
Targeting DDX3X Helicase Activity with BA103 Shows Promising Therapeutic Effects in Preclinical Glioblastoma Models

 , , , , ,
, , , , ,  , , , , , and
, , , , , and
Abstract
:Simple Summary
Abstract
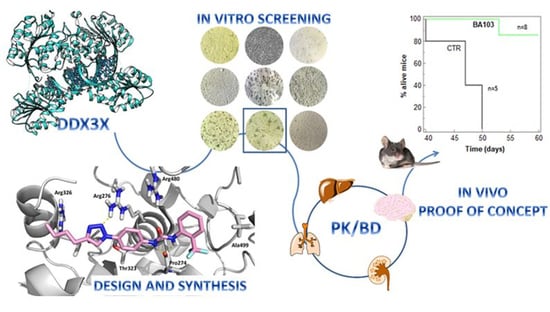
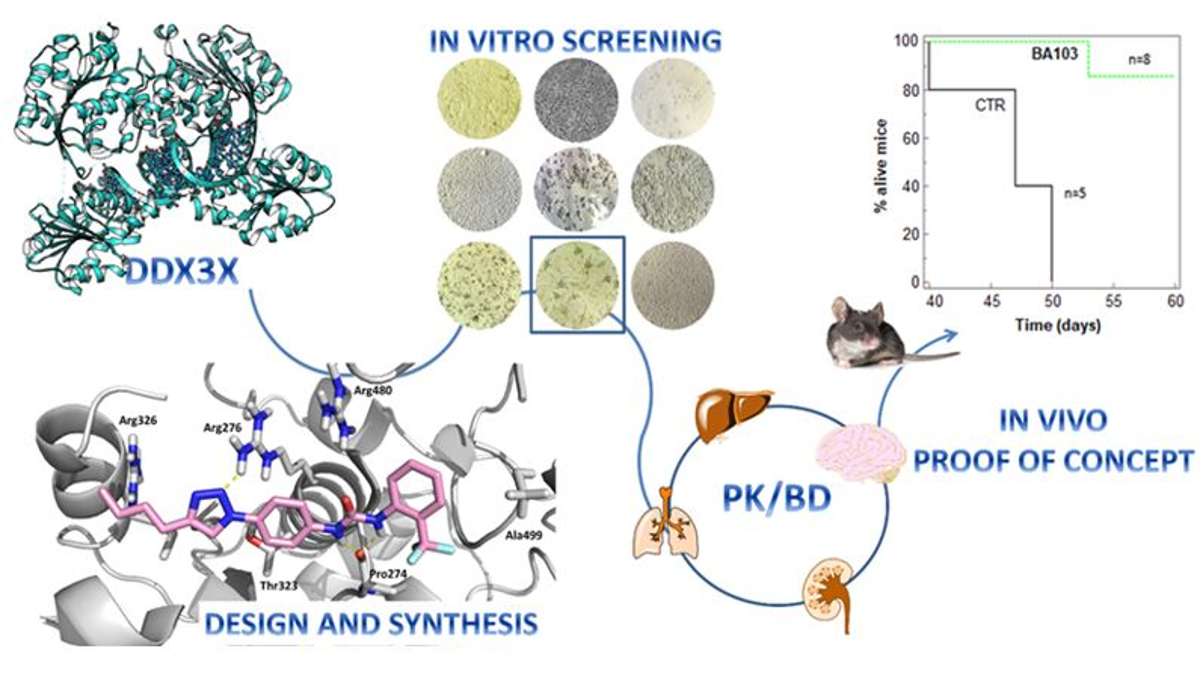
1. Introduction
2. Materials and Methods
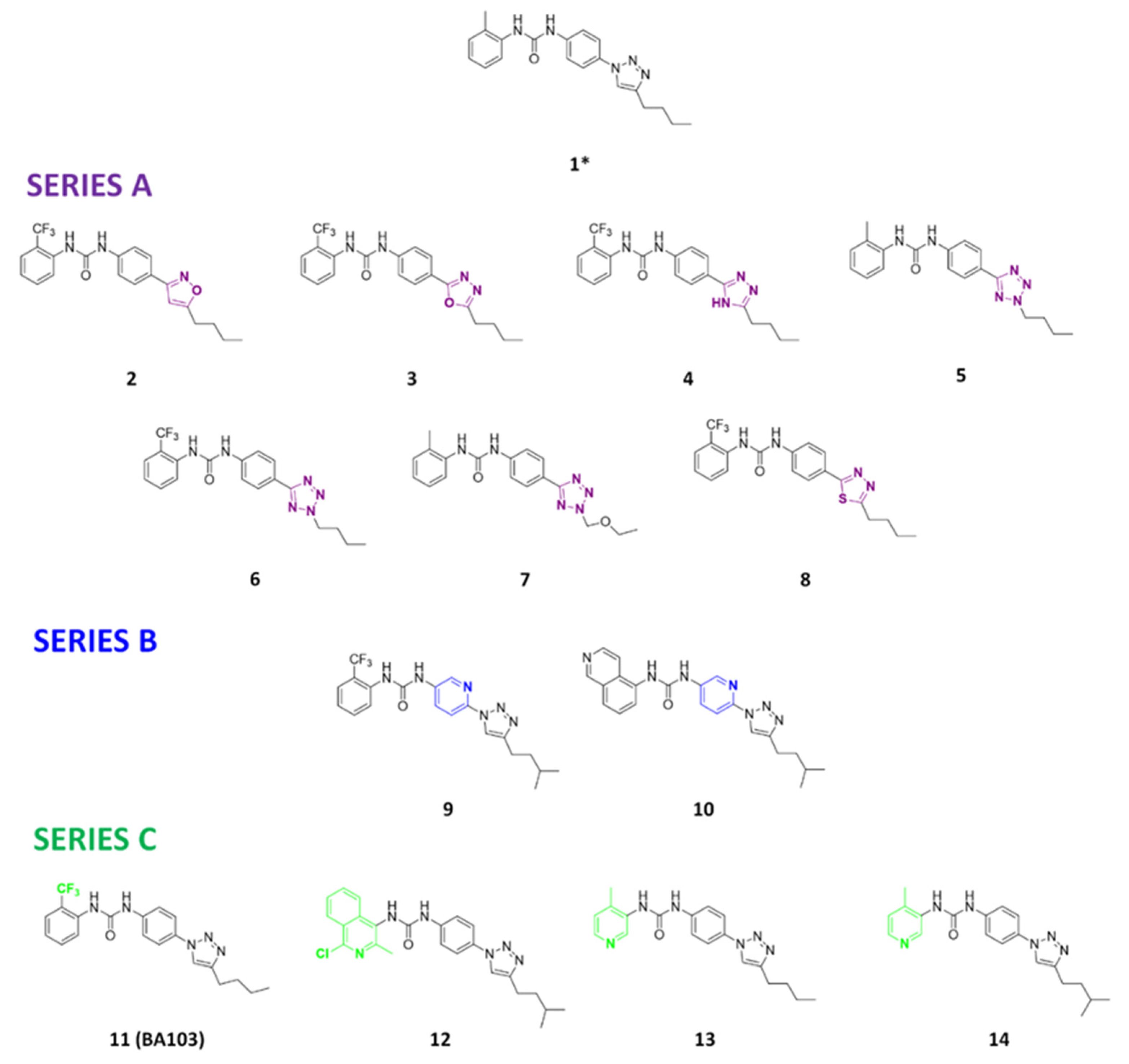
2.1. Chemistry
2.1.1. General and Materials
2.1.2. Instrumentation
2.1.3. Synthesis of Final Compounds
