
The Role of ROS as a Double-Edged Sword in (In)Fertility: The Impact of Cancer Treatment

 , , and
, , and
Abstract
:Simple Summary
Abstract

1. Introduction
2. Oxidative Stress and Fertility
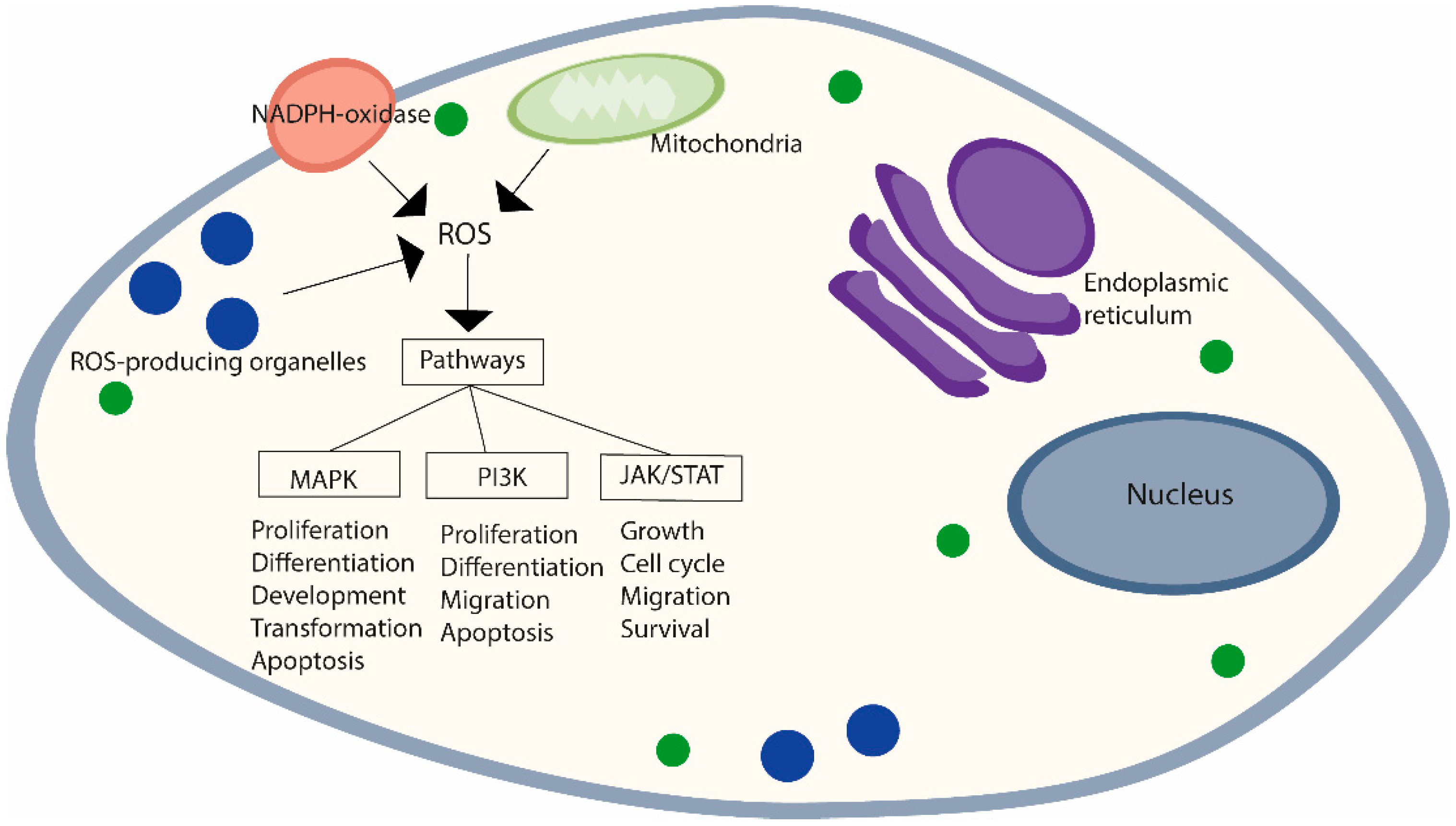
2.1. Reactive Oxygen Species and Oxidative Stress
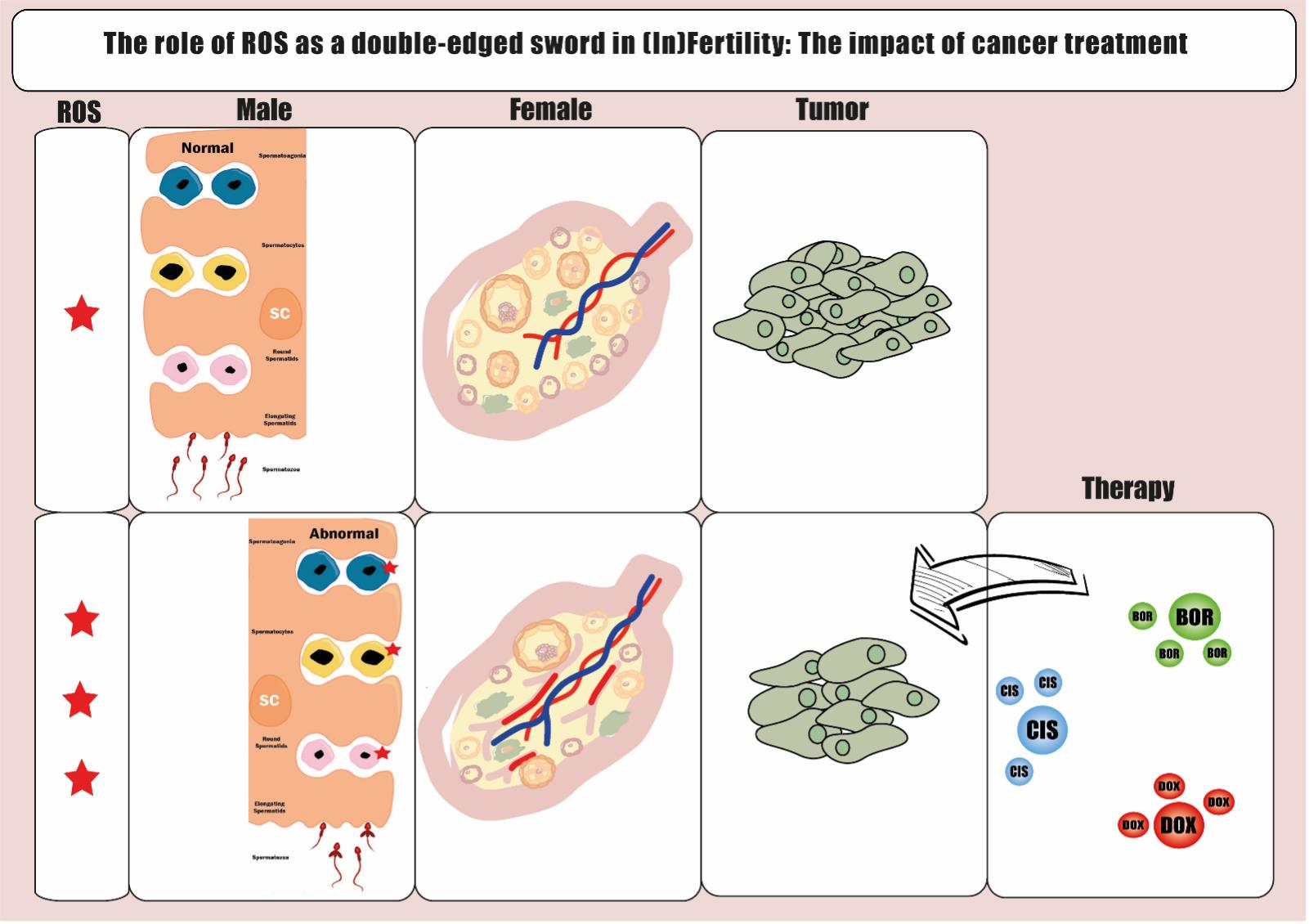
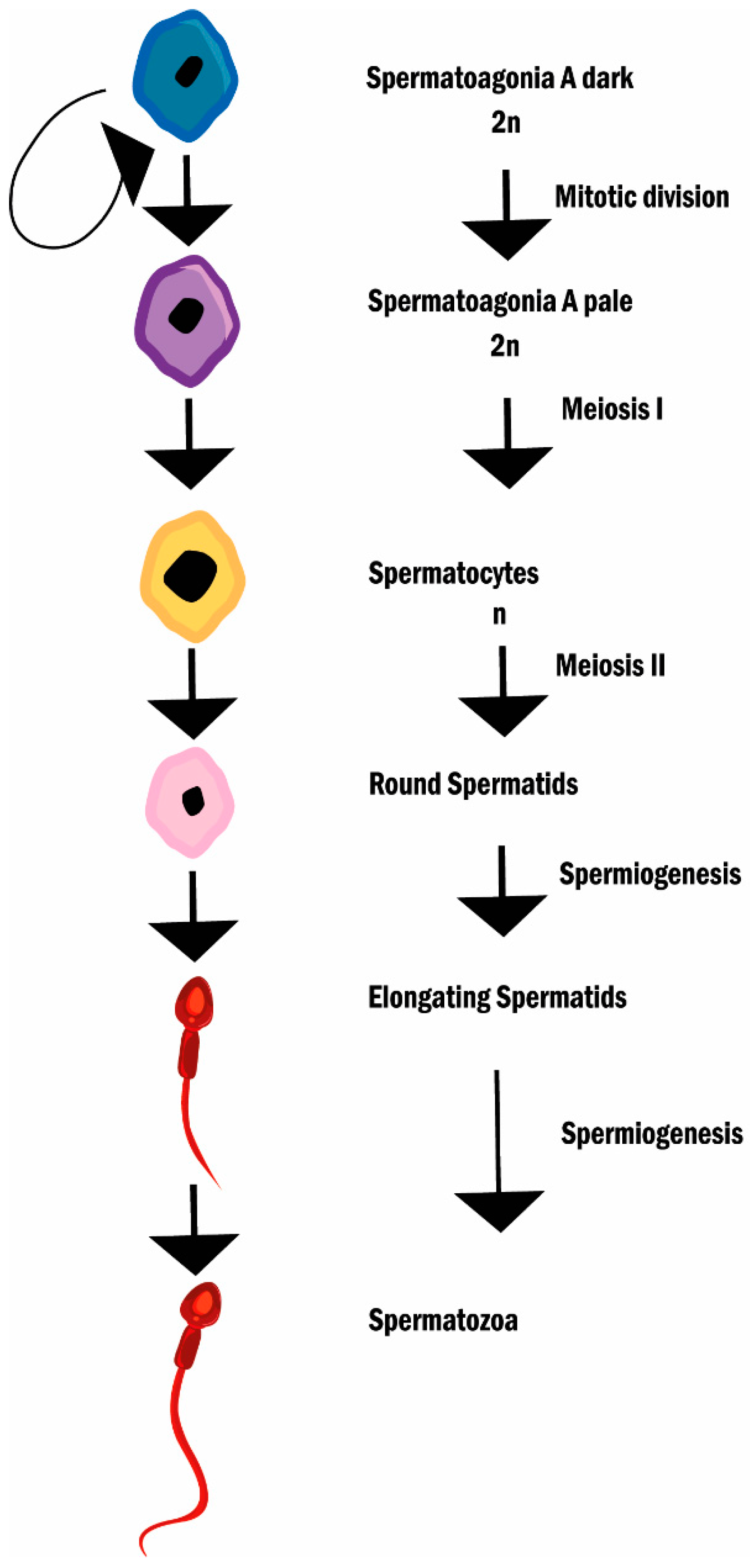
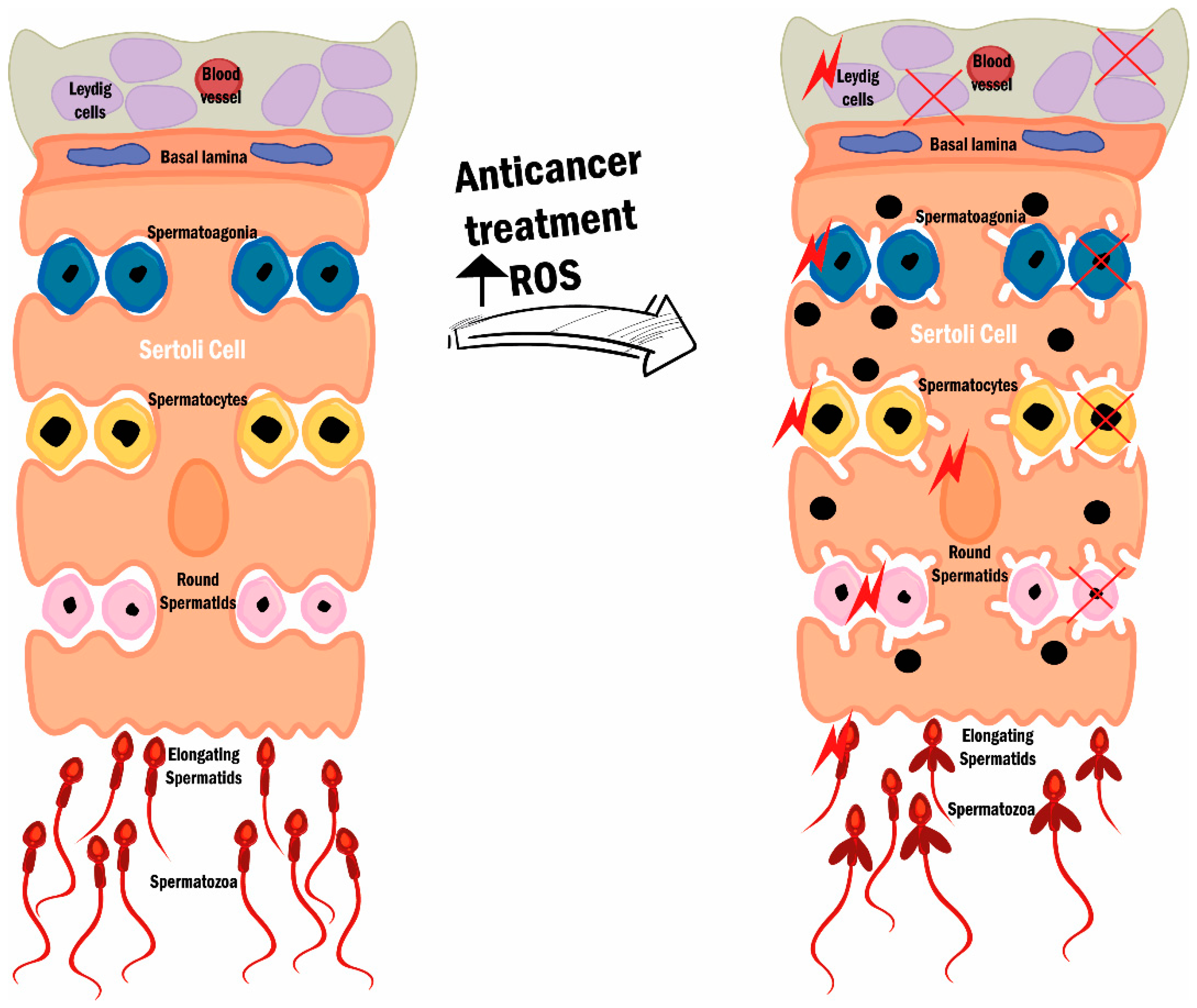
2.2. Reactive Oxygen Species, Oxidative Stress, and Male (In)Fertility
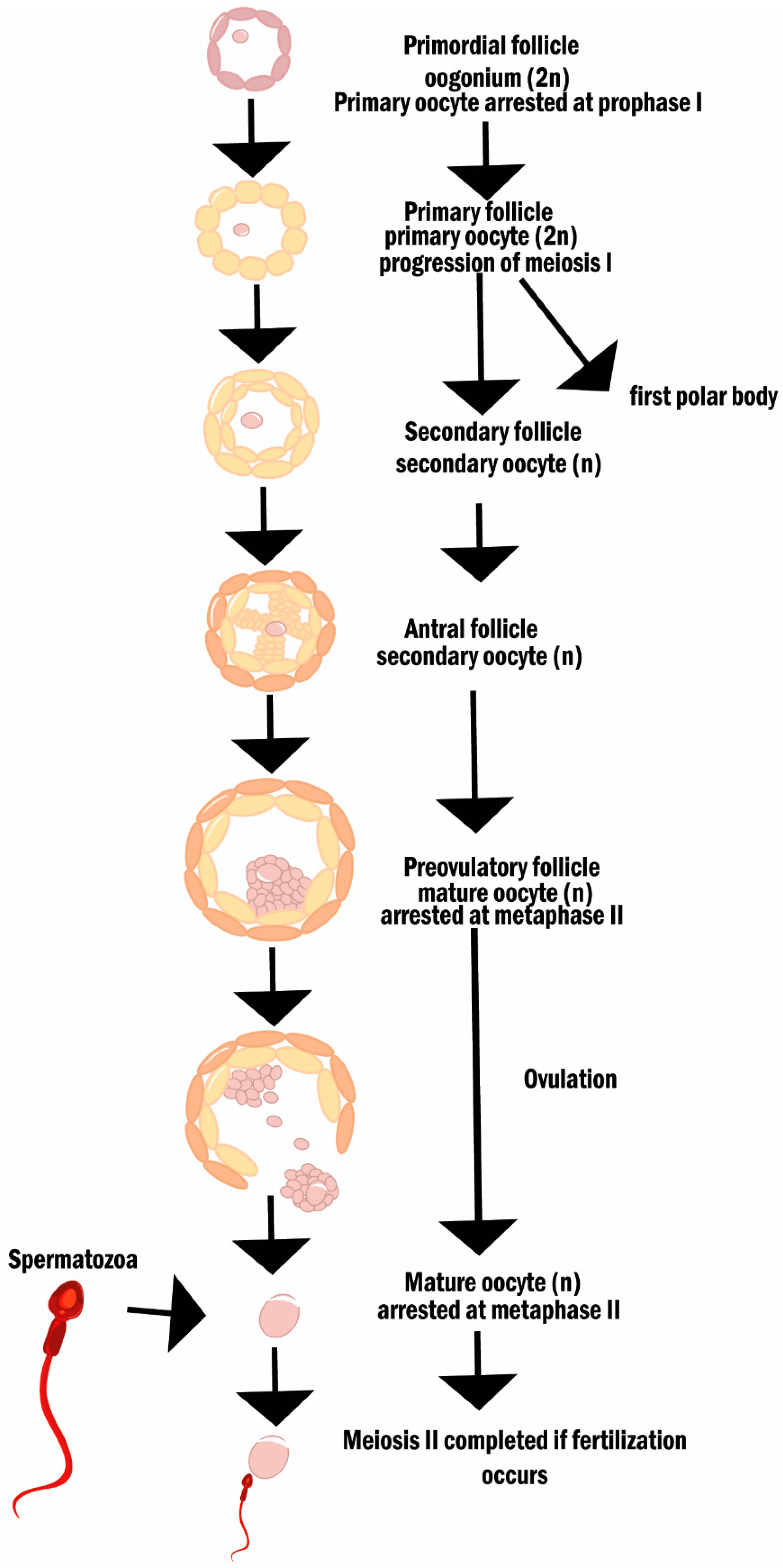
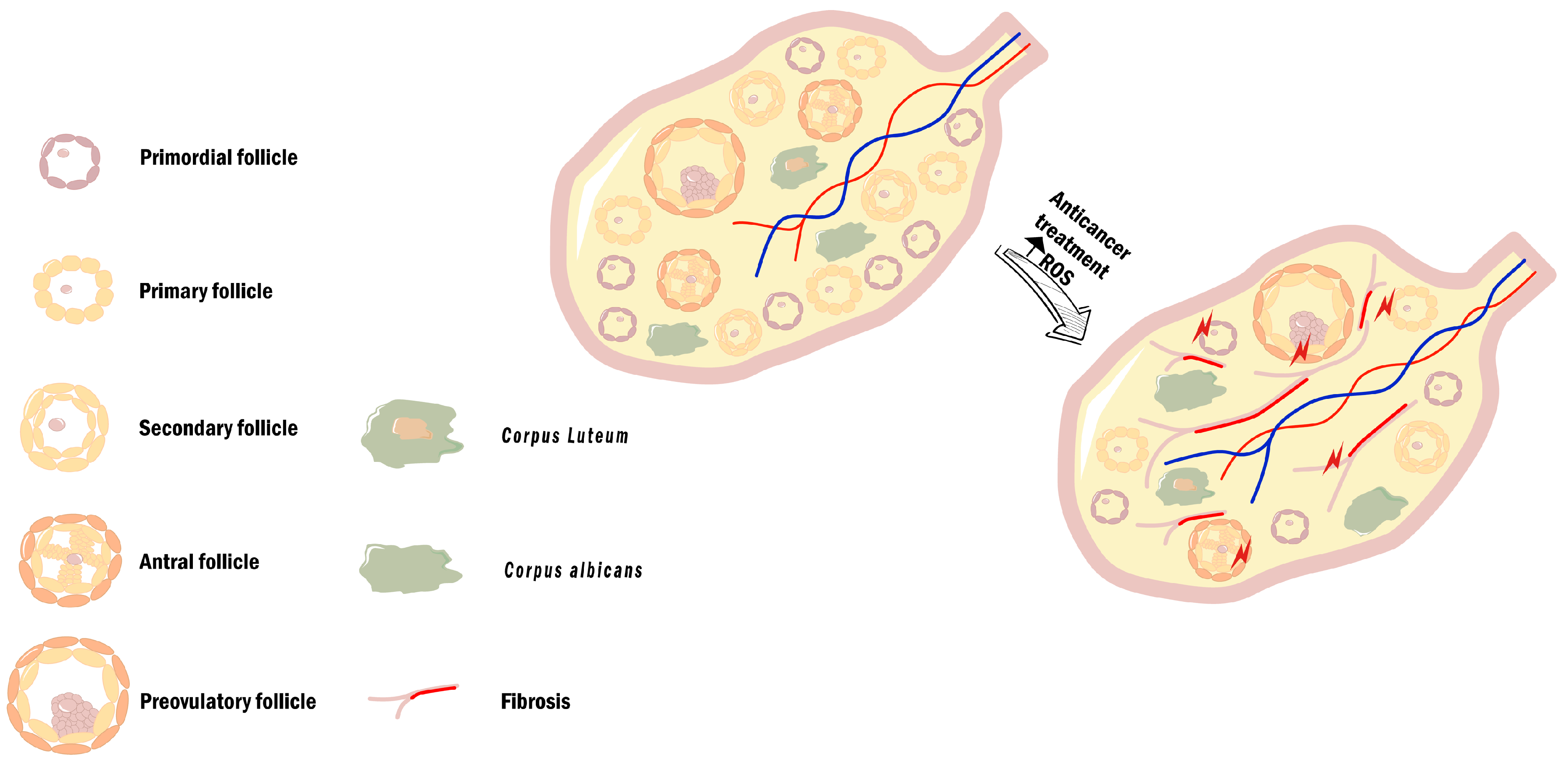
2.3. Reactive Oxygen Species, Oxidative Stress, and Female (In)Fertility
3. Current Evidence of OS-Mediated Effects on Fertility Derived from Cancer Therapies
3.1. Carcinogenesis, Anticancer Therapies and Oxidative Stress
3.2. Treatment-Induced OS and Its Impact on Fertility
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chabner, B.; Roberts, T. Timeline: Chemotherapy and the War on Cancer. Nat. Rev. Cancer 2005, 5, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Kim, H.S.; Seo, Y.R. Understanding of ROS-Inducing Strategy in Anticancer Therapy. Oxidative Med. Cell. Longev. 2019, 2019, 5381692. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, B.; Luo, X.; Rao, H.; Li, Q.; Shan, N.; Liu, X.; Qi, H. Oxidative stress-induced C/EBPbeta inhibits beta-catenin signaling molecule involving in the pathology of Preeclampsia. Placenta 2015, 36, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Panji, M.; Behmard, V.; Zare, Z.; Malekpour, M.; Nejadbiglari, H.; Yavari, S.; Nayerpour, T.; Safaeian, A.; Maleki, N.; Abbasi, M.; et