Impact of MYC on Anti-Tumor Immune Responses in Aggressive B Cell Non-Hodgkin Lymphomas: Consequences for Cancer Immunotherapy

 ,
,  , and
, and 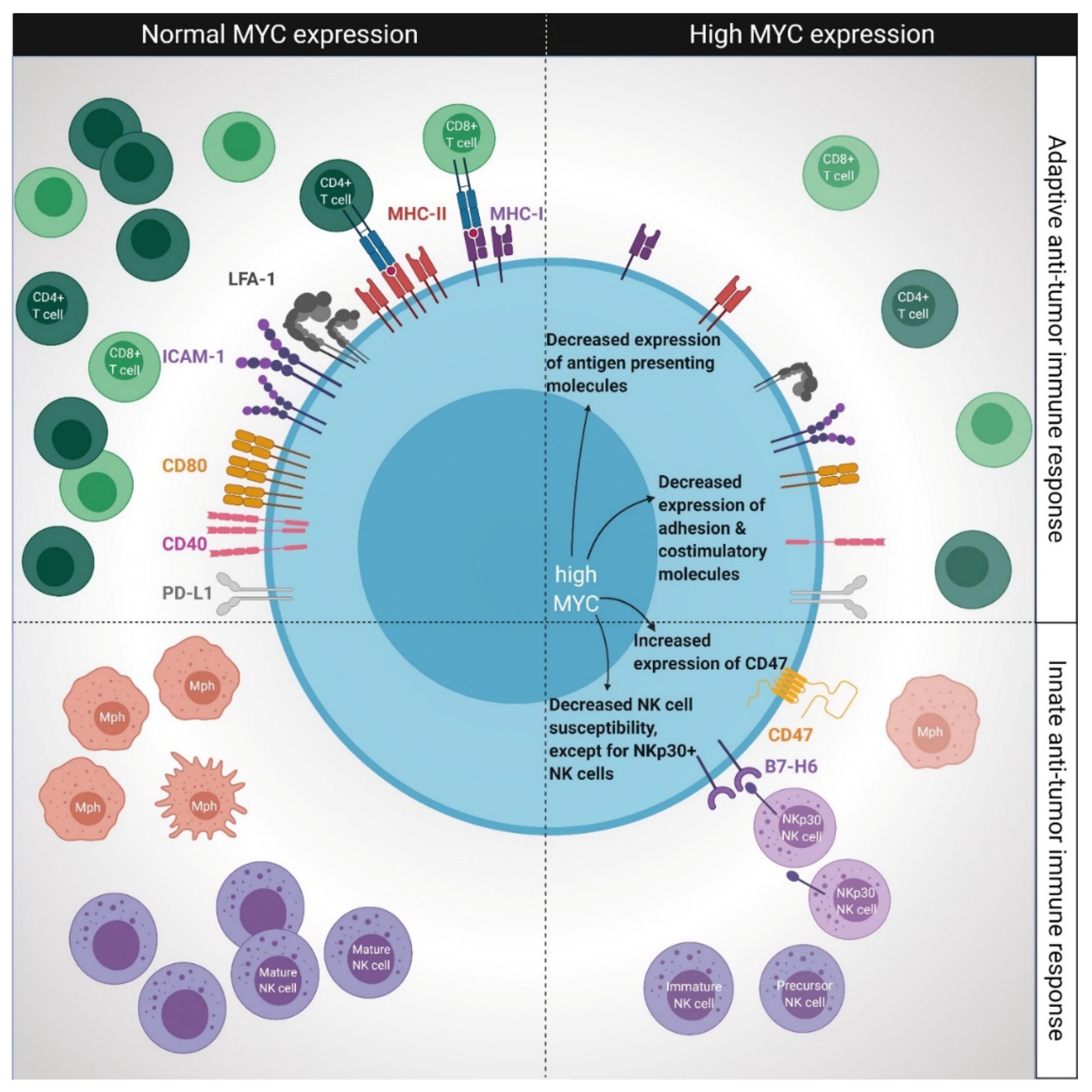{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. The Role of MYC in Normal B Cell Development
3. The Role of MYC Overexpression on the Immune System in Lymphoid Malignancies
3.1. Impact of MYC Overexpression for Adaptive Immunity
3.1.1. The Effect of MYC Overexpression on Antigen Presentation
3.1.2. Consequences of MYC Overexpression on the Expression of Adhesion and Costimulatory Molecules
3.1.3. The Role of MYC Overexpression on PD-L1-Mediated T Cell Tolerance
3.2. Impact of MYC Overexpression for Innate Immunity
3.3. Impact of MYC Overexpression on Apoptosis of Tumor Cells
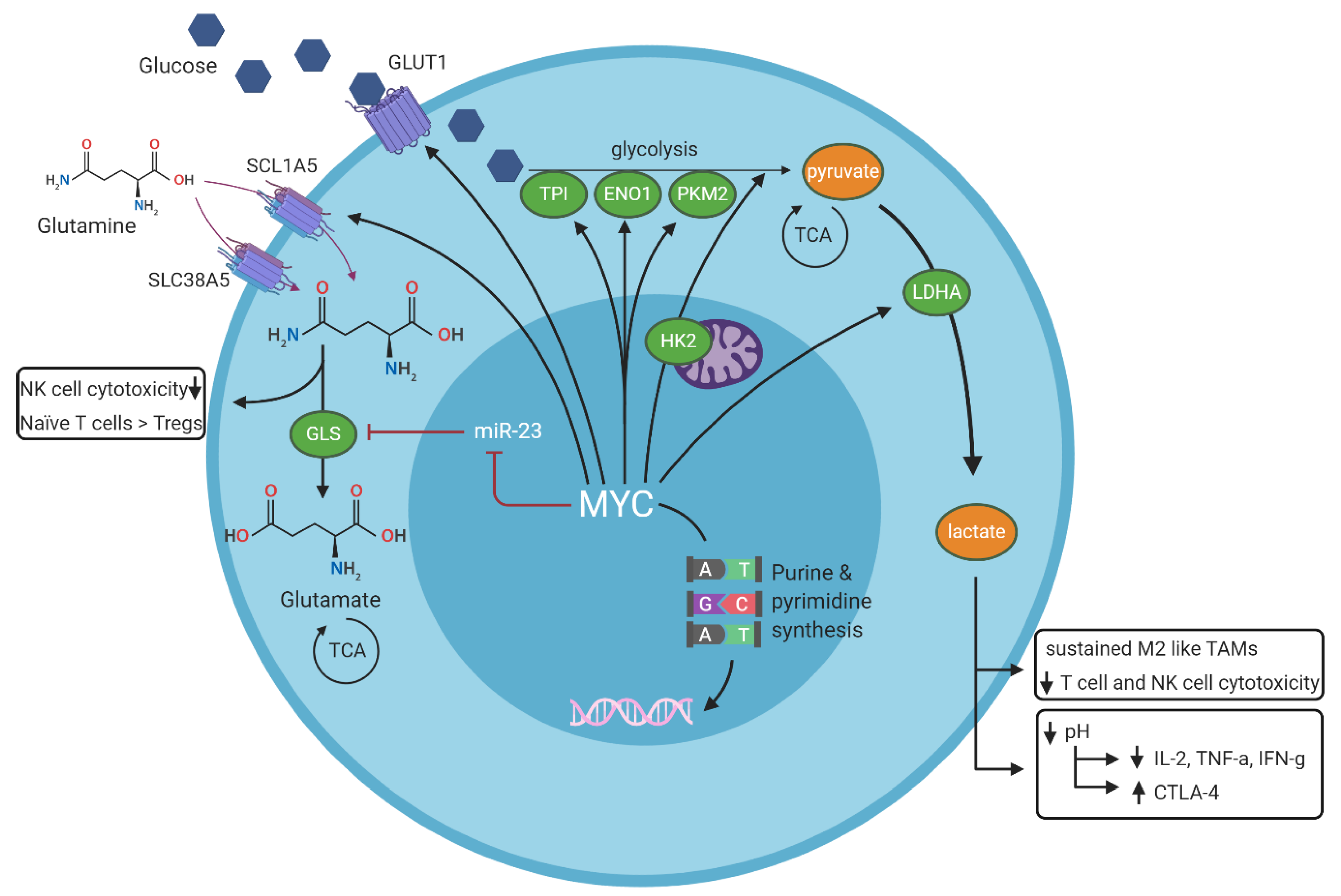
3.4. MYC-Dependent Regulation of Metabolism and Interplay with the Immune Microenvironment
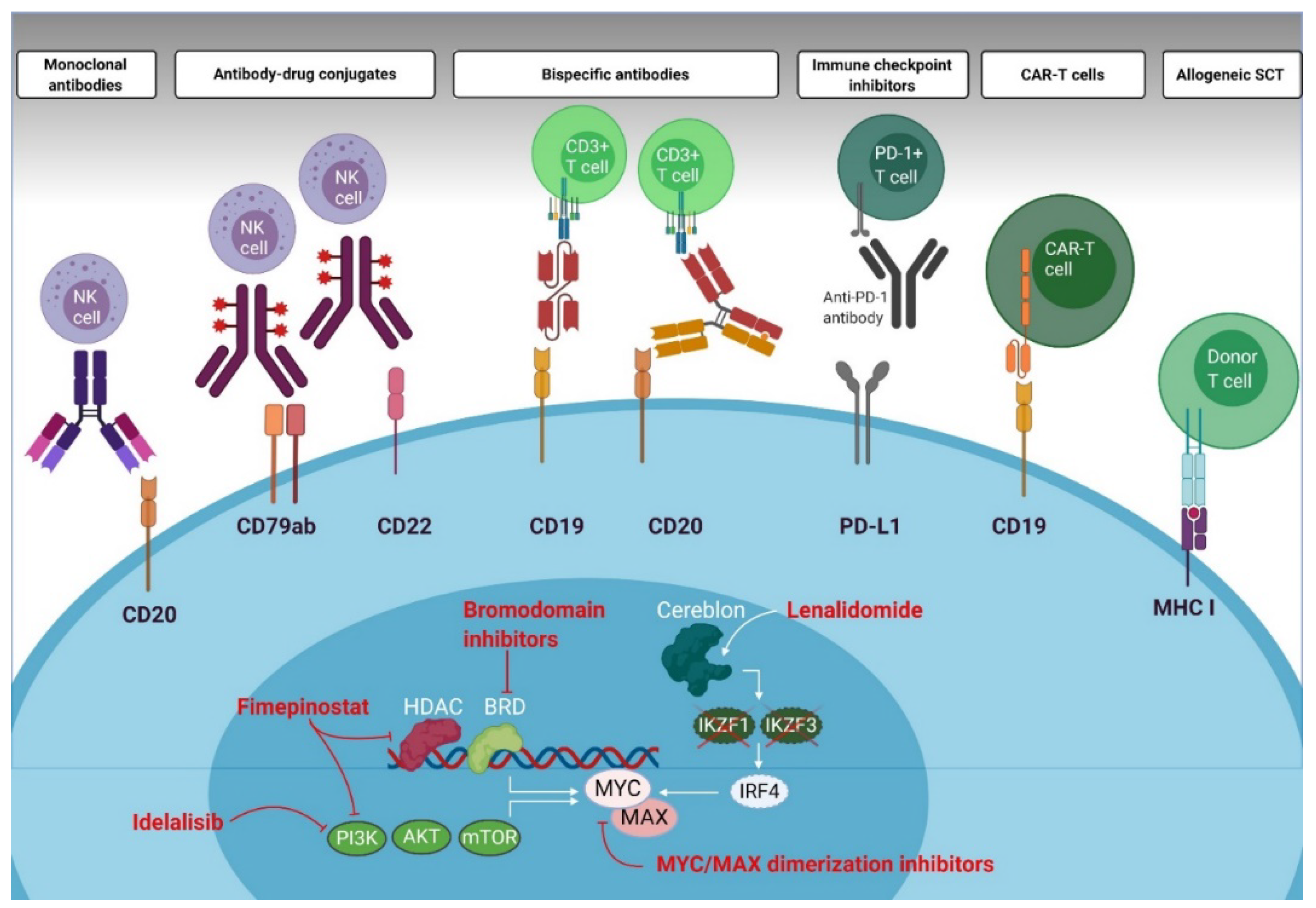
4. Direct and Indirect MYC Modulating Drugs and Consequences for Immune Effector Cells
4.1. Bromodomain Inhibitors
4.2. PI3K Inhibitors
4.3. MYC/MAX Dimerization Inhibitors
4.4. Immunomodulatory Drugs
5. Immunotherapies in DLBCL, with Special Emphasis on Patients with MYC Overexpression
5.1. T Cell Engaging and Modulating Therapies
5.2. Cell-Based Immunotherapies
6. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerhan, J.R. Aggressive Lymphomas; Lenz, G., Salles, G., Eds.; Springer: München, Germany, 2019. [Google Scholar]
- Horn, H.; Ziepert, M.; Becher, C.; Barth, T.F.; Bernd, H.W.; Feller, A.C.; Klapper, W.; Hummel, M.; Stein, H.; Hansmann, M.L.; et al. MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood 2013, 121, 2253–2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sewastianik, T.; Prochorec-Sobieszek, M.; Chapuy, B.; Juszczynski, P. MYC deregulation in lymphoid tumors: Molecular mechanisms, clinical consequences and therapeutic implications. Biochim. Biophys. Acta 2014, 1846, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Aukema, S.M.; Siebert, R.; Schuuring, E.; van Imhoff, G.W.; Kluin-Nelemans, H.C.; Boerma, E.J.; Kluin, P.M. Double-hit B-cell lymphomas. Blood 2011, 117, 2319–2331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boerma, E.G.; Siebert, R.; Kluin, P.M.; Baudis, M. Translocations involving 8q24 in Burkitt lymphoma and other malignant lymphomas: A historical review of cytogenetics in the light of todays knowledge. Leukemia 2009, 23, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Riedell, P.A.; Smith, S.M. Double hit and double expressors in lymphoma: Definition and treatment. Cancer 2018, 124, 4622–4632. [Google Scholar] [CrossRef] [Green Version]
- Ennishi, D.; Jiang, A.; Boyle, M.; Collinge, B.; Grande, B.M.; Ben-Neriah, S.; Rushton, C.; Tang, J.; Thomas, N.; Slack, G.W.; et al. Double-Hit Gene Expression Signature Defines a Distinct Subgroup of Germinal Center B-Cell-Like Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2018, 37, 190–201. [Google Scholar] [CrossRef]
- Sha, C.; Barrans, S.; Cucco, F.; Bently, M.A.; Care, M.A.; Cummin, T.; Kennedy, H.; Thompson, J.S.; Uddin, R.; Worrillow, L.; et al. Molecular High-Grade B-Cell Lymphoma: Defining a Poor-Risk Group That Requires Different Approaches to Therapy. J. Clin. Oncol. 2018, 37, 202–212. [Google Scholar] [CrossRef]
- Coiffier, B.L.E.; Briere, J.; Herbrecht, R.; Tilly, H.; Bouabdallah, R.; Morel, P.; Van den Neste, E.; Salles, G.; Gaulard, P.; Reyes, F.; et al. CHOP Chemotherapy Plus Rituximab Compared with CHOP Alone in Elderly Patients with Diffuse Large B-Cell Kymphoma. N. Engl. J. Med. 2002, 346, 235–242. [Google Scholar] [CrossRef]
- Savage, K.J.; Johnson, N.A.; Ben-Neriah, S.; Connors, J.M.; Sehn, L.H.; Farinha, P.; Horsman, D.E.; Gascoyne, R.D. MYC gene rearrangements are associated with a poor prognosis in diffuse large B-cell lymphoma patients treated with R-CHOP chemotherapy. Blood 2009, 114, 3533–3537. [Google Scholar] [CrossRef] [Green Version]
- Barrans, S.; Crouch, S.; Smith, A.; Turner, K.; Owen, R.; Patmore, R.; Roman, E.; Jack, A. Rearrangement of MYC is associated with poor prognosis in patients with diffuse large B-cell lymphoma treated in the era of rituximab. J. Clin. Oncol. 2010, 28, 3360–3365. [Google Scholar] [CrossRef] [PubMed]
- Oki, Y.; Noorani, M.; Lin, P.; Davis, R.E.; Neelapu, S.S.; Ma, L.; Ahmed, M.; Rodriguez, M.A.; Hagemeister, F.B.; Fowler, N.; et al. Double hit lymphoma: The MD Anderson Cancer Center clinical experience. Br. J. Haematol. 2014, 166, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Petrich, A.M.; Gandhi, M.; Jovanovic, B.; Castillo, J.J.; Rajguru, S.; Yang, D.T.; Shah, K.A.; Whyman, J.D.; Lansigan, F.; Hernandez-Ilizaliturri, F.J.; et al. Impact of induction regimen and stem cell transplantation on outcomes in double-hit lymphoma: A multicenter retrospective analysis. Blood 2014, 124, 2354–2361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunleavy, K.; Fanale, M.A.; Abramson, J.S.; Noy, A.; Caimi, P.F.; Pittaluga, S.; Parekh, S.; Lacasce, A.; Hayslip, J.W.; Jagadeesh, D.; et al. Dose-adjusted EPOCH-R (etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, and rituximab) in untreated aggressive diffuse large B-cell lymphoma with MYC rearrangement: A prospective, multicentre, single-arm phase 2 study. Lancet Haematol. 2018, 5, e609–e617. [Google Scholar] [CrossRef]
- Rosenwald, A.; Bens, S.; Advani, R.; Barrans, S.; Copie-Bergman, C.; Elsensohn, M.-H.; Natkunam, Y.; Calaminici, M.; Sander, B.; Baia, M.; et al. Prognostic Significance of MYC Rearrangement and Translocation Partner in Diffuse Large B-Cell Lymphoma: A Study by the Lunenburg Lymphoma Biomarker Consortium. J. Clin. Oncol. 2019, 37, 3359–3368. [Google Scholar] [CrossRef] [Green Version]