2.1.4. General Procedure for the Preparation of Compounds 11–14
2.2. In Silico Studies
2.2.1. Docking Studies
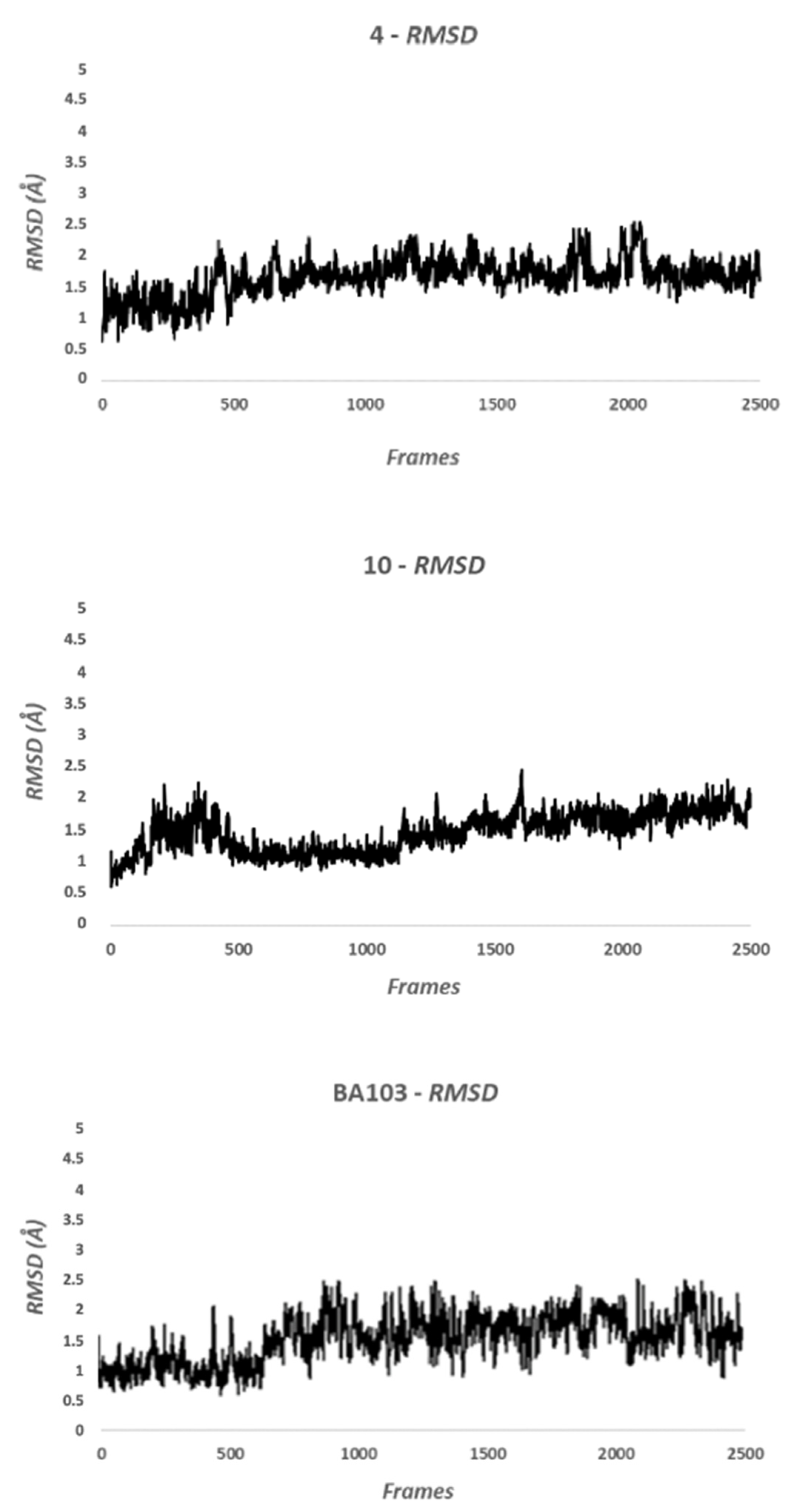
2.2.2. Molecular Dynamic Methods
2.3. Enzymatic Assays
2.3.1. Protein Expression and Purification
2.3.2. Helicase Assay Based on Fluorescence Resonance Energy Transfer (FRET)
- Fluo-FAM
- 5′ UUUUUUUUUUUUUUAGUACCGCCACCCUCAGAACC 3′
- Qu-BHQ1
- 5′ GGUUCUGAGGGUGGCGGUACUA 3′
- DNA capture
- 5′ TAGTACCGCCACCCTCAGAACC 3′.