al. Suppressing effects of green tea extract and Epigallocatechin-3-gallate (EGCG) on TGF-β- induced Epitheli-al-to-mesenchymal transition via ROS/Smad signaling in human cervical cancer cells. Gene 2021, 794, 145774. [Google Scholar] [CrossRef]
- Piktel, E.; Ościłowska, I.; Suprewicz, Ł.; Depciuch, J.; Marcińczyk, N.; Chabielska, E.; Wolak, P.; Wollny, T.; Janion, M.; Parlinska-Wojtan, M.; et al. ROS-Mediated Apoptosis and Autophagy in Ovarian Cancer Cells Treated with Peanut-Shaped Gold Nano-particles. Int. J. Nanomed. 2021, 16, 1993–2011. [Google Scholar] [CrossRef]
- Cheng, Q.; Bao, L.; Li, M.; Chang, K.; Yi, X. Erastin synergizes with cisplatin via ferroptosis to inhibit ovarian cancer growth in vitro and in vivo. J. Obstet. Gynaecol. Res. 2021, 47, 2481–2491. [Google Scholar] [CrossRef]
- Angsutararux, P.; Luanpitpong, S.; Issaragrisil, S. Chemotherapy-Induced Cardiotoxicity: Overview of the Roles of Oxidative Stress. Oxidative Med. Cell. Longev. 2015, 2015, 795602. [Google Scholar] [CrossRef] [Green Version]
- Karasawa, T.; Steyger, P.S. An integrated view of cisplatin-induced nephrotoxicity and ototoxicity. Toxicol. Lett. 2015, 237, 219–227. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Bhatti, M.T.; Salama, A. Neuro-ophthalmic side effects of molecularly targeted cancer drugs. Eye 2017, 32, 287–301. [Google Scholar] [CrossRef]
- Hansel, T.T.; Kropshofer, H.; Singer, T.; Mitchell, J.A.; George, A.J.T. The safety and side effects of monoclonal antibodies. Nat. Rev. Drug Discov. 2010, 9, 325–338. [Google Scholar] [CrossRef]