- Davies, A. Tailoring front-line therapy in diffuse large B-cell lymphoma: Who should we treat differently? Hematol. Am. Soc. Hematol. Educ. Program 2017, 1, 284–294. [Google Scholar] [CrossRef] [Green Version]
- Sesques, P.; Johnson, N.A. Approach to the diagnosis and treatment of high-grade B-cell lymphomas with MYC and BCL2 and/or BCL6 rearrangements. Blood 2017, 129, 280–288. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, C.A.; Armand, P. Immunotherapy in aggressive B-cell lymphomas. Best Pract. Res. Clin. Haematol. 2018, 31, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Love, C.; Sun, Z.; Jima, D.; Li, G.; Zhang, J.; Miles, R.; Richards, K.L.; Dunphy, C.H.; Choi, W.W.L.; Srivastava, G.; et al. The genetic landscape of mutations in Burkitt lymphoma. Nat. Genet. 2012, 44, 1321–1325. [Google Scholar] [CrossRef] [Green Version]
- Blackwell, T.K.; Kretzner, L.; Blackwood, E.M.; Eisenman, R.N.; Weintraub, H. Sequence-specific DNA binding by the c-Myc protein. Science 1990, 250, 1149–1151. [Google Scholar] [CrossRef]
- Patel, J.H.; Loboda, A.P.; Showe, M.K.; Showe, L.C.; McMahon, S.B. Analysis of genomic targets reveals complex functions of MYC. Nat. Rev. 2004, 4, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; O’Donnell, K.A.; Zeller, K.I.; Nguyen, T.; Osthus, R.C.; Li, F. The c-Myc target gene network. Semin. Cancer Biol. 2006, 16, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and cancer metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amati, B.; Dalton, S.; Brooks, M.W.; Littlewood, T.D.; Evan, G.I.; Land, H. Transcriptational Activity by the Human c-Myc Oncoprotein in Yeast Requires Interaction with Max. Nature 1992, 359, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Siska, P.J.; Rathmell, J.C. T cell metabolic fitness in antitumor immunity. Trends Immunol. 2015, 36, 257–264. [Google Scholar] [CrossRef] [Green Version]
- Gnanaprakasam, J.N.; Wang, R. MYC in Regulating Immunity: Metabolism and Beyond. Genes 2017, 8, 88. [Google Scholar] [CrossRef] [Green Version]
- Gnanaprakasam, J.N.R.; Sherman, J.W.; Wang, R. MYC and HIF in shaping immune response and immune metabolism. Cytokine Growth Factor Rev. 2017, 35, 63–70. [Google Scholar] [CrossRef]
- Kim, T.W.; Hong, S.; Lin, Y.; Murat, E.; Joo, H.; Kim, T.; Pascual, V.; Liu, Y.J. Transcriptional Repression of IFN Regulatory Factor 7 by MYC Is Critical for Type I IFN Production in Human Plasmacytoid Dendritic Cells. J. Immunol. 2016, 197, 3348–3359. [Google Scholar] [CrossRef] [Green Version]
- Kurosaki, T. Regulation of B cell fates by BCR signaling components. Curr. Opin. Immunol. 2002, 14, 341–347. [Google Scholar] [CrossRef]
- Kraus, M.; Alimzhanov, M.B.; Rajewsky, N.; Rajewsky, K. Survival of resting mature B lymphocytes depends on BCR signaling via the Igalpha/beta heterodimer. Cell 2004, 117, 787–800. [Google Scholar] [CrossRef] [Green Version]
- Küppers, R. Mechanisms of B-cell lymphoma pathogenesis. Nat. Rev. Cancer 2005, 5, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Leone, G.; DeGregori, J.; Sears, R.; Jakoi, L.; Nevins, J.R. Myc and Ras collaborate in inducing accumulation of active cyclin E/Cdk2 and E2F. Nature 1997, 387, 422–426. [Google Scholar] [CrossRef]
- Iritani, B.M.; Eisenman, R.N. c-MYC enhances protein synthesis and cell size during B lymphocyte development. Proc. Natl. Acad. Sci. USA 1999, 96, 13180–13185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habib, T.; Park, H.; Tsang, M.; de Alboran, I.M.; Nicks, A.; Wilson, L.; Knoepfler, P.S.; Andrews, S.; Rawlings, D.J.; Eisenman, R.N.; et al. Myc stimulates B lymphocyte differentiation and amplifies calcium signaling. J. Cell Biol. 2007, 179, 717–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno de Alboran, I.; O’Hagan, R.C.; Gärtner, F.; Malynn, B.; Davidson, L.; Rickert, R.; Rajewsky, K.; DePinho, R.A.; Alt, F.W. Analysis of C-MYC function in Normal Cells via Conditional Gene-Targeted Mutation. Immunity 2001, 14, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Vallespinos, M.; Fernandez, D.; Rodriguez, L.; Alvaro-Blanco, J.; Baena, E.; Ortiz, M.; Dukovska, D.; Martinez, D.; Rojas, A.; Campanero, M.R.; et al. B Lymphocyte commitment program is driven by the proto-oncogene c-Myc. J. Immunol. 2011, 186, 6726–6736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez-Sola, D.; Victora, G.D.; Ying, C.Y.; Phan, R.T.; Saito, M.; Nussenzweig, M.C.; Dalla-Favera, R. The proto-oncogene MYC is required for selection in the germinal center and cyclic reentry. Nat. Immunol. 2012, 13, 1083–1091. [Google Scholar] [CrossRef] [Green Version]
- de Barrios, O.; Meler, A.; Parra, M. MYC’s Fine Line Between B Cell Development and Malignancy. Cells 2020, 9, 523. [Google Scholar] [CrossRef] [Green Version]
- Murn, J.; Mlinaric-Rascan, I.; Vaigot, P.; Alibert, O.; Frouin, V.; Gidrol, X. A Myc-regulated transcriptional network controls B-cell fate in response to BCR triggering. BMC Genom. 2009, 10, 323. [Google Scholar] [CrossRef] [Green Version]
- Korac, P.; Dotlic, S.; Matulic, M.; Zajc Petranovic, M.; Dominis, M. Role of MYC in B Cell Lymphomagenesis. Genes 2017, 8, 115. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Wong, K.K.; Calame, K. Repression of c-myc Transcription by Blimp-1, an Inducer of Terminal B Cell Differentiation. Science 1997, 267, 596–599. [Google Scholar] [CrossRef] [PubMed]
- Rooney, C.M.; Rowe, M.; Wallace, L.E.; Rickinson, A.B. Epstein-Barr virus-positive Burkitt’s lymphoma cells not recognized by virus-specific T cell surveillance. Nature 1985, 317, 629–631. [Google Scholar] [CrossRef] [PubMed]
- Torsteinsdottir, S.; Masucci, M.G.; Ehlin-Henrikson, B.; Brautbar, C.; Ben Bassat, H.; Klein, G.; Klein, E. Differentiation-dependent sensitivity of human B-cell-derived lines to major histocompatibility complex-restricted T-cell cytotoxicity. Proc. Natl. Acad. Sci. USA 1986, 83, 5620–5624. [Google Scholar] [CrossRef] [Green Version]