2.3.3. Kinetic Analysis
2.3.4. Cell Extracts and DDX3X Quantification
2.4. Cellular Assay
2.4.1. Cell Cultures and Reagents
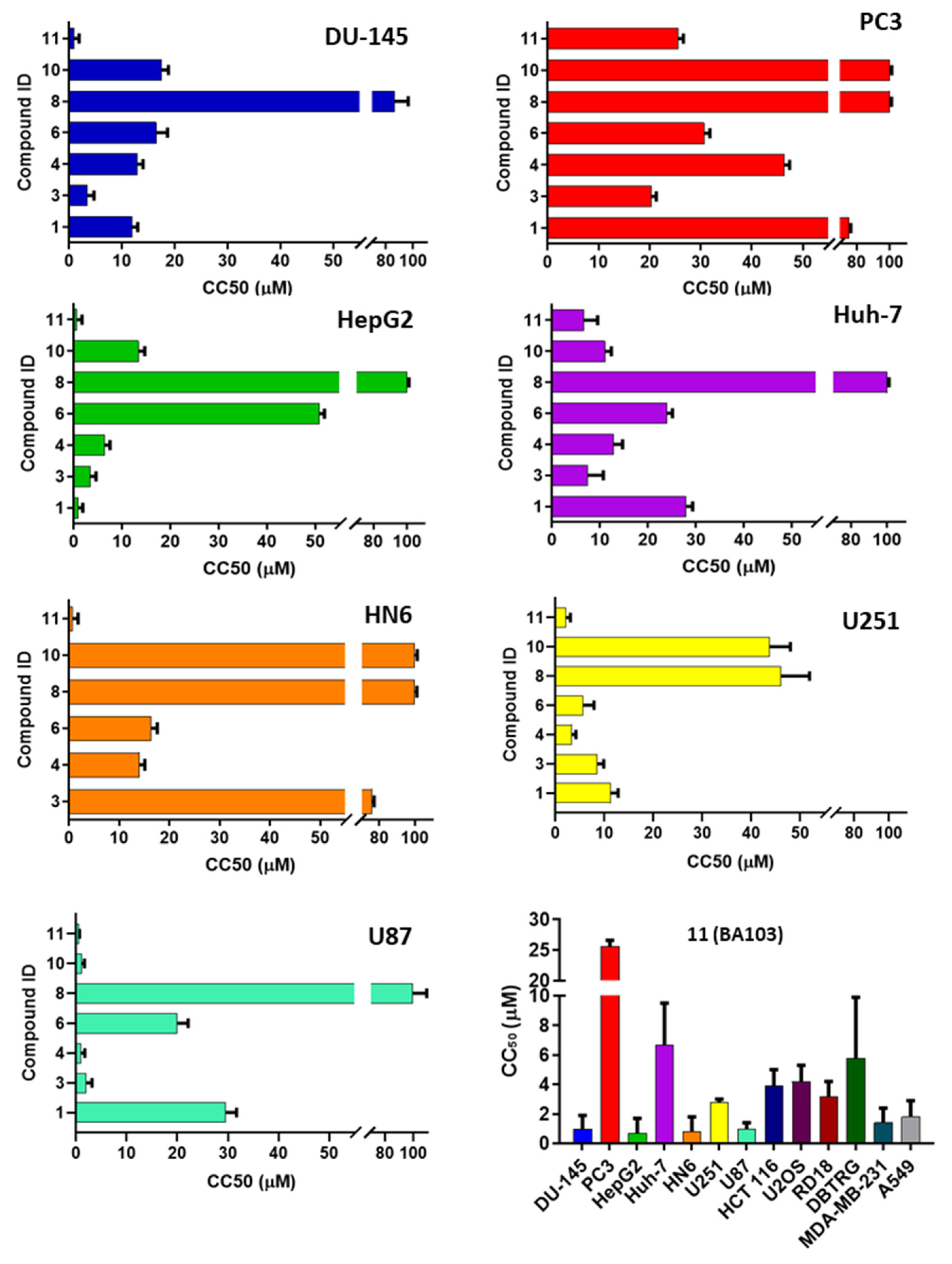
2.4.2. Cytotoxicity Assay
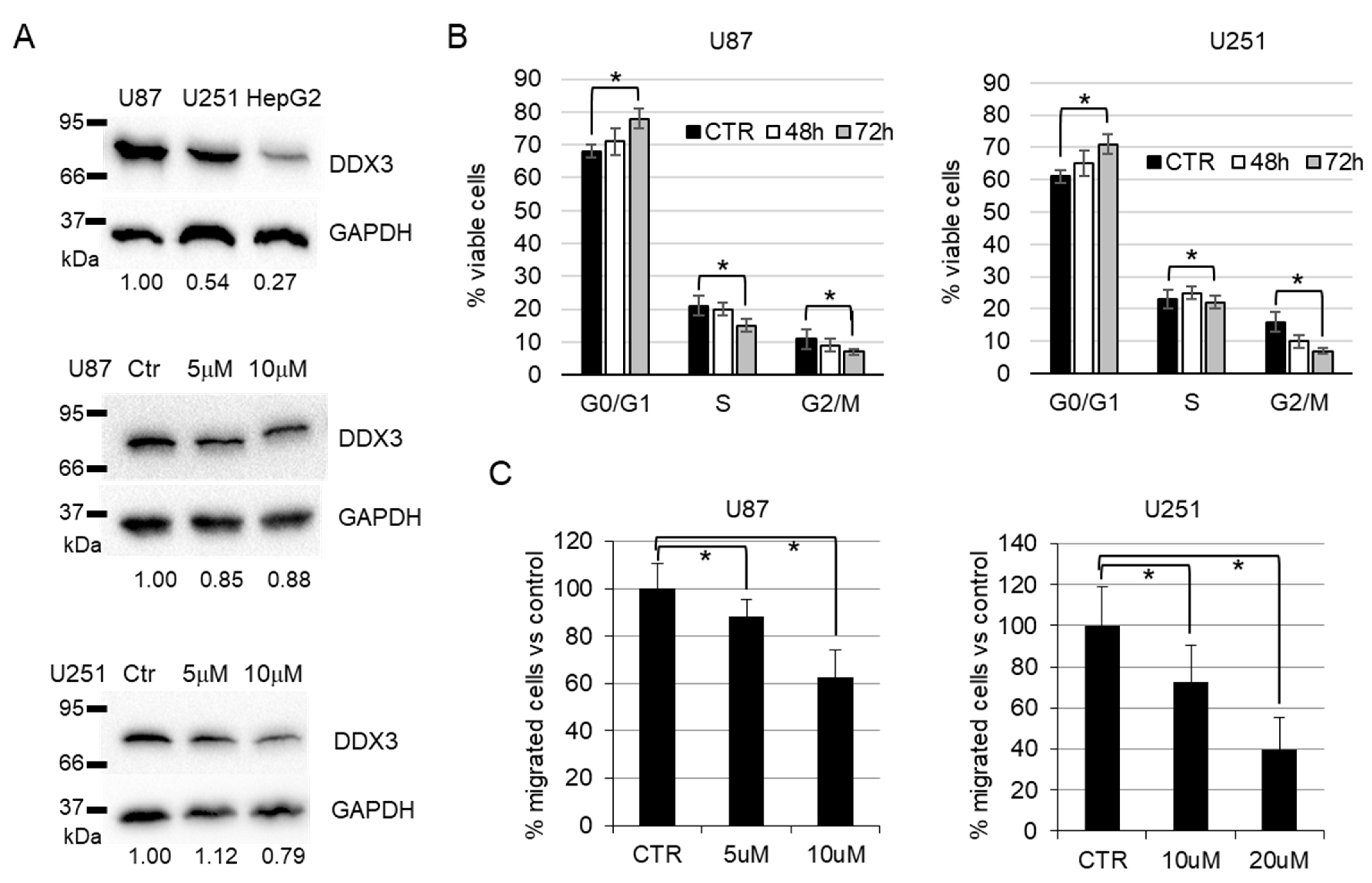