- Kroschinsky, F.; on behalf of the Intensive Care in Hematological and Oncological Patients (iCHOP) Collaborative Group; Stölzel, F.; von Bonin, S.; Beutel, G.; Kochanek, M.; Kiehl, M.; Schellongowski, P. New drugs, new toxicities: Severe side effects of modern targeted and immunotherapy of cancer and their management. Crit. Care 2017, 21, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raso, V. Antibodies in diagnosis and therapy. The magic bullet--nearing the century mark. Semin. Cancer Biol. 1990, 1, 227–242. [Google Scholar] [PubMed]
- Dimitrov, D.S.; Marks, J.D. Therapeutic antibodies: Current state and future trends—Is a paradigm change coming soon? Methods Mol. Biol. 2009, 525, 1–27. [Google Scholar] [PubMed]
- Teppo, H.-R.; Soini, Y.; Karihtala, P. Reactive Oxygen Species-Mediated Mechanisms of Action of Targeted Cancer Therapy. Oxidative Med. Cell. Longev. 2017, 2017, 1485283. [Google Scholar] [CrossRef] [PubMed]
- Meistrich, M.L. Effects of chemotherapy and radiotherapy on spermatogenesis in humans. Fertil. Steril. 2013, 100, 1180–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabanegh, E.S.; Ragheb, A.M. Male Fertility after Cancer. Urology 2009, 73, 225–231. [Google Scholar] [CrossRef]
- Meirow, D.; Nugent, D. The effects of radiotherapy and chemotherapy on female reproduction. Hum. Reprod. Update 2001, 7, 535–543. [Google Scholar] [CrossRef] [Green Version]
- Meirow, D.; Biederman, H.; Anderson, R.A.; Wallace, W.H.B. Toxicity of Chemotherapy and Radiation on Female Reproduction. Clin. Obstet. Gynecol. 2010, 53, 727–739. [Google Scholar] [CrossRef]
