022. Online ahead of print. [Google Scholar] [CrossRef]
- Kumari, A.; Kristensen, K.K.; Ploug, M.; Winther, A.L. The Importance of Lipoprotein Lipase Regulation in Atherosclerosis. Biomedicines 2021, 9, 782. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R. Lipid and Lipoprotein Metabolism. Endocrinol. Metab. Clin. N. Am. 2022, 51, 437–458. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.; Contini, C. LDL retention time in plasma can be -based on causation- estimated by the lipid composition of LDL and other lipoproteins. PLoS ONE 2022, 17, e0272050. [Google Scholar] [CrossRef] [PubMed]
- Kuklenyik, Z.; Jones, J.I.; Gardner, M.S.; Schieltz, D.M.; Parks, B.A.; Toth, C.A.; Rees, J.C.; Andrews, M.L.; Carter, K.; Lehtikoski, A.K.; et al. Core lipid, surface lipid and apolipoprotein composition analysis of lipoprotein particles as a function of particle size in one workflow integrating asymmetric flow field-flow fractionation and liquid chromatography-tandem mass spectrometry. PLoS ONE 2018, 13, e0194797. [Google Scholar] [CrossRef]
- Morton, R.E.; Liu, Y. The lipid transfer properties of CETP define the concentration and composition of plasma lipoproteins. J. Lipid Res. 2020, 61, 1168–1179. [Google Scholar] [CrossRef]
- Bjornson, E.; Packard, C.J.; Adiels, M.; Andersson, L.; Matikainen, N.; Soderlund, S.; Kahri, J.; Sihlbom, C.; Thorsell, A.; Zhou, H.; et al. Investigation of human apoB48 metabolism using a new, integrated non-steady-state model of apoB48 and apoB100 kinetics. J. Intern. Med. 2019, 285, 562–577. [Google Scholar] [CrossRef]
- Higgins, V.; Adeli, K. Postprandial Dyslipidemia: Pathophysiology and Cardiovascular Disease Risk Assessment. EJIFCC 2017, 28, 168–184. [Google Scholar]
- Levy, E.; Poinsot, P.; Spahis, S. Chylomicron retention disease: Genetics, biochemistry, and clinical spectrum. Curr. Opin. Lipidol. 2019, 30, 134–139. [Google Scholar] [CrossRef]
- Lo, C.C.; Coschigano, K.T. ApoB48 as an Efficient Regulator of Intestinal Lipid Transport. Front. Physiol. 2020, 11, 796. [Google Scholar] [CrossRef]
- Varbo, A.; Nordestgaard, B.G. Remnant lipoproteins. Curr. Opin. Lipidol. 2017, 28, 300–307. [Google Scholar] [CrossRef]
- Laufs, U.; Parhofer, K.G.; Ginsberg, H.N.; Hegele, R.A. Clinical review on triglycerides. Eur. Heart J. 2020, 41, 99–109c. [Google Scholar] [CrossRef] [PubMed]
- Tomo, S.; Sankanagoudar, S.; Shukla, R.; Sharma, P. Calculated small dense LDL-cholesterol and its correlation with the atherogenic index of plasma. Ann. Clin. Biochem. 2022, 59, 454–456. [Google Scholar] [CrossRef]
- Havel, R.J. Remnant lipoproteins as therapeutic targets. Curr. Opin. Lipidol. 2000, 11, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.; Ginsberg, H.N.; Schaefer, E.J. Relative atherogenicity and predictive value of non-high-density lipoprotein cholesterol for coronary heart disease. Am. J. Cardiol. 2008, 101, 1003–1008. [Google Scholar] [CrossRef]
- Sniderman, A.D.; Thanassoulis, G.; Glavinovic, T.; Navar, A.M.; Pencina, M.; Catapano, A.; Ference, B.A. Apolipoprotein B Particles and Cardiovascular Disease: A Narrative Review. JAMA Cardiol. 2019, 4, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Cantey, E.P.; Wilkins, J.T. Discordance between lipoprotein particle number and cholesterol content: An update. Curr. Opin. Endocrinol. Diabetes Obes. 2018, 25, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, T.A. Opening a new lipid “apo-thecary”: Incorporating apolipoproteins as potential risk factors and treatment targets to reduce cardiovascular risk. Mayo Clin. Proc. 2011, 86, 762–780. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R.; Chait, A. Approach to patients with elevated low-density lipoprotein cholesterol levels. Best Pract. Res. Clin. Endocrinol. Metab. 2023, 37, 101658. [Google Scholar] [CrossRef]