2.4.3. Cell Cycle Analysis
2.4.4. Migration Assay
2.4.5. Western Blot
2.4.6. Immunofluorescence Assay
2.5. In Vivo Studies
2.5.1. Subcutaneous Xenograft
2.5.2. Immunohistochemistry
2.5.3. Orthotopic Xenograft
2.6. Data Analysis and Statistics
3. Results
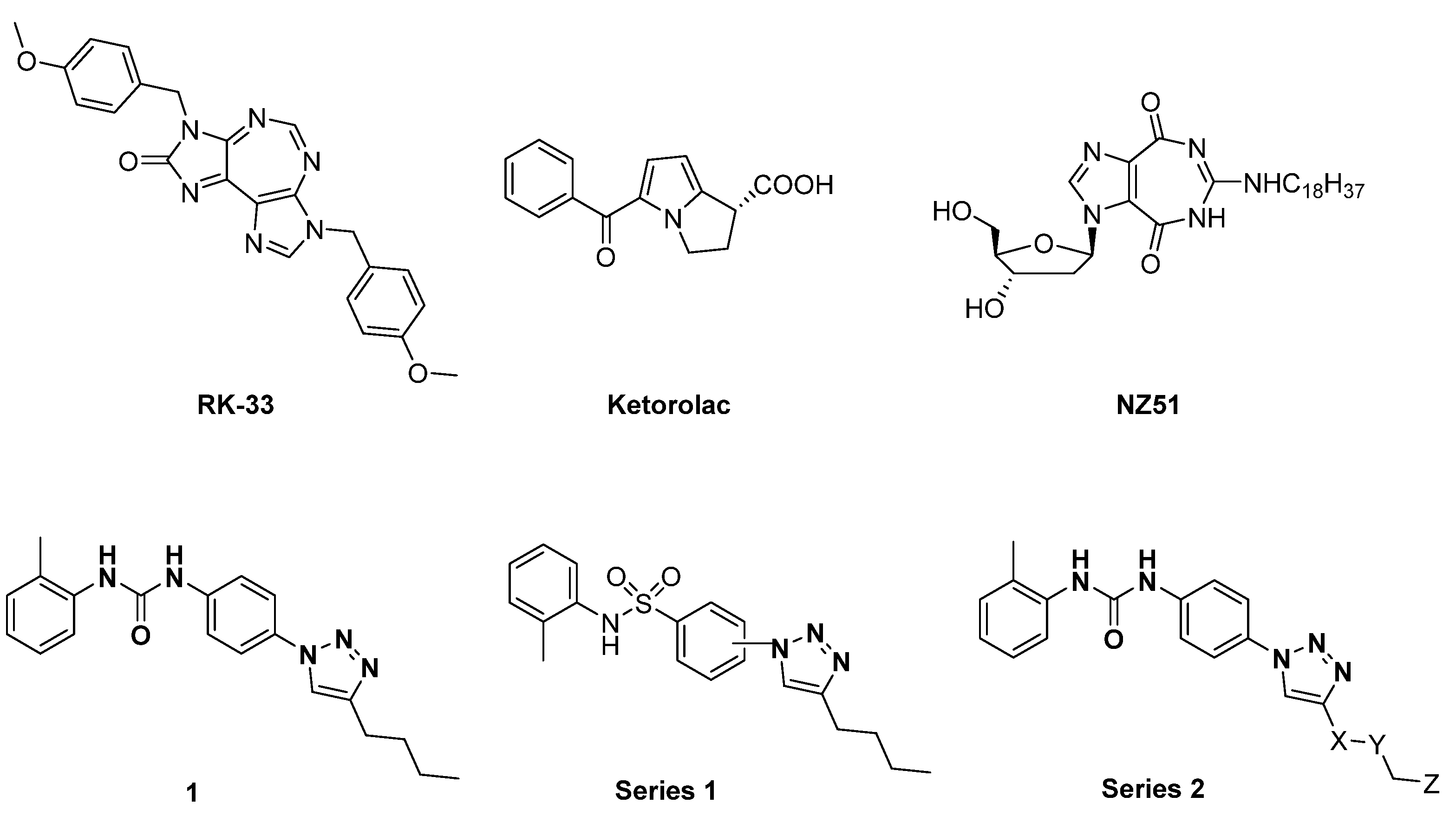
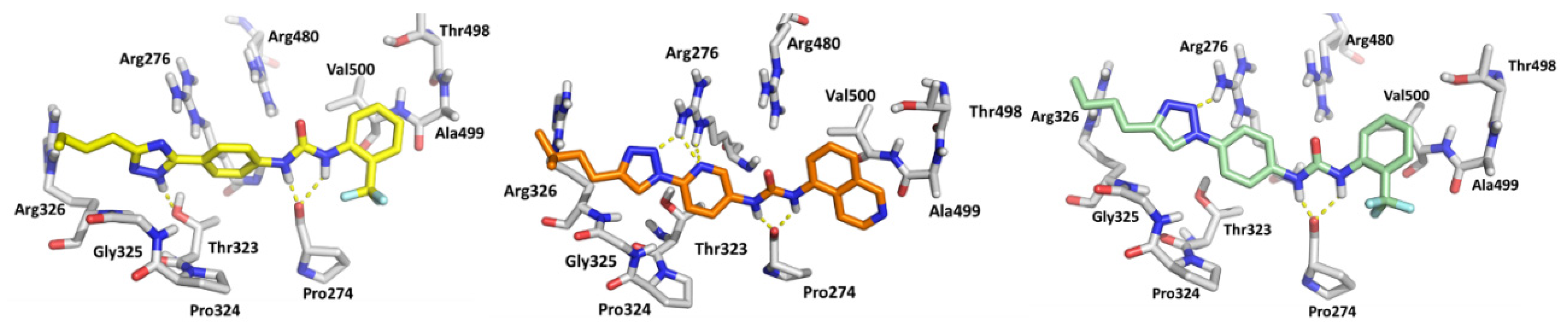
3.1. Design and Synthesis of Compounds
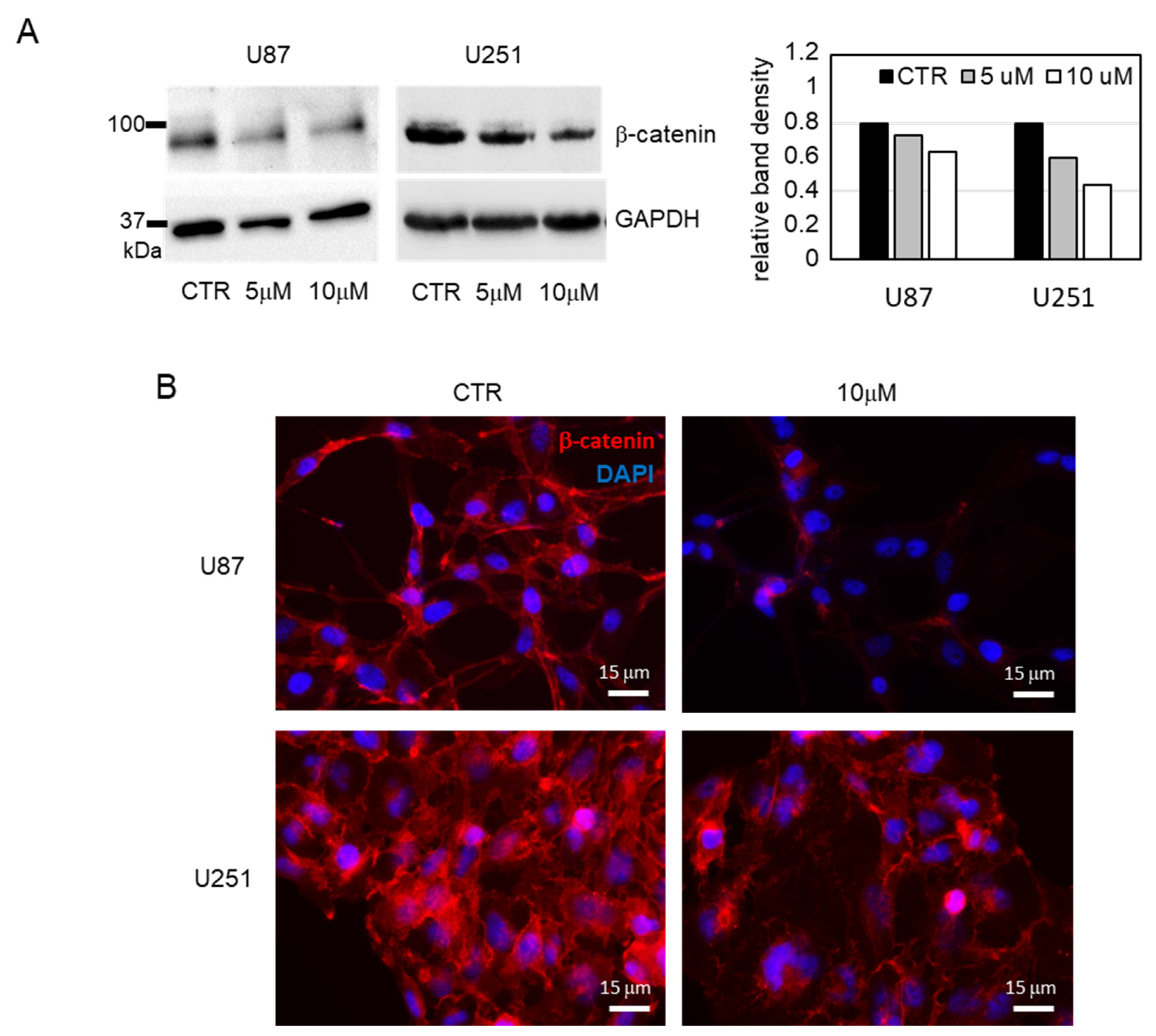
3.2. Biological Evaluation
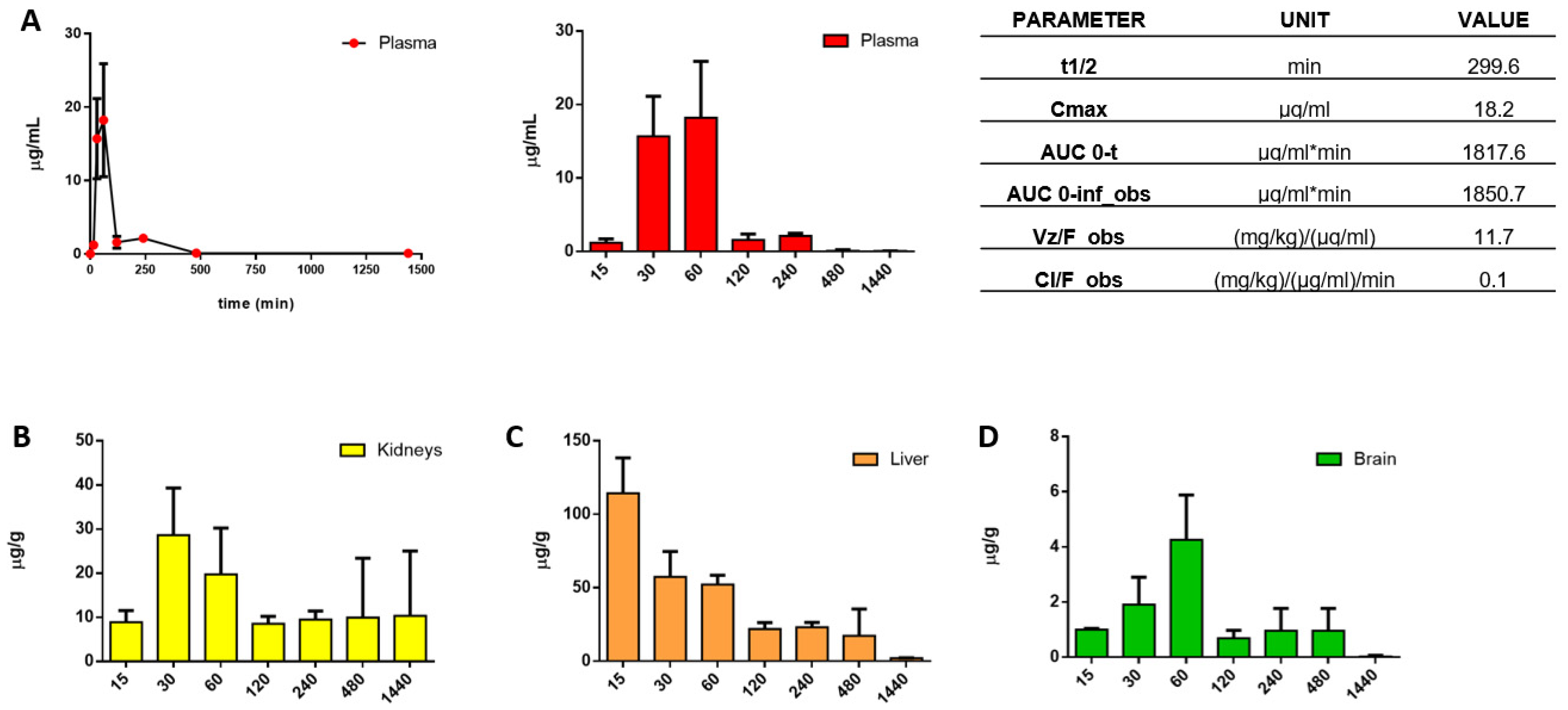
3.3. In Vitro ADME