- Chasle, S.; How, C.C. The effect of cytotoxic chemotherapy on female fertility. Eur. J. Oncol. Nurs. 2003, 7, 91–98. [Google Scholar] [CrossRef]
- Nudell, D.M.; Monoski, M.M.; Lipshultz, L.I. Common medications and drugs: How they affect male fertility. Urol. Clin. 2002, 29, 965–973. [Google Scholar] [CrossRef]
- Green, D.M.; Kawashima, T.; Stovall, M.; Leisenring, W.; Sklar, C.A.; Mertens, A.C.; Donaldson, S.S.; Byrne, J.; Robison, L.L. Fertility of Female Survivors of Childhood Cancer: A Report From the Childhood Cancer Survivor Study. J. Clin. Oncol. 2009, 27, 2677–2685. [Google Scholar] [CrossRef] [PubMed]
- Fleischer, R.T.; Vollenhoven, B.J.; Weston, G.C. The Effects of Chemotherapy and Radiotherapy on Fertility in Premenopausal Women. Obstet. Gynecol. Surv. 2011, 66, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Trottmann, M.; Becker, A.J.; Stadler, T.; Straub, J.; Soljanik, I.; Schlenker, B.; Stief, C.G. Semen Quality in Men with Malignant Diseases before and after Therapy and the Role of Cryopreservation. Eur. Urol. 2007, 52, 355–367. [Google Scholar] [CrossRef]
- Choy, J.T.; Brannigan, R.E. The determination of reproductive safety in men during and after cancer treatment. Fertil. Steril. 2013, 100, 1187–1191. [Google Scholar] [CrossRef]
- Kim, S.-Y.; Cho, G.J.; Davis, J.S. Consequences of chemotherapeutic agents on primordial follicles and future clinical applica-tions. Obstet. Gynecol. Sci. 2019, 62, 382–390. [Google Scholar] [CrossRef]