- Staege, M.S.; Lee, S.P.; Frisan, T.; Mautner, J.; Scholz, S.; Pajic, A.; Rickinson, A.; Masucci, M.G.; Polack, A.; Bornkamm, G.W. MYC overexpression imposes a nonimmunogenic phenotype on Epstein-Barr virus-nfected B cells. Proc. Natl. Acad. Sci. USA 2002, 99, 4550–4555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Versteeg, R.; Noordermeer, I.A.; Krüse-Wolters, M.; Ruiter, D.J.; Schier, P.I. C-myc down-regulates class I HLA expression in human melanomas. EMBO J. 1988, 7, 1023–1029. [Google Scholar] [CrossRef]
- Peltenburg, L.T.C.; Dee, R.; Schier, P.I. Downregulation HLA Class I expression by c-MYC in human melanoma is independent of enhancer A. Nucleic Acids Res. 1993, 21, 1179–1185. [Google Scholar] [CrossRef] [Green Version]
- Ock, C.Y.; Hwang, J.E.; Keam, B.; Kim, S.B.; Shim, J.J.; Jang, H.J.; Park, S.; Sohn, B.H.; Cha, M.; Ajani, J.A.; et al. Genomic landscape associated with potential response to anti-CTLA-4 treatment in cancers. Nat. Commun. 2017, 8, 1050. [Google Scholar] [CrossRef]
- Kortlever, R.M.; Sodir, N.M.; Wilson, C.H.; Burkhart, D.L.; Pellegrinet, L.; Brown Swigart, L.; Littlewood, T.D.; Evan, G.I. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 2017, 171, 1301–1315.e1314. [Google Scholar] [CrossRef]
- Topper, M.J.; Vaz, M.; Chiappinelli, K.B.; DeStefano Shields, C.E.; Niknafs, N.; Yen, R.C.; Wenzel, A.; Hicks, J.; Ballew, M.; Stone, M.; et al. Epigenetic Therapy Ties MYC Depletion to Reversing Immune Evasion and Treating Lung Cancer. Cell 2017, 171, 1284–1300 e1221. [Google Scholar] [CrossRef] [Green Version]
- God, J.M.; Cameron, C.; Figueroa, J.; Amria, S.; Hossain, A.; Kempkes, B.; Bornkamm, G.W.; Stuart, R.K.; Blum, J.S.; Haque, A. Elevation of c-MYC disrupts HLA class II-mediated immune recognition of human B cell tumors. J. Immunol. 2015, 194, 1434–1445. [Google Scholar] [CrossRef] [Green Version]
- Walling, B.L.; Kim, M. LFA-1 in T Cell Migration and Differentiation. Front. Immunol. 2018, 9, 952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inghirami, G.; Grignani, F.; Sternas, L.; Lombardi, L.; Knowles, D.M.; Dalla-Favera, R. Down-regulation of LFA-1 Adhesion Receptors by C-myc oncogene in Human B Lymphoblastoid Cells. Science 1990, 250, 682–686. [Google Scholar] [CrossRef] [PubMed]
- Polack, A.; Hörtnagel, K.; Pajic, A.; Christoph, B.; Baier, B.; Falk, M.; Mautner, J.; Geltinger, C.; Bornkamm, G.W.; Kempkes, B. c-myc activation renders proliferation of Epstein-Barr virus (EBV)-transformed cells independent of EBV nuclear antigen 2 and latent membrane protein 1. Proc. Natl. Acad. Sci. USA 1996, 93, 10411–10416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, A.; Arnaud, N.; Fradet, M.; Lascaux, H.; Ouk-Martin, C.; Gachard, N.; Zimber-Strobl, U.; Feuillard, J.; Faumont, N. c-Myc dysregulation is a co-transforming event for nuclear factor-kappaB activated B cells. Haematologica 2017, 102, 883–894. [Google Scholar] [CrossRef] [Green Version]
- Schlee, M.; Schuhmacher, M.; Hölzel, M.; Laux, G.; Bornkamm, G.W. c-MYC Impairs Immunogenicity of Human B Cells. Adv. Cancer Res. 2007, 97, 167–188. [Google Scholar] [CrossRef]
- Pajic, A.; Staege, M.S.; Dudziak, D.; Schuhmacher, M.; Spitkovsky, D.; Eissner, G.; Brielmeier, M.; Polack, A.; Bornkamm, G.W. Antagonistic effects of c-myc and Epstein-barr virus latent genes on the phenotype of human B cells. Int. J. Cancer 2001, 93. [Google Scholar] [CrossRef]
- Elgueta, R.; Benson, M.J.; de Vries, V.C.; Wasiuk, A.; Guo, Y.; Noelle, R.J. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev. 2009, 229, 152–172. [Google Scholar] [CrossRef] [Green Version]
- Bromley, S.K.; Iaboni, A.; Davis, S.J.; Whitty, A.; Green, J.M.; Shaw, A.S.; Weiss, A.; Dustin, M.L. The immunological synapse and CD28-CD80 interactions. Nat. Immunol. 2001, 2, 1159–1166. [Google Scholar] [CrossRef]
- Xu, P.P.; Sun, C.; Cao, X.; Zhao, X.; Dai, H.J.; Lu, S.; Guo, J.J.; Fu, S.J.; Liu, Y.X.; Li, S.C.; et al. Immune Characteristics of Chinese Diffuse Large B-Cell Lymphoma Patients: Implications for Cancer Immunotherapies. EBioMedicine 2018, 33, 94–104. [Google Scholar] [CrossRef]
- Chen, L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat. Rev. Immunol. 2004, 4, 336–347. [Google Scholar] [CrossRef]
- Kiyasu, J.; Miyoshi, H.; Hirata, A.; Arakawa, F.; Ichikawa, A.; Niino, D.; Sugita, Y.; Yufu, Y.; Choi, I.; Abe, Y.; et al. Expression of programmed cell death ligand 1 is associated with poor overall survival in patients with diffuse large B-cell lymphoma. Blood 2015, 126, 2193–2201. [Google Scholar] [CrossRef]
- Hu, L.Y.; Xu, X.L.; Rao, H.L.; Chen, J.; Lai, R.C.; Huang, H.Q.; Jiang, W.Q.; Lin, T.Y.; Xia, Z.J.; Cai, Q.Q. Expression and clinical value of programmed cell death-ligand 1 (PD-L1) in diffuse large B cell lymphoma: A retrospective study. Chin. J. Cancer 2017, 36, 94. [Google Scholar] [CrossRef] [PubMed]
- Elbaek, M.V.; Pedersen, M.O.; Breinholt, M.F.; Reddy, A.; Love, C.; Clasen-Linde, E.; Knudsen, H.; Nielsen, S.L.; Gang, A.O.; Hogdall, E.; et al. PD-L1 expression is low in large B-cell lymphoma with MYC or double-hit translocation. Hematol. Oncol. 2019, 37, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Sun, R.; Miao, Y.; Tran, T.; Adams, L.; Roscoe, N.; Xu, B.; Manyam, G.C.; Tan, X.; Zhang, H.; et al. PD-1/PD-L1 expression and interaction by automated quantitative immunofluorescent analysis show adverse prognostic impact in patients with diffuse large B-cell lymphoma having T-cell infiltration: A study from the International DLBCL Consortium Program. Mod. Pathol. 2019, 32, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, J.; Tumuluru, S.; Bao, R.; Leukam, M.; Venkataraman, G.; Phillip, J.; Fitzpatrick, C.; McElherne, J.; MacNabb, B.W.; Orlowski, R.; et al. PD-L1 gene alterations identify a subset of diffuse large B-cell lymphoma harboring a T-cell inflamed phenotype. Blood 2019, 133, 2279–2290. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.G.; Jiang, X.N.; Sheng, D.; Sun, C.B.; Lee, J.; Zhou, X.Y.; Li, X.Q. PD-L1 over-expression is driven by B-cell receptor signaling in diffuse large B-cell lymphoma. Lab Investig. 2019, 99, 1418–1427. [Google Scholar] [CrossRef] [PubMed]