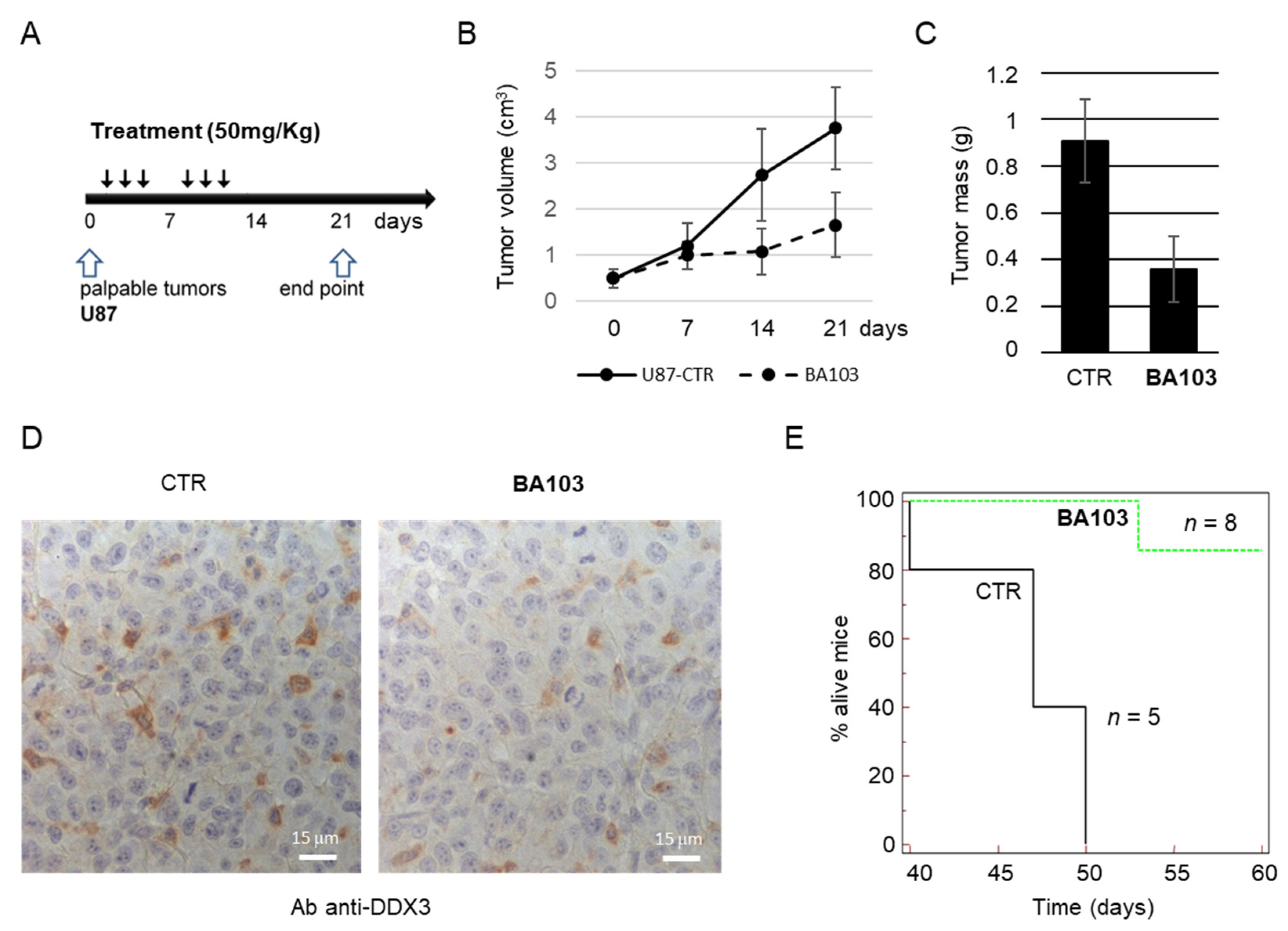
3.4. In Vivo Experiments
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cancer. Available online: https://www.who.int/en/news-room/fact-sheets/detail/cancer (accessed on 9 December 2019).