- Stefansdottir, A.; Johnston, Z.C.; Powles-Glover, N.; Anderson, R.A.; Adams, I.R.; Spears, N. Etoposide damages female germ cells in the developing ovary. BMC Cancer 2016, 16, 482. [Google Scholar] [CrossRef] [Green Version]
- Van Der Kaaij, M.A.; van Echten-Arends, J.; Simmons, A.H.M.; Kluin-Nelemans, H.C. Fertility preservation after chemotherapy for Hodgkin lymphoma. Hematol. Oncol. 2010, 28, 168–179. [Google Scholar] [CrossRef]
- Sigman, M. Introduction: Cancer treatment and male fertility: Effects of therapy and current and future management options. Fertil. Steril. 2013, 100, 1179. [Google Scholar] [CrossRef]
- Forman, E.J.; Anders, C.K.; Behera, M.A. A nationwide survey of oncologists regarding treatment-related infertility and fertility preservation in female cancer patients. Fertil. Steril. 2010, 94, 1652–1656. [Google Scholar] [CrossRef]
- Meistrich, M.L. Male gonadal toxicity. Pediatr. Blood Cancer 2009, 53, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Wallace, W.H.B.; Anderson, R.A.; Irvine, D.S. Fertility preservation for young patients with cancer: Who is at risk and what can be offered? Lancet Oncol. 2005, 6, 209–218. [Google Scholar] [CrossRef]
- Grigg, A. The Impact of Conventional and High-Dose Therapy for Lymphoma on Fertility. Clin. Lymphoma 2004, 5, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Schrader, M.; Müller, M.; Straub, B.; Miller, K. The impact of chemotherapy on male fertility: A survey of the biologic basis and clinical aspects. Reprod. Toxicol. 2001, 15, 611–617. [Google Scholar] [CrossRef]