- Xing, W.; Dresser, K.; Zhang, R.; Evens, A.M.; Yu, H.; Woda, B.A.; Chen, B.J. PD-L1 expression in EBV-negative diffuse large B-cell lymphoma: Clinicopathologic features and prognostic implications. Oncotarget 2016, 7, 55976–55986. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Poggio, M.; Jin, H.Y.; Shi, Z.; Forester, C.M.; Wang, Y.; Stumpf, C.R.; Xue, L.; Devericks, E.; So, L.; et al. Translation control of the immune checkpoint in cancer and its therapeutic targeting. Nat. Med. 2019, 25, 301–311. [Google Scholar] [CrossRef]
- Pan, Y.; Fei, Q.; Xiong, P.; Yang, J.; Zhang, Z.; Lin, X.; Pan, M.; Lu, F.; Huang, H. Synergistic inhibition of pancreatic cancer with anti-PD-L1 and c-Myc inhibitor JQ1. Oncoimmunology 2019, 8, e1581529. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.Y.; Kim, A.; Kim, S.K.; Chang, Y.S. MYC expression correlates with PD-L1 expression in non-small cell lung cancer. Lung Cancer 2017, 110, 63–67. [Google Scholar] [CrossRef]
- Liang, M.Q.; Yu, F.Q.; Chen, C. C-Myc regulates PD-L1 expression in esophageal squamous cell carcinoma. Am. J. Transl. Res. 2020, 12, 379–388. [Google Scholar] [PubMed]
- Wang, J.; Jia, Y.; Zhao, S.; Zhang, X.; Wang, X.; Han, X.; Wang, Y.; Ma, M.; Shi, J.; Liu, L. BIN1 reverses PD-L1-mediated immune escape by inactivating the c-MYC and EGFR/MAPK signaling pathways in non-small cell lung cancer. Oncogene 2017, 36, 6235–6243. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, Q.; Chen, B.; Zhao, Y.; Shen, B.; Wang, H.; Xu, J.; Zhu, M.; Zhao, X.; Xu, C.; et al. Gastric cancer mesenchymal stem cells derived IL-8 induces PD-L1 expression in gastric cancer cells via STAT3/mTOR-c-Myc signal axis. Cell Death Dis. 2018, 9, 928. [Google Scholar] [CrossRef] [PubMed]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gutgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogg, S.J.; Vervoort, S.J.; Deswal, S.; Ott, C.J.; Li, J.; Cluse, L.A.; Beavis, P.A.; Darcy, P.K.; Martin, B.P.; Spencer, A.; et al. BET-Bromodomain Inhibitors Engage the Host Immune System and Regulate Expression of the Immune Checkpoint Ligand PD-L1. Cell Rep. 2017, 18, 2162–2174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durand-Panteix, S.; Farhat, M.; Youlyouz-Marfak, I.; Rouaud, P.; Ouk-Martin, C.; David, A.; Faumont, N.; Feuillard, J.; Jayat-Vignoles, C. B7-H1, which represses EBV-immortalized B cell killing by autologous T and NK cells, is oppositely regulated by c-Myc and EBV latency III program at both mRNA and secretory lysosome levels. J. Immunol. 2012, 189, 181–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlee, M.; Holzel, M.; Bernard, S.; Mailhammer, R.; Schuhmacher, M.; Reschke, J.; Eick, D.; Marinkovic, D.; Wirth, T.; Rosenwald, A.; et al. c-MYC activation impairs the NF-kappaB and the interferon response: Implications for the pathogenesis of Burkitt’s lymphoma. Int. J. Cancer 2007, 120, 1387–1395. [Google Scholar] [CrossRef]
- Ramana, C.V.; Grammatikakis, N.; Chernov, M.; Nguyen, H.; Goh, K.C.; Williams, B.R.G.; Stark, G.R. Regulation of c-myc expression by IFN-ɣ through Stat1-dependent and -independent pathways. EMBO J. 2000, 19, 263–272. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Gupta, S.K.; Han, W.; Kundson, R.A.; Nelson, S.; Knutson, D.; Greipp, P.T.; Elsawa, S.F.; Sotomayor, E.M.; Gupta, M. Targeting MYC activity in double-hit lymphoma with MYC and BCL2 and/or BCL6 rearrangements with epigenetic bromodomain inhibitors. J. Hematol. Oncol. 2019, 12, 73. [Google Scholar] [CrossRef] [Green Version]
- Xue, T.; Wang, W.G.; Zhou, X.Y.; Li, X.Q. EBV-positive diffuse large B-cell lymphoma features PD-L1 protein but not mRNA overexpression. Pathology 2018, 50, 725–729. [Google Scholar] [CrossRef]
- Zuo, J.; Currin, A.; Griffin, B.D.; Shannon-Lowe, C.; Thomas, W.A.; Ressing, M.E.; Wiertz, E.J.; Rowe, M. The Epstein-Barr virus G-protein-coupled receptor contributes to immune evasion by targeting MHC class I molecules for degradation. PLoS Pathog. 2009, 5, e1000255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, M.; Zuo, J. Immune responses to Epstein-Barr virus: Molecular interactions in the virus evasion of CD8+ T cell immunity. Microbes Infect. 2010, 12, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.; Kim, S.; Kim, P.J.; Go, H.; Nam, S.J.; Paik, J.H.; Kim, Y.A.; Kim, T.M.; Heo, D.S.; Kim, C.W.; et al. Clinicopathological analysis of programmed cell death 1 and programmed cell death ligand 1 expression in the tumour microenvironments of diffuse large B cell lymphomas. Histopathology 2016, 68, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadou, E.; Stroopinsky, D.; Alimperti, S.; Jiao, A.L.; Pyzer, A.R.; Cippitelli, C.; Pepe, G.; Severa, M.; Rosenblatt, J.; Etna, M.P.; et al. Epstein-Barr virus-encoded EBNA2 alters immune checkpoint PD-L1 expression by downregulating miR-34a in B-cell lymphomas. Leukemia 2019, 33, 132–147. [Google Scholar] [CrossRef] [Green Version]
- Kärre, K.; Ljunggren, H.G.; Piontek, G.; Kiessling, R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 1986, 319, 675–678. [Google Scholar] [CrossRef]
- Bern, M.D.; Parikh, B.A.; Yang, L.; Beckman, D.L.; Poursine-Laurent, J.; Yokoyama, W.M. Inducible down-regulation of MHC class I results in natural killer cell tolerance. J. Exp. Med. 2019, 216, 99–116. [Google Scholar] [CrossRef]