- Liu, H.; Li, J.; Liu, F.; Huang, K.; Cao, J.; Chen, S.; Li, H.; Shen, C.; Hu, D.; Huang, J.; et al. Efficacy and safety of low levels of low-density lipoprotein cholesterol: Trans-ancestry linear and non-linear Mendelian randomization analyses. Eur. J. Prev. Cardiol. 2023; Online ahead of print. [Google Scholar] [CrossRef]
- White-Al Habeeb, N.M.A.; Higgins, V.; Wolska, A.; Delaney, S.R.; Remaley, A.T.; Beriault, D.R. The Present and Future of Lipid Testing in Cardiovascular Risk Assessment. Clin. Chem. 2023, 69, 456–469. [Google Scholar] [CrossRef]
- Groener, J.E.; Van Rozen, A.J.; Erkelens, D.W. Cholesteryl ester transfer activity. Localization and role in distribution of cholesteryl ester among lipoproteins in man. Atherosclerosis 1984, 50, 261–271. [Google Scholar] [CrossRef]
- Nelson, A.J.; Sniderman, A.D.; Ditmarsch, M.; Dicklin, M.R.; Nicholls, S.J.; Davidson, M.H.; Kastelein, J.J.P. Cholesteryl Ester Transfer Protein Inhibition Reduces Major Adverse Cardiovascular Events by Lowering Apolipoprotein B Levels. Int. J. Mol. Sci. 2022, 23, 9417. [Google Scholar] [CrossRef] [PubMed]
- Calvier, L.; Herz, J.; Hansmann, G. Interplay of Low-Density Lipoprotein Receptors, LRPs, and Lipoproteins in Pulmonary Hypertension. JACC Basic Transl. Sci. 2022, 7, 164–180. [Google Scholar] [CrossRef] [PubMed]
- Bakkeren, H.F.; Kuipers, F.; Vonk, R.J.; Van Berkel, T.J. Evidence for reverse cholesterol transport in vivo from liver endothelial cells to parenchymal cells and bile by high-density lipoprotein. Biochem. J. 1990, 268, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, C.C.; Halloran, L.G.; Vlahcevic, Z.R.; Gregory, D.H.; Swell, L. Preferential utilization of free cholesterol from high-density lipoproteins for biliary cholesterol secretion in man. Science 1978, 200, 62–64. [Google Scholar] [CrossRef] [PubMed]
- Pieters, M.N.; Schouten, D.; Bakkeren, H.F.; Esbach, B.; Brouwer, A.; Knook, D.L.; van Berkel, T.J. Selective uptake of cholesteryl esters from apolipoprotein-E-free high-density lipoproteins by rat parenchymal cells in vivo is efficiently coupled to bile acid synthesis. Biochem. J. 1991, 280, 359–365. [Google Scholar] [CrossRef]
- Sniderman, A.; McQueen, M.; Contois, J.; Williams, K.; Furberg, C.D. Why is non-high-density lipoprotein cholesterol a better marker of the risk of vascular disease than low-density lipoprotein cholesterol? J. Clin. Lipidol. 2010, 4, 152–155. [Google Scholar] [CrossRef]
- Balling, M.; Langsted, A.; Afzal, S.; Varbo, A.; Davey Smith, G.; Nordestgaard, B.G. A third of nonfasting plasma cholesterol is in remnant lipoproteins: Lipoprotein subclass profiling in 9293 individuals. Atherosclerosis 2019, 286, 97–104. [Google Scholar] [CrossRef]
- Brown, W.V.; Myers, G.L.; Sniderman, A.D.; Stein, E. Should we use apoB for risk assessment and as a target for treatment? J. Clin. Lipidol. 2010, 4, 144–151. [Google Scholar] [CrossRef]
- Langlois, M.R.; Sniderman, A.D. Non-HDL Cholesterol or apoB: Which to Prefer as a Target for the Prevention of Atherosclerotic Cardiovascular Disease? Curr. Cardiol. Rep. 2020, 22, 67. [Google Scholar] [CrossRef]
- Babiak, J.; Rudel, L.L. Lipoproteins and atherosclerosis. Baillieres Clin. Endocrinol. Metab. 1987, 1, 515–550. [Google Scholar] [CrossRef]
- Potts, J.L.; Fisher, R.M.; Humphreys, S.M.; Gibbons, G.F.; Frayn, K.N. Separation of lipoprotein fractions by ultracentrifugation: Investigation of analytical recovery with sequential flotation and density gradient procedures. Clin. Chim. Acta 1994, 230, 215–220. [Google Scholar] [CrossRef]