- Lambertini, M.; Del Mastro, L.; Pescio, M.C.; Andersen, C.Y.; Azim, H.A., Jr.; Peccatori, F.A.; Costa, M.; Revelli, A.; Salvagno, F.; Gennari, A.; et al. Cancer and fertility preservation: International recommendations from an expert meeting. BMC Med. 2016, 14, 1. [Google Scholar] [CrossRef]
- Rabaca, A.; Sousa, M.; Alves, M.G.; Oliveira, P.F.; Sá, R. Novel drug therapies for fertility preservation in men undergoing chemotherapy: Clinical relevance of pro-tector agents. Curr. Med. Chem. 2015, 3347–3369. [Google Scholar] [CrossRef]
- Blumenfeld, Z. Chemotherapy and fertility. Best Pract. Res. Clin. Obstet. Gynaecol. 2012, 26, 379–390. [Google Scholar] [CrossRef]
- Ahmed, M.R.; Edmund, S.S., Jr. Male fertility-implications of anticancer treatment and strategies to mitigate gonadotoxicity. Anti-Cancer Agents Med. Chem. 2010, 10, 92–102. [Google Scholar]
- Puscheck, E.; Philip, P.A.; Jeyendran, R.S. Male fertility preservation and cancer treatment. Cancer Treat. Rev. 2004, 30, 173–180. [Google Scholar] [CrossRef]
- Hobbie, W.L.; Ogle, S.K.; Ginsberg, J.P. Fertility Concerns for Young Males Undergoing Cancer Therapy. Semin. Oncol. Nurs. 2009, 25, 245–250. [Google Scholar] [CrossRef]
- Kohen, R.; Nyska, A. Invited Review: Oxidation of Biological Systems: Oxidative Stress Phenomena, Antioxidants, Redox Reactions, and Methods for Their Quantification. Toxicol. Pathol. 2002, 30, 620–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halliwell, B.; Cross, C.E. Oxygen-derived species: Their relation to human disease and environmental stress. Environ. Health Perspect. 1994, 102, 5–12. [Google Scholar]
- Kehrer, J.P. The Haber-Weiss reaction and mechanisms of toxicity. Toxicology 2000, 149, 43–50. [Google Scholar] [CrossRef]