- Swaminathan, S.; Hansen, A.S.; Heftdal, L.D.; Dhanasekaran, R.; Deutzmann, A.; Fernandez, W.D.M.; Liefwalker, D.F.; Horton, C.; Mosley, A.; Liebersbach, M.; et al. MYC functions as a switch for natural killer cell-mediated immune surveillance of lymphoid malignancies. Nat. Commun. 2020, 11, 2860. [Google Scholar] [CrossRef]
- Textor, S.; Bossler, F.; Henrich, K.O.; Gartlgruber, M.; Pollmann, J.; Fiegler, N.; Arnold, A.; Westermann, F.; Waldburger, N.; Breuhahn, K.; et al. The proto-oncogene Myc drives expression of the NK cell-activating NKp30 ligand B7-H6 in tumor cells. Oncoimmunology 2016, 5, e1116674. [Google Scholar] [CrossRef]
- Yetil, A.; Anchang, B.; Gouw, A.M.; Adam, S.J.; Zabuawala, T.; Parameswaran, R.; Van Riggelen, J.; Plevritis, S.; Felsher, D.W. p19ARF is a critical mediator of both cellular senescence and an innate immune response associated with MYC inactivation in mouse model of acute leukemia. Oncotarget 2015, 6, 3563–3577. [Google Scholar] [CrossRef] [Green Version]
- Jaime-Sanchez, P.; Catalan, E.; Uranga-Murillo, I.; Aguilo, N.; Santiago, L.; Lanuza, P.M.; de Miguel, D.; Arias, M.A.; Pardo, J. Antigen-specific primed cytotoxic T cells eliminate tumour cells in vivo and prevent tumour development, regardless of the presence of anti-apoptotic mutations conferring drug resistance. Cell Death Differ. 2018, 25, 1536–1548. [Google Scholar] [CrossRef]
- Klapproth, K.; Wirth, T. Advances in the understanding of MYC-induced lymphomagenesis. Br. J. Haematol. 2010, 149, 484–497. [Google Scholar] [CrossRef] [PubMed]
- Askew, D.S.; Ashmun, R.A.; Simmons, B.C.; Cleveland, J.L. Constitutive c-myc expression in an IL-3-dependent myeloid cell line suppresses cell cycle arrest and accelerates apoptosis. Oncogene 1991, 6, 1915–1922. [Google Scholar] [PubMed]
- Evan, G.I.; Wyllie, A.H.; Gilbert, C.S.; Littlewood, T.D.; Land, H.; Brooks, M.; Waters, C.M.; Penn, L.Z.; Hancock, D.C. Induction of apoptosis in fibroblasts by c-myc protein. Cell 1992, 69, 119–128. [Google Scholar] [CrossRef]
- Vecchio, E.; Fiume, G.; Correnti, S.; Romano, S.; Iaccino, E.; Mimmi, S.; Maisano, D.; Nistico, N.; Quinto, I. Insights about MYC and Apoptosis in B-Lymphomagenesis: An Update from Murine Models. Int. J. Mol. Sci. 2020, 21, 4265. [Google Scholar] [CrossRef]
- Eischen, C.M.; Weber, J.D.; Roussel, M.F.; Sherr, C.J.; Cleveland, J.L. Disrupton of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999, 13, 2658–2669. [Google Scholar] [CrossRef] [Green Version]
- McMahon, S.B. MYC and the control of apoptosis. Cold Spring Harb. Perspect Med. 2014, 4, a014407. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.V.; Vandenberg, C.J.; Ng, A.P.; Robati, M.R.; Anstee, A.S.; Rimes, J.; Hawkins, E.D.; Cory, S. Development and survival of MYC-driven lymphomas require the MYC antagonist MNT to curb MYC-induced apoptosis. Blood 2020, 135, 1019–1031. [Google Scholar] [CrossRef]
- Grabow, S.; Delbridge, A.R.; Aubrey, B.J.; Vandenberg, C.J.; Strasser, A. Loss of a Single Mcl-1 Allele Inhibits MYC-Driven Lymphomagenesis by Sensitizing Pro-B Cells to Apoptosis. Cell Rep. 2016, 14, 2337–2347. [Google Scholar] [CrossRef] [Green Version]
- Ott, G.; Rosenwald, A.; Campo, E. Understanding MYC-driven aggressive B-cell lymphomas: Pathogenesis and classification. Blood 2013, 122, 3884–3891. [Google Scholar] [CrossRef] [Green Version]
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012, 15, 110–121. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.W.; Gao, P.; Liu, Y.C.; Semenza, G.L.; Dang, C.V. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol. Cell Biol. 2007, 27, 7381–7393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mushtaq, M.; Darekar, S.; Klein, G.; Kashuba, E. Different Mechanisms of Regulation of the Warburg Effect in Lymphoblastoid and Burkitt Lymphoma Cells. PLoS ONE 2015, 10, e0136142. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Zeller, K.I.; Wang, Y.; Jegga, A.G.; Aronow, B.J.; O’Donnell, K.A.; Dang, C.V. Evaluation of myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol. Cell Biol. 2004, 24, 5923–5936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osthus, R.C.; Shim, H.; Kim, S.; Li, Q.; Reddy, R.; Mukherjee, M.; Xu, Y.; Wonsey, D.; Lee, L.A.; Dang, C.V. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J. Biol. Chem. 2000, 275, 21797–21800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, H.; Dolde, C.; Lewis, B.C.; Wu, C.-S.; Dang, G.; Jungmann, R.A.; Dalla-Favera, R.; Dang, C.V. c-Myc transactivation of LHD-A: Implications for tumor metabolism and growth. Proc. Natl. Acad. Sci. USA 1997, 94, 6658–6663. [Google Scholar] [CrossRef] [Green Version]
- Shim, H.; Chun, Y.S.; Lewis, B.C.; Dang, C.V. A unique glucose-dependent apoptotic pathway induced by c-Myc. Proc. Natl. Acad. Sci. USA 1998, 95, 1511–1516. [Google Scholar] [CrossRef] [Green Version]
- Lewis, B.C.; Prescott, J.E.; Campbell, S.E.; Shim, H.; Orlowski, R.Z.; Dang, C.V. Tumor Induction by the c-Myc Target Genes rcl and Lactate Dehydrogenase A. Cancer Res. 2000, 60, 6178–6183. [Google Scholar]
- David, C.J.; Chen, M.; Assanah, M.; Canoll, P.; Manley, J.L. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 2010, 463, 364–368. [Google Scholar] [CrossRef]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [Green Version]
- Wise, D.R.; DeBerardinis, R.J.; Manusco, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.C.; Li, F.; Handler, J.; Huang, C.R.; Xiang, Y.; Neretti, N.; Sedivy, J.M.; Zeller, K.I.; Dang, C.V. Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS ONE 2008, 3, e2722. [Google Scholar] [CrossRef] [PubMed]
- Mannava, S.; Grachtchouk, V.; Wheeler, L.J.; Im, M.; Zhuang, D.; Slavina, E.G.; Mathews, C.K.; Shewach, D.S.; Nikiforov, M.A. Direct role of nucleotide metabolism in C-MYC-dependent proliferation of melanoma cells. Cell Cycle 2008, 7, 2392–2400. [Google Scholar] [CrossRef] [PubMed]