- Baudino, T.A. Targeted Cancer Therapy: The Next Generation of Cancer Treatment. Curr. Drug Discov. Technol. 2015, 12, 3–20. [Google Scholar] [CrossRef]
- Doi, T.; Shitara, K.; Naito, Y.; Shimomura, A.; Fujiwara, Y.; Yonemori, K.; Shimizu, C.; Shimoi, T.; Kuboki, Y.; Matsubara, N.; et al. Safety, pharmacokinetics, and antitumour activity of trastuzumab deruxtecan (DS-8201), a HER2-targeting antibody-drug conjugate, in patients with advanced breast and gastric or gastro-oesophageal tumours: A phase 1 dose-escalation study. Lancet Oncol. 2017, 18, 1512–1522. [Google Scholar] [CrossRef]
- Ascierto, P.A.; McArthur, G.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef]
- Manier, S.; Huynh, D.; Shen, Y.J.; Zhou, J.; Yusufzai, T.; Salem, K.Z.; Ebright, R.Y.; Shi, J.; Park, J.; Glavey, S.V.; et al. Inhibiting the oncogenic translation program is an effective therapeutic strategy in multiple myeloma. Sci. Transl. Med. 2017, 9, eaal2668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Voss, M.R.h.; van Diest, P.J.; Raman, V. Targeting RNA helicases in cancer: The translation trap. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 510–520. [Google Scholar] [CrossRef]
- Soto-Rifo, R.; Ohlmann, T. The role of the DEAD-box RNA helicase DDX3 in mRNA metabolism. Wiley Interdiscip. Rev. RNA 2013, 4, 369–385. [Google Scholar] [CrossRef]
- Ariumi, Y. Multiple functions of DDX3 RNA helicase in gene regulation, tumorigenesis, and viral infection. Front. Genet. 2014, 5, 423. [Google Scholar] [CrossRef] [Green Version]
- He, T.-Y.; Wu, D.-W.; Lin, P.-L.; Wang, L.; Huang, C.-C.; Chou, M.-C.; Lee, H. DDX3 promotes tumor invasion in colorectal cancer via the CK1ε/Dvl2 axis. Sci. Rep. 2016, 6, 21483. [Google Scholar] [CrossRef] [Green Version]
- Botlagunta, M.; Vesuna, F.; Mironchik, Y.; Raman, A.; Lisok, A.; Winnard, P.; Mukadam, S.; Van Diest, P.; Chen, J.H.; Farabaugh, P.; et al. Oncogenic role of DDX3 in breast cancer biogenesis. Oncogene 2008, 27, 3912–3922. [Google Scholar] [CrossRef] [Green Version]
- Bol, G.M.; Vesuna, F.; Xie, M.; Zeng, J.; Aziz, K.; Gandhi, N.; Levine, A.; Irving, A.; Korz, D.; Tantravedi, S.; et al. Targeting DDX3 with a small molecule inhibitor for lung cancer therapy. EMBO Mol. Med. 2015, 7, 648–669. [Google Scholar] [CrossRef]
- Zhao, L.; Mao, Y.; Zhou, J.; Zhao, Y.; Cao, Y.; Chen, X. Multifunctional DDX3: Dual roles in various cancer development and its related signaling pathways. Am. J. Cancer Res. 2016, 6, 387–402. [Google Scholar]
- Sun, M.; Song, L.; Zhou, T.; Gillespie, G.Y.; Jope, R.S. The role of DDX3 in regulating Snail. Biochim. Biophys. Acta 2011, 1813, 438–447. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Zhou, T.; Jonasch, E.; Jope, R.S. DDX3 regulates DNA damage-induced apoptosis and p53 stabilization. Biochim. Biophys. Acta-Mol. Cell Res. 2013, 1833, 1489–1497. [Google Scholar] [CrossRef] [Green Version]
- Wilky, B.A.; Kim, C.; McCarty, G.; Montgomery, E.A.; Kammers, K.; DeVine, L.R.; Cole, R.N.; Raman, V.; Loeb, D.M. RNA helicase DDX3: A novel therapeutic target in Ewing sarcoma. Oncogene 2016, 35, 2574–2583. [Google Scholar] [CrossRef]
- Samal, S.K.; Routray, S.; Veeramachaneni, G.K.; Dash, R.; Botlagunta, M. Ketorolac salt is a newly discovered DDX3 inhibitor to treat oral cancer. Sci. Rep. 2015, 5, 9982. [Google Scholar] [CrossRef]
- Xie, M.; Vesuna, F.; Botlagunta, M.; Bol, G.M.; Irving, A.; Bergman, Y.; Hosmane, R.S.; Kato, Y.; Winnard, P.T.; Raman, V. NZ51, a ring-expanded nucleoside analog, inhibits motility and viability of breast cancer cells by targeting the RNA helicase DDX3. Oncotarget 2015, 6, 29901–29913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccosanti, F.; Di Rienzo, M.; Romagnoli, A.; Colavita, F.; Refolo, G.; Castilletti, C.; Agrati, C.; Brai, A.; Manetti, F.; Botta, L.; et al. Proteomic Analysis Identifies the RNA Helicase DDX3X as a Host Target Against SARS-CoV-2 Infection. Antivir. Res. 2021, 190, 105064. [Google Scholar] [CrossRef]
- Brai, A.; Fazi, R.; Tintori, C.; Zamperini, C.; Bugli, F.; Sanguinetti, M.; Stigliano, E.; Esté, J.; Badia, R.; Franco, S.; et al. Human DDX3 protein is a valuable target to develop broad spectrum antiviral agents. Proc. Natl. Acad. Sci. USA 2016, 113, 5388–5393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brai, A.; Riva, V.; Saladini, F.; Zamperini, C.; Trivisani, C.I.; Garbelli, A.; Pennisi, C.; Giannini, A.; Boccuto, A.; Bugli, F.; et al. DDX3X inhibitors, an effective way to overcome HIV-1 resistance targeting host proteins. Eur. J. Med. Chem. 2020, 200. [Google Scholar] [CrossRef] [PubMed]
- Brai, A.; Boccuto, A.; Monti, M.; Marchi, S.; Vicenti, I.; Saladini, F.; Trivisani, C.I.; Pollutri, A.; Trombetta, C.M.; Montomoli, E.; et al. Exploring the Implication of DDX3X in DENV Infection: Discovery of the First-in-Class DDX3X Fluorescent Inhibitor. ACS Med. Chem. Lett. 2020, 11, 956–962. [Google Scholar] [CrossRef]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, I.; Mahfooz, S.; Elbasan, E.B.; Karacam, B.; Oztanir, M.N.; Hatiboglu, M.A. Targeting Glioblastoma: The Current State of Different Therapeutic Approaches. Curr. Neuropharmacol. 2021, 19. [Google Scholar] [CrossRef] [PubMed]
- Ou, A.; Yung, W.K.A.; Majd, N. Molecular Mechanisms of Treatment Resistance in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 351. [Google Scholar] [CrossRef]
- Rominiyi, O.; Vanderlinden, A.; Clenton, S.J.; Bridgewater, C.; Al-Tamimi, Y.; Collis, S.J. Tumour treating fields therapy for glioblastoma: Current advances and future directions. Br. J. Cancer 2021, 124, 697. [Google Scholar] [CrossRef]
- Brai, A.; Martelli, F.; Riva, V.; Garbelli, A.; Fazi, R.; Zamperini, C.; Pollutri, A.; Falsitta, L.; Ronzini, S.; Maccari, L.; et al. DDX3X Helicase Inhibitors as a New Strategy To Fight the West Nile Virus Infection. J. Med. Chem. 2019, 62, 2333–2347. [Google Scholar] [CrossRef]
- Gravina, G.L.; Mancini, A.; Colapietro, A.; Delle Monache, S.; Sferra, R.; Pompili, S.; Vitale, F.; Martellucci, S.; Marampon, F.; Mattei, V.; et al. The Brain Penetrating and Dual TORC1/TORC2 Inhibitor, RES529, Elicits Anti-Glioma Activity and Enhances the Therapeutic Effects of Anti-Angiogenetic Compounds in Preclinical Murine Models. Cancers (Basel) 2019, 11, 1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Bioinforma. 2004, 55, 351–367. [Google Scholar] [CrossRef] [Green Version]
- QikProp; Schrödinger, LLC: New York, NY, USA, 2019.