- Redgrave, T.G.; Roberts, D.C.; West, C.E. Separation of plasma lipoproteins by density-gradient ultracentrifugation. Anal. Biochem. 1975, 65, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.L.; Kammerer, C.M.; Lowe, W.F.; Dyke, B.; VandeBerg, J.L. Method for quantitating cholesterol in subfractions of serum lipoproteins separated by gradient gel electrophoresis. Biochem. Genet. 1988, 26, 657–681. [Google Scholar] [CrossRef] [PubMed]
- Otvos, J. Measurement of triglyceride-rich lipoproteins by nuclear magnetic resonance spectroscopy. Clin. Cardiol. 1999, 22, II21–II27. [Google Scholar] [CrossRef]
- Hirowatari, Y.; Yoshida, H. Innovatively Established Analysis Method for Lipoprotein Profiles Based on High-Performance Anion-Exchange Liquid Chromatography. J. Atheroscler. Thromb. 2019, 26, 1027–1040. [Google Scholar] [CrossRef] [PubMed]
- Friedewald, W.T.; Levy, R.I.; Fredrickson, D.S. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin. Chem. 1972, 18, 499–502. [Google Scholar] [CrossRef]
- Martins, J.; Rossouw, H.M.; Pillay, T.S. How should low-density lipoprotein cholesterol be calculated in 2022? Curr. Opin. Lipidol. 2022, 33, 237–256. [Google Scholar] [CrossRef] [PubMed]
- Kolovou, G.D.; Watts, G.F.; Mikhailidis, D.P.; Perez-Martinez, P.; Mora, S.; Bilianou, H.; Panotopoulos, G.; Katsiki, N.; Ooi, T.C.; Lopez-Miranda, J.; et al. Postprandial Hypertriglyceridaemia Revisited in the Era of Non-Fasting Lipid Profile Testing: A 2019 Expert Panel Statement, Narrative Review. Curr. Vasc. Pharmacol. 2019, 17, 515–537. [Google Scholar] [CrossRef] [PubMed]
- Kolovou, G.D.; Watts, G.F.; Mikhailidis, D.P.; Perez-Martinez, P.; Mora, S.; Bilianou, H.; Panotopoulos, G.; Katsiki, N.; Ooi, T.C.; Lopez-Miranda, J.; et al. Postprandial Hypertriglyceridaemia Revisited in the Era of Non-Fasting Lipid Profile Testing: A 2019 Expert Panel Statement, Main Text. Curr. Vasc. Pharmacol. 2019, 17, 498–514. [Google Scholar] [CrossRef]
- Liu, M.M.; Peng, J.; Cao, Y.X.; Guo, Y.L.; Wu, N.Q.; Zhu, C.G.; Gao, Y.; Li, J.J. The difference between fasting and non-fasting lipid measurements is not related to statin treatment. Ann. Transl. Med. 2021, 9, 386. [Google Scholar] [CrossRef]
- Nakajima, K.; Tokita, Y.; Sakamaki, K.; Shimomura, Y.; Kobayashi, J.; Kamachi, K.; Tanaka, A.; Stanhope, K.L.; Havel, P.J.; Wang, T.; et al. Triglyceride content in remnant lipoproteins is significantly increased after food intake and is associated with plasma lipoprotein lipase. Clin. Chim. Acta 2017, 465, 45–52. [Google Scholar] [CrossRef] [PubMed]
- D’Aquila, T.; Hung, Y.H.; Carreiro, A.; Buhman, K.K. Recent discoveries on absorption of dietary fat: Presence, synthesis, and metabolism of cytoplasmic lipid droplets within enterocytes. Biochim. Biophys. Acta 2016, 1861, 730–747. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.S.; Blaha, M.J.; Elshazly, M.B.; Toth, P.P.; Kwiterovich, P.O.; Blumenthal, R.S.; Jones, S.R. Comparison of a novel method vs the Friedewald equation for estimating low-density lipoprotein cholesterol levels from the standard lipid profile. JAMA 2013, 310, 2061–2068. [Google Scholar] [CrossRef] [PubMed]