- Zeller, K.I.; Zhao, X.; Lee, C.W.H.; Chiu, K.P.; Yao, F.; Yustein, J.; Ooi, H.S.; Orlov, Y.L.; Shahab, A.; Yong, H.C.; et al. Global mapping of c-Myc binding sites and target gene networks in human B cells. Proc. Natl. Acad. Sci. USA 2006, 103, 17834–17839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [Green Version]
- Bohn, T.; Rapp, S.; Luther, N.; Klein, M.; Bruehl, T.J.; Kojima, N.; Aranda Lopez, P.; Hahlbrock, J.; Muth, S.; Endo, S.; et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat. Immunol. 2018, 19, 1319–1329. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Yu, Y.R.; Ho, P.C. Sculpting tumor microenvironment with immune system: From immunometabolism to immunoediting. Clin. Exp. Immunol. 2019, 197, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Calcinotto, A.; Filipazzi, P.; Grioni, M.; Iero, M.; De Milito, A.; Ricupito, A.; Cova, A.; Canese, R.; Jachetti, E.; Rossetti, M.; et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 2012, 72, 2746–2756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klysz, D.; Tai, X.; Robert, P.; Craveiro, M.; Cretenet, G.; Oburoglu, L.; Mongellaz, C.; Floess, S.; Fritz, V.; Matias, M.I.; et al. Glutamine-dependent alpha-ketoglutarate production regulates teh balance between T helper 1 cell and regulatory T cell generation. Sci. Signal. 2015, 8, ra97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzler, B.; Gfeller, P.; Guinet, E. Restricting Glutamine or Glutamine-Dependent Purine and Pyrimidine Syntheses Promotes Human T Cells with High FOXP3 Expression and Regulatory Properties. J. Immunol. 2016, 196, 3618–3630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loftus, R.M.; Assmann, N.; Kedia-Mehta, N.; O’Brien, K.L.; Garcia, A.; Gillespie, C.; Hukelmann, J.L.; Oefner, P.J.; Lamond, A.I.; Gardiner, C.M.; et al. Amino acid-dependent cMyc expression is essential for NK cell metabolic and functional responses in mice. Nat. Commun. 2018, 9, 2341. [Google Scholar] [CrossRef]
- McKeown, M.R.; Bradner, J.E. Therapeutic strategies to inhibit MYC. Cold Spring Harb. Perspect Med. 2014, 4. [Google Scholar] [CrossRef]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal. Transduct Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Allen-Petersen, B.L.; Sears, R.C. Mission Possible: Advances in MYC Therapeutic Targeting in Cancer. BioDrugs 2019, 33, 539–553. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Barta, S.K. Diffuse large B-cell lymphoma: 2019 update on diagnosis, risk stratification, and treatment. Am. J. Hematol. 2019, 94, 604–616. [Google Scholar] [CrossRef] [Green Version]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [Green Version]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [Green Version]
- Abramson, J.S.; Blum, K.A.; Flinn, I.W.; Gutierrez, M.; Goy, A.; Maris, M.; Cooper, M.; O’Meara, M.; Borger, D.; Mertz, J.; et al. BET Inhibitor CPI-0610 Is Well Tolerated and Induces Responses in Diffuse Large B-Cell Lymphoma and Follicular Lymphoma: Preliminary Analysis of an Ongoing Phase 1 Study. Blood 2015, 126, 1491. [Google Scholar] [CrossRef]
- Bandukwala, H.S.; Gagnon, J.; Togher, S.; Greenbaum, J.A.; Lamperti, E.D.; Parr, N.J.; Molesworth, A.M.; Smithers, N.; Lee, K.; Witherington, J.; et al. Selective inhibition of CD4+ T-cell cytokine production and autoimmunity by BET protein and c-Myc inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 14532–14537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgiev, P.; Wang, Y.; Muise, E.S.; Bandi, M.L.; Blumenschein, W.; Sathe, M.; Pinheiro, E.M.; Shumway, S.D. BET Bromodomain Inhibition Suppresses Human T Cell Function. Immunohorizons 2019, 3, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, H.R.; Mi, D.J.; Farley, V.M.; Esmond, T.; Kaood, M.B.; Aune, T.M. Bromodomain inhibitor JQ1 reversibly blocks IFN-gamma production. Sci. Rep. 2019, 9, 10280. [Google Scholar] [CrossRef] [PubMed]
- Kagoya, Y.; Nakatsugawa, M.; Yamashita, Y.; Ochi, T.; Guo, T.; Anczurowski, M.; Saso, K.; Butler, M.O.; Arrowsmith, C.H.; Hirano, N. BET bromodomain inhibition enhances T cell persistence and function in adoptive immunotherapy models. J. Clin. Investig. 2016, 126, 3479–3494. [Google Scholar] [CrossRef] [Green Version]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Xie, S.; Chen, M.; Yan, B.; He, X.; Chen, X.; Li, D. Identification of a role for the PI3K/AKT/mTOR signaling pathway in innate immune cells. PLoS ONE 2014, 9, e94496. [Google Scholar] [CrossRef]
- Han, J.M.; Patterson, S.J.; Levings, M.K. The Role of the PI3K Signaling Pathway in CD4(+) T Cell Differentiation and Function. Front. Immunol. 2012, 3, 245. [Google Scholar] [CrossRef] [Green Version]
- So, L.; Fruman, D.A. PI3K signalling in B- and T-lymphocytes: New developments and therapeutic advances. Biochem. J. 2012, 442, 465–481. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.K.; Nandagopal, N.; Lee, S.H. IL-15-PI3K-AKT-mTOR: A Critical Pathway in the Life Journey of Natural Killer Cells. Front. Immunol. 2015, 6, 355. [Google Scholar] [CrossRef] [Green Version]
- Deng, C.; Lipstein, M.R.; Scotto, L.; Jireau Serrano, X.O.; Mangone, M.A.; Li, S.; Vendome, J.; Hao, Y.; Xu, X.; Deng, S.X.; et al. Silencing c-Myc Translation as a Therapeutic Strategy through Targeting PI3K Delta and CK1 Epsilon in Hematological Malignancies. Blood 2016, 129, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Atoyan, R.; Borek, M.A.; Dellarocca, S.; Samson, M.E.; Ma, A.W.; Xu, G.X.; Patterson, T.; Tuck, D.P.; Viner, J.L.; et al. Dual HDAC and PI3K Inhibitor CUDC-907 Downregulates MYC and Suppresses Growth of MYC-dependent Cancers. Mol. Cancer Ther. 2017, 16, 285–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oki, Y.; Kelly, K.R.; Flinn, I.; Patel, M.R.; Gharavi, R.; Ma, A.; Parker, J.; Hafeez, A.; Tuck, D.; Younes, A. CUDC-907 in relapsed/refractory diffuse large B-cell lymphoma, including patients with MYC-alterations: Results from an expanded phase I trial. Haematologica 2017, 102, 1923–1930. [Google Scholar] [CrossRef] [PubMed]