- Castro, J.P.; Jung, T.; Grune, T.; Almeida, H. Actin carbonylation: From cell dysfunction to organism disorder. J. Proteom. 2013, 92, 171–180. [Google Scholar] [CrossRef]
- Orient, A.; Donkó, S.A.; Leto, T.L.; Geiszt, M. Novel sources of reactive oxygen species in the human body. Nephrol. Dial. Transplant. 2007, 22, 1281–1288. [Google Scholar] [CrossRef] [Green Version]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Mironczuk-Chodakowska, I.; Witkowska, A.; Zujko, M.E. Endogenous non-enzymatic antioxidants in the human body. Adv. Med. Sci. 2018, 63, 68–78. [Google Scholar] [CrossRef]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell. Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-mediated cellular signaling. Oxidative Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantley, L.C. The Phosphoinositide 3-Kinase Pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clermont, Y. The cycle of the seminiferous epithelium in man. Am. J. Anat. 1963, 112, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Siu, M.K.Y.; Cheng, C.Y. Extracellular Matrix and Its Role in Spermatogenesis. Mol. Mech. Spermatogenesis 2009, 636, 74–91. [Google Scholar]
- Phillips, B.T.; Gassei, K.; Orwig, K.E. Spermatogonial stem cell regulation and spermatogenesis. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 1663–1678. [Google Scholar] [CrossRef] [Green Version]
- Hess, R.A.; Franca, L.R.D. Spermatogenesis and cycle of the seminiferous epithelium. Adv. Exp. Med. Biol. 2008, 636, 1–15. [Google Scholar]