- Sampson, M.; Ling, C.; Sun, Q.; Harb, R.; Ashmaig, M.; Warnick, R.; Sethi, A.; Fleming, J.K.; Otvos, J.D.; Meeusen, J.W.; et al. A New Equation for Calculation of Low-Density Lipoprotein Cholesterol in Patients With Normolipidemia and/or Hypertriglyceridemia. JAMA Cardiol. 2020, 5, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.G.; Myers, G.L.; Sakurabayashi, I.; Bachmann, L.M.; Caudill, S.P.; Dziekonski, A.; Edwards, S.; Kimberly, M.M.; Korzun, W.J.; Leary, E.T.; et al. Seven direct methods for measuring HDL and LDL cholesterol compared with ultracentrifugation reference measurement procedures. Clin. Chem. 2010, 56, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Koren, E.; Corder, C.; Mueller, G.; Centurion, H.; Hallum, G.; Fesmire, J.; McConathy, W.D.; Alaupovic, P. Triglyceride enriched lipoprotein particles correlate with the severity of coronary artery disease. Atherosclerosis 1996, 122, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Jae, S.Y.; Kim, H.J.; Kunutsor, S.K.; Bunsawat, K.; Kurl, S.; Laukkanen, J.A.; Choi, Y.H. Associations of Cardiorespiratory Fitness With Estimated Remnant Cholesterol and Non-High-Density Lipoprotein Cholesterol in Healthy Men. Am. J. Cardiol. 2023, 186, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Wu, W.; Qin, L.; Yu, X.; Cai, L.; Wang, H.; Zhang, Z. Prognostic value of remnant cholesterol in patients with coronary heart disease: A systematic review and meta-analysis of cohort studies. Front. Cardiovasc. Med. 2022, 9, 951523. [Google Scholar] [CrossRef]
- Yang, J.; Wang, Y.; Xi, Z.; Ma, Y.; Shao, C.; Wang, W.; Tang, Y.D. Remnant-Like Particle Cholesterol and the Risk of Major Adverse Cardiovascular Events: A Systematic Review and Meta-Analysis. J. Cardiovasc. Dev. Dis. 2022, 9, 452. [Google Scholar] [CrossRef]
- Moon, J.H.; Kim, K.; Choi, S.H. Lipoprotein Lipase: Is It a Magic Target for the Treatment of Hypertriglyceridemia. Endocrinol. Metab. 2022, 37, 575–586. [Google Scholar] [CrossRef]
- Su, X. ANGPLT3 in cardio-metabolic disorders. Mol. Biol. Rep. 2021, 48, 2729–2739. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, H.; Gotoda, T.; Ogura, M.; Ishibashi, S.; Inagaki, K.; Daida, H.; Hayashi, T.; Hori, M.; Masuda, D.; Matsuki, K.; et al. Current Diagnosis and Management of Primary Chylomicronemia. J. Atheroscler. Thromb. 2021, 28, 883–904. [Google Scholar] [CrossRef] [PubMed]
- Paquette, M.; Amyot, J.; Fantino, M.; Baass, A.; Bernard, S. Rare Variants in Triglycerides-Related Genes Increase Pancreatitis Risk in Multifactorial Chylomicronemia Syndrome. J. Clin. Endocrinol. Metab. 2021, 106, e3473–e3482. [Google Scholar] [CrossRef] [PubMed]
- Steyn, N.; Muller Rossouw, H.; Pillay, T.S.; Martins, J. Comparability of calculated LDL-C with directly measured LDL-C in selected paediatric and adult cohorts. Clin. Chim. Acta 2022, 537, 158–166. [Google Scholar] [CrossRef]
- Varbo, A.; Nordestgaard, B.G. Directly measured vs. calculated remnant cholesterol identifies additional overlooked individuals in the general population at higher risk of myocardial infarction. Eur. Heart J. 2021, 42, 4833–4843. [Google Scholar] [CrossRef] [PubMed]
- Avenell, A.; Robertson, C.; Skea, Z.; Jacobsen, E.; Boyers, D.; Cooper, D.; Aceves-Martins, M.; Retat, L.; Fraser, C.; Aveyard, P.; et al. Bariatric surgery, lifestyle interventions and orlistat for severe obesity: The REBALANCE mixed-methods systematic review and economic evaluation. Health Technol. Assess. 2018, 22, 1–246. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Chauhan, S. Pancreatic lipase inhibitors: The road voyaged and successes. Life Sci. 2021, 271, 119115. [Google Scholar] [CrossRef]
- Paccosi, S.; Cresci, B.; Pala, L.; Rotella, C.M.; Parenti, A. Obesity Therapy: How and Why? Curr. Med. Chem. 2020, 27, 174–186. [Google Scholar] [CrossRef]