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. Amber 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Riva, V.; Garbelli, A.; Brai, A.; Casiraghi, F.; Fazi, R.; Trivisani, C.I.; Boccuto, A.; Saladini, F.; Vicenti, I.; Martelli, F.; et al. Unique Domain for a Unique Target: Selective Inhibitors of Host Cell DDX3X to Fight Emerging Viruses. J. Med. Chem. 2020, 63, 9876–9887. [Google Scholar] [CrossRef]
- Małecki, P.H.; Rüger, N.; Roatsch, M.; Krylova, O.; Link, A.; Jung, M.; Heinemann, U.; Weiss, M.S. Structure-Based Screening of Tetrazolylhydrazide Inhibitors versus KDM4 Histone Demethylases. ChemMedChem 2019, 14, 1828–1839. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.H.; Yu, H.I.; Cho, W.C.; Tarn, W.Y. DDX3 modulates cell adhesion and motility and cancer cell metastasis via Rac1-mediated signaling pathway. Oncogene 2015, 34, 2790–2800. [Google Scholar] [CrossRef] [PubMed]
- Hueng, D.Y.; Tsai, W.C.; Chiou, H.Y.C.; Feng, S.W.; Lin, C.; Li, Y.F.; Huang, L.C.; Lin, M.H. DDX3X biomarker correlates with poor survival in human gliomas. Int. J. Mol. Sci. 2015, 16, 15578–15591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Fang, E.; Mei, H.; Chen, Y.; Li, H.; Li, D.; Song, H.; Wang, J.; Hong, M.; Xiao, W.; et al. Cis-Acting circ-CTNNB1 Promotes β-Catenin Signaling and Cancer Progression via DDX3-Mediated Transactivation of YY1. Cancer Res. 2019, 79, 557–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd. ID | IC50 ± SD(μM) [a,b] |
|---|---|
 1[c] | 0.30 ± 0.07 |
 2 | nd [d] |
 3 | 1.0 ± 0.2 |
 4 | 0.07 ± 0.02 |
 5 | 4.9 ± 0.5 |
 6 | 0.8 ± 0.2 |
 7 | 14 ± 2.0 |
 8 | 50 ± 7 |
 9 | 1.1 ± 0.2 |
 10 | 0.10 ± 0.03 |
 11/BA 103 | 0.40 ± 0.05 |
 12 | 10 ± 1.0 |
 13 | 1.5 ± 0.3 |
 14 | 2.0 ± 0.3 |
| Cpd ID | ATPase DDX3X IC50 ± SD, µM | DDX1 IC50 ± SD, µM | SI |
|---|---|---|---|
| 1 * | >200 [a] | >200 [a] | >666 |
| 4 | NT | 29.29 ± 16.75 | 418 |
| 10 | NT | >100 | >1000 |
| BA103 | >200 [a] | >100 | >250 |
| Cell Line | U2OS | A549 | Huh-7 | DU-145 | HepG2 |
|---|---|---|---|---|---|
| DDX3X ± SD (nM) | 174 ± 20 | 103 ± 10 | 755 ± 75 | 372 ± 20 | 538 ± 33 |
| Cpd. ID | AppP [b] | Memb. [c] Ret. % | LogS [d] | HLM Stability ± SD [e] |
|---|---|---|---|---|
| 1 | 2.86 × 10−6 | 19.1 | −7.05 | 99.0 ± 0.6 |
| 3 | 9.75 × 10−6 | 10.3 | <−7.60 | 88.9 ± 0.9 |
| 4 | <0.1 × 10−6 | 2.3 | −6.56 | 97.9 ± 0.3 |
| 6 | 10.23 × 10−6 | 21.6 | <−7.60 | 90.32 ± 0.7 |
| 10 | 1.21 × 10−6 | 11.3 | <−7.44 | 91.31 ± 0.1 |
| 11/BA103 | 7.47 × 10−6 | 23.9 | −7.43 | 97.0 ± 0.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brai, A.; Riva, V.; Clementi, L.; Falsitta, L.; Zamperini, C.; Sinigiani, V.; Festuccia, C.; Sabetta, S.; Aiello, D.; Roselli, C.; et al. Targeting DDX3X Helicase Activity with BA103 Shows Promising Therapeutic Effects in Preclinical Glioblastoma Models. Cancers 2021, 13, 5569. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13215569
Brai A, Riva V, Clementi L, Falsitta L, Zamperini C, Sinigiani V, Festuccia C, Sabetta S, Aiello D, Roselli C, et al. Targeting DDX3X Helicase Activity with BA103 Shows Promising Therapeutic Effects in Preclinical Glioblastoma Models. Cancers. 2021; 13(21):5569. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13215569
Chicago/Turabian StyleBrai, Annalaura, Valentina Riva, Letizia Clementi, Lucia Falsitta, Claudio Zamperini, Virginia Sinigiani, Claudio Festuccia, Samantha Sabetta, Davide Aiello, Camilla Roselli, and et al. 2021. "Targeting DDX3X Helicase Activity with BA103 Shows Promising Therapeutic Effects in Preclinical Glioblastoma Models" Cancers 13, no. 21: 5569. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13215569