- Jackstadt, R.; Hermeking, H. MicroRNAs as regulators and mediators of c-MYC function. Biochim. Biophys. Acta 2015, 1849, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Soucek, L.; Helmer-Citterich, M.; Sacco, A.; Jucker, R.; Cesareni, G.; Nasi, S. Design and properties of a Myc derivative that efficiently homodimerizes. Oncogene 1998, 17, 2463–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaulieu, M.E.; Jauset, T.; Masso-Valles, D.; Martinez-Martin, S.; Rahl, P.; Maltais, L.; Zacarias-Fluck, M.F.; Casacuberta-Serra, S.; Serrano Del Pozo, E.; Fiore, C.; et al. Intrinsic cell-penetrating activity propels Omomyc from proof of concept to viable anti-MYC therapy. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Masso-Valles, D.; Soucek, L. Blocking Myc to Treat Cancer: Reflecting on Two Decades of Omomyc. Cells 2020, 9, 883. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Giap, C.; Lazo, J.S.; Prochownik, E.V. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene 2003, 22, 6151–6159. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Curet, I.; Perkins, R.S.; Bennett, R.; Feidler, K.L.; Dunn, S.P.; Krueger, L.J. c-Myc inhibition negatively impacts lymphoma growth. J. Pediatr. Surg. 2006, 41, 207–211. [Google Scholar] [CrossRef]
- Struntz, N.B.; Chen, A.; Deutzmann, A.; Wilson, R.M.; Stefan, E.; Evans, H.L.; Ramirez, M.A.; Liang, T.; Caballero, F.; Wildschut, M.H.E.; et al. Stabilization of the Max Homodimer with a Small Molecule Attenuates Myc-Driven Transcription. Cell Chem. Biol. 2019, 26, 711–723.e714. [Google Scholar] [CrossRef]
- Gallant, P.; Steiger, D. Myc’s secret life without Max. Cell Cycle 2009, 8, 3848–3853. [Google Scholar] [CrossRef] [Green Version]
- Han, H.; Jain, A.D.; Truica, M.I.; Izquierdo-Ferrer, J.; Anker, J.F.; Lysy, B.; Sagar, V.; Luan, Y.; Chalmers, Z.R.; Unno, K.; et al. Small-Molecule MYC Inhibitors Suppress Tumor Growth and Enhance Immunotherapy. Cancer Cell 2019, 36, 483–497.e415. [Google Scholar] [CrossRef]
- Quach, H.; Ritchie, D.; Stewart, A.K.; Neeson, P.; Harrison, S.; Smyth, M.J.; Prince, H.M. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia 2010, 24, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gribben, J.G.; Fowler, N.; Morschhauser, F. Mechanisms of Action of Lenalidomide in B-Cell Non-Hodgkin Lymphoma. J. Clin. Oncol. 2015, 33, 2803–2811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Girona, A.; Heintel, D.; Zhang, L.H.; Mendy, D.; Gaidarova, S.; Brady, H.; Bartlett, J.B.; Schafer, P.H.; Schreder, M.; Bolomsky, A.; et al. Lenalidomide downregulates the cell survival factor, interferon regulatory factor-4, providing a potential mechanistic link for predicting response. Br. J. Haematol. 2011, 154, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.K.; Bradner, J.E.; Kaelin, W.G., Jr. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamuleau, M.E.D.; Burggraaff, C.N.; Nijland, M.; Bakunina, K.; Mous, R.; Lugtenburg, P.J.; Dierickx, D.; van Imhoff, G.W.; Vermaat, J.S.P.; Marijt, E.A.F.; et al. Treatment of patients with MYC rearrangement positive large B-cell lymphoma with R-CHOP plus lenalidomide: Results of a multicenter HOVON phase II trial. Haematologica 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef] [PubMed]
- Goebeler, M.E.; Knop, S.; Viardot, A.; Kufer, P.; Topp, M.S.; Einsele, H.; Noppeney, R.; Hess, G.; Kallert, S.; Mackensen, A.; et al. Bispecific T-Cell Engager (BiTE) Antibody Construct Blinatumomab for the Treatment of Patients With Relapsed/Refractory Non-Hodgkin Lymphoma: Final Results From a Phase I Study. J. Clin. Oncol. 2016, 34, 1104–1111. [Google Scholar] [CrossRef]
- Viardot, A.; Goebeler, M.E.; Hess, G.; Neumann, S.; Pfreundschuh, M.; Adrian, N.; Zettl, F.; Libicher, M.; Sayehli, C.; Stieglmaier, J.; et al. Phase 2 study of the bispecific T-cell engager (BiTE) antibody blinatumomab in relapsed/refractory diffuse large B-cell lymphoma. Blood 2016, 127, 1410–1416. [Google Scholar] [CrossRef]
- Buhmann, R.; Simoes, B.; Stanglmaier, M.; Yang, T.; Faltin, M.; Bund, D.; Lindhofer, H.; Kolb, H.J. Immunotherapy of recurrent B-cell malignancies after allo-SCT with Bi20 (FBTA05), a trifunctional anti-CD3 x anti-CD20 antibody and donor lymphocyte infusion. Bone Marrow Transplant. 2009, 43, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younes, A.; Santoro, A.; Shipp, M.; Zinzani, P.L.; Timmerman, J.M.; Ansell, S.; Armand, P.; Fanale, M.; Ratanatharathorn, V.; Kuruvilla, J.; et al. Nivolumab for classical Hodgkin’s lymphoma after failure of both autologous stem-cell transplantation and brentuximab vedotin: A multicentre, multicohort, single-arm phase 2 trial. Lancet Oncol. 2016, 17, 1283–1294. [Google Scholar] [CrossRef] [Green Version]
- Ansell, S.M.; Minnema, M.C.; Johnson, P.; Timmerman, J.M.; Armand, P.; Shipp, M.A.; Rodig, S.J.; Ligon, A.H.; Roemer, M.G.M.; Reddy, N.; et al. Nivolumab for Relapsed/Refractory Diffuse Large B-Cell Lymphoma in Patients Ineligible for or Having Failed Autologous Transplantation: A Single-Arm, Phase II Study. J. Clin. Oncol. 2019, 37. [Google Scholar] [CrossRef]
- Smith, S.D.; Till, B.G.; Shadman, M.S.; Lynch, R.C.; Cowan, A.J.; Wu, Q.W.; Voutsinas, J.; Rasmussen, H.A.; Blue, K.; Ujjani, C.S.; et al. Pembrolizumab with R-CHOP in previously untreated diffuse large B-cell lymphoma: Potential for biomarker driven therapy. Br. J. Haematol. 2020. [Google Scholar] [CrossRef]
- Advani, R.; Flinn, I.; Popplewell, L.; Forero, A.; Bartlett, N.L.; Ghosh, N.; Kline, J.; Roschewski, M.; LaCasce, A.; Collins, G.P.; et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 379, 1711–1721. [Google Scholar] [CrossRef]