- Arai, H.; Yamashita, S.; Yokote, K.; Araki, E.; Suganami, H.; Ishibashi, S.; Group, K.S. Efficacy and safety of K-877, a novel selective peroxisome proliferator-activated receptor alpha modulator (SPPARMalpha), in combination with statin treatment: Two randomised, double-blind, placebo-controlled clinical trials in patients with dyslipidaemia. Atherosclerosis 2017, 261, 144–152. [Google Scholar] [CrossRef]
- Orringer, C.E. Icosapent ethyl: Where will it fit into guideline-based medical therapy for high risk atherosclerotic cardiovascular disease? Trends Cardiovasc. Med. 2020, 30, 151–157. [Google Scholar] [CrossRef]
- Rodriguez-Saldana, J.; Padilla-Padilla, F.; Cardona-Munoz, E.G.; Romero-Antonio, Y.; Arguedas-Nunez, M.M.; Sander-Padilla, J.G.; Martinez-Munoz, A.; Lugo-Sanchez, L.A.; Rodriguez-Vazquez, I.C.; Gonzalez-Canudas, J. Real-World Evidence Evaluation on the Lipid Profile, Therapeutic Goals, and Safety of the Fixed-Dose Combination of Rosuvastatin/Ezetimibe (Trezete(R)) in Dyslipidemia Patients. Cardiol. Res. Pract. 2022, 2022, 9464733. [Google Scholar] [CrossRef] [PubMed]
- Sudhop, T.; Reber, M.; Tribble, D.; Sapre, A.; Taggart, W.; Gibbons, P.; Musliner, T.; von Bergmann, K.; Lutjohann, D. Changes in cholesterol absorption and cholesterol synthesis caused by ezetimibe and/or simvastatin in men. J. Lipid Res. 2009, 50, 2117–2123. [Google Scholar] [CrossRef] [PubMed]
- Carr, S.S.; Hooper, A.J.; Sullivan, D.R.; Burnett, J.R. Non-HDL-cholesterol and apolipoprotein B compared with LDL-cholesterol in atherosclerotic cardiovascular disease risk assessment. Pathology 2019, 51, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Johannesen, C.D.L.; Mortensen, M.B.; Langsted, A.; Nordestgaard, B.G. Apolipoprotein B and Non-HDL Cholesterol Better Reflect Residual Risk Than LDL Cholesterol in Statin-Treated Patients. J. Am. Coll. Cardiol. 2021, 77, 1439–1450. [Google Scholar] [CrossRef]
- Miida, T.; Nishimura, K.; Okamura, T.; Hirayama, S.; Ohmura, H.; Yoshida, H.; Miyashita, Y.; Ai, M.; Tanaka, A.; Sumino, H.; et al. A multicenter study on the precision and accuracy of homogeneous assays for LDL-cholesterol: Comparison with a beta-quantification method using fresh serum obtained from non-diseased and diseased subjects. Atherosclerosis 2012, 225, 208–215. [Google Scholar] [CrossRef]
- Wolska, A.; Remaley, A.T. LDL Cholesterol: What Is the Best Way to Measure It? Clin. Chem. 2019, 65, 1067–1069. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Total C | LDL-C | TRL-C | Therapy |
|---|---|---|---|
| Normal | Elevated | Low | LDL-C lowering |
| Normal | Low | Elevated | TG-lowering |
| Elevated | Elevated | Normal | LDL-C lowering |
| Elevated | Normal | Elevated | TG-lowering |
| Elevated | Elevated | Elevated | LDL-C and TG-lowering |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lütjohann, D.; Klör, H.-U.; Stellaard, F. Measurement of Serum Low Density Lipoprotein Cholesterol and Triglyceride-Rich Remnant Cholesterol as Independent Predictors of Atherosclerotic Cardiovascular Disease: Possibilities and Limitations. Nutrients 2023, 15, 2202. https://0-doi-org.brum.beds.ac.uk/10.3390/nu15092202
Lütjohann D, Klör H-U, Stellaard F. Measurement of Serum Low Density Lipoprotein Cholesterol and Triglyceride-Rich Remnant Cholesterol as Independent Predictors of Atherosclerotic Cardiovascular Disease: Possibilities and Limitations. Nutrients. 2023; 15(9):2202. https://0-doi-org.brum.beds.ac.uk/10.3390/nu15092202
Chicago/Turabian StyleLütjohann, Dieter, Hans-Ulrich Klör, and Frans Stellaard. 2023. "Measurement of Serum Low Density Lipoprotein Cholesterol and Triglyceride-Rich Remnant Cholesterol as Independent Predictors of Atherosclerotic Cardiovascular Disease: Possibilities and Limitations" Nutrients 15, no. 9: 2202. https://0-doi-org.brum.beds.ac.uk/10.3390/nu15092202