- Kawashima, I.; Inamoto, Y.; Maeshima, A.M.; Nomoto, J.; Tajima, K.; Honda, T.; Shichijo, T.; Kawajiri, A.; Takemura, T.; Onishi, A.; et al. Double-Expressor Lymphoma Is Associated with Poor Outcomes after Allogeneic Hematopoietic Cell Transplantation. Biol. Blood Marrow Transplant. 2018, 24, 294–300. [Google Scholar] [CrossRef] [Green Version]
- Herrera, A.F.; Rodig, S.J.; Song, J.Y.; Kim, Y.; Griffin, G.K.; Yang, D.; Nikolaenko, L.; Mei, M.; Bedell, V.; Dal Cin, P.; et al. Outcomes after Allogeneic Stem Cell Transplantation in Patients with Double-Hit and Double-Expressor Lymphoma. Biol. Blood Marrow Transplant. 2018, 24, 514–520. [Google Scholar] [CrossRef] [Green Version]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. Car T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.; Lin, J.; Zhong, Y.; Burcul, A.; Mohan, P.; Jiang, M.; Sun, L.; Yong-Gonzalez, V.; Viale, A.; Cross, J.R.; et al. c-MYC regulates mRNA translation efficiency and start-site selection in lymphoma. J. Exp. Med. 2019, 216, 1509–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jager, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020. [Google Scholar] [CrossRef]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.; Niu, Q.; Deng, B.; Liu, S.; Wu, T.; Gao, Z.; Liu, Z.; Zhang, Y.; Qu, X.; Zhang, Y.; et al. CD22 CAR T-cell therapy in refractory or relapsed B acute lymphoblastic leukemia. Leukemia 2019, 33, 2854–2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.M.; Wu, Z.Q.; Wang, Y.; Guo, Y.L.; Dai, H.R.; Wang, X.H.; Li, X.; Zhang, Y.J.; Zhang, W.Y.; Chen, M.X.; et al. Autologous T Cells Expressing CD30 Chimeric Antigen Receptors for Relapsed or Refractory Hodgkin Lymphoma: An Open-Label Phase I Trial. Clin. Cancer Res. 2017, 23, 1156–1166. [Google Scholar] [CrossRef] [Green Version]
- Ramos, C.A.; Ballard, B.; Zhang, H.; Dakhova, O.; Gee, A.P.; Mei, Z.; Bilgi, M.; Wu, M.F.; Liu, H.; Grilley, B.; et al. Clinical and immunological responses after CD30-specific chimeric antigen receptor-redirected lymphocytes. J. Clin. Investig. 2017, 127, 3462–3471. [Google Scholar] [CrossRef]
- Ormhøj, M.; Scarfo, I.; Cabral, M.L.; Bailey, S.R.; Lorrey, S.J.; Bouffard, A.A.; Castano, A.P.; Larson, R.C.; Riley, L.S.; Schmidts, A.; et al. Chimeric Antigen Receptor T Cells Targeting CD79b Show Efficacy in Lymphoma with or without Cotargeting CD19. Clin. Cancer Res. 2019, 25, 7046–7057. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Tian, X.; Bian, X.; Zhu, T.; Qin, H.; Zhang, R.; Xu, Y.; Pan, Z.; Huang, H.; Fu, J.; et al. T cells redirected against Igbeta for the immunotherapy of B cell lymphoma. Leukemia 2020, 34, 821–830. [Google Scholar] [CrossRef]
- Shimizu, K.; Iyoda, T.; Yamasaki, S.; Kadowaki, N.; Tojo, A.; Fujii, S. NK and NKT Cell-Mediated Immune Surveillance against Hematological Malignancies. Cancers 2020, 12, 817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casey, S.C.; Baylot, V.; Felsher, D.W. The MYC oncogene is a global regulator of the immune response. Blood 2018, 131, 2007–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dose, M.; Khan, I.; Guo, Z.; Kovalovsky, D.; Krueger, A.; von Boehmer, H.; Khazaie, K.; Gounari, F. c-Myc mediates pre-TCR-induced proliferation but not developmental progression. Blood 2006, 108, 2669–2677. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Jiao, A.; Dai, M.; Wiest, D.L.; Zhuang, Y. Id3 Restricts gammadelta NKT Cell Expansion by Controlling Egr2 and c-Myc Activity. J. Immunol. 2018, 201, 1452–1459. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Trumpp, A.; Refaeli, Y.; Oskarsson, T.; Gasser, S.; Murphy, M.; Martin, G.R.; Bishop, J.M. c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 2001, 414, 768–773. [Google Scholar] [CrossRef]
- Cichocki, F.; Hanson, R.J.; Lenvik, T.; Pitt, M.; McCullar, V.; Li, H.; Anderson, S.K.; Miller, J.S. The transcription factor c-Myc enhances KIR gene transcription through direct binding to an upstream distal promoter element. Blood 2009, 113, 3245–3253. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Adams, N.M.; Xu, Y.; Cao, J.; Allan, D.S.J.; Carlyle, J.R.; Chen, X.; Sun, J.C.; Glimcher, L.H. The IRE1 endoplasmic reticulum stress sensor activates natural killer cell immunity in part by regulating c-Myc. Nat. Immunol. 2019, 20, 865–878. [Google Scholar] [CrossRef]
- Abramson, J.S.; Siddiqi, T.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Arnason, J.E.; Wang, M.; Forero-Torres, A.; Albertson, T.; Dehner, C.; et al. High Durable CR Rates and Preliminary Safety Profile for JCAR017 in R/R Aggressive B-NHL (TRANSCEND NHL 001 Study): A Defined Composition CD19-Directed CAR T Cell Product with Potential for Outpatient Administration. Biol. Blood Marrow Transplant. 2018, 24, S25. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Jonge, A.V.; Mutis, T.; Roemer, M.G.M.; Scheijen, B.; Chamuleau, M.E.D. Impact of MYC on Anti-Tumor Immune Responses in Aggressive B Cell Non-Hodgkin Lymphomas: Consequences for Cancer Immunotherapy. Cancers 2020, 12, 3052. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12103052
de Jonge AV, Mutis T, Roemer MGM, Scheijen B, Chamuleau MED. Impact of MYC on Anti-Tumor Immune Responses in Aggressive B Cell Non-Hodgkin Lymphomas: Consequences for Cancer Immunotherapy. Cancers. 2020; 12(10):3052. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12103052
Chicago/Turabian Stylede Jonge, A. Vera, Tuna Mutis, Margaretha G. M. Roemer, Blanca Scheijen, and Martine E. D. Chamuleau. 2020. "Impact of MYC on Anti-Tumor Immune Responses in Aggressive B Cell Non-Hodgkin Lymphomas: Consequences for Cancer Immunotherapy" Cancers 12, no. 10: 3052. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12103052