- Griswold, M.D. Spermatogenesis: The Commitment to Meiosis. Physiol. Rev. 2016, 96, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Hao, S.-L.; Ni, F.-D.; Yang, W.-X. The dynamics and regulation of chromatin remodeling during spermiogenesis. Gene 2019, 706, 201–210. [Google Scholar] [CrossRef]
- Yoshinaga, K.; Toshimori, K. Organization and modifications of sperm acrosomal molecules during spermatogenesis and epi-didymal maturation. Microsc. Res. Tech. 2003, 61, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Griswold, M.D. The central role of Sertoli cells in spermatogenesis. Semin. Cell Dev. Biol. 1998, 9, 411–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aitken, R.J.; Buckingham, D.W.; West, K.M. Reactive oxygen species and human spermatozoa: Analysis of the cellular mecha-nisms involved in luminol- and lucigenin-dependent chemiluminescence. J. Cell. Physiol. 1992, 151, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Gavella, M.; Lipovac, V. Nadh-Dependent Oxidoreductase (Diaphorase) Activity and Isozyme Pattern of Sperm in Infertile Men. Arch. Androl. 1992, 28, 135–141. [Google Scholar] [CrossRef] [Green Version]
- Gil-Guzman, E.; Ollero, M.; Lopez, M.C.; Sharma, R.K.; Alvarez, J.G.; Thomas, A.J., Jr.; Agarwal, A. Differential production of reactive oxygen species by subsets of human spermatozoa at different stages of maturation. Hum. Reprod. 2001, 16, 1922–1930. [Google Scholar] [CrossRef] [Green Version]
- Fisher, H.M.; Aitken, R.J. Comparative analysis of the ability of precursor germ cells and epididymal spermatozoa to generate reactive oxygen metabolites. J. Exp. Zool. 1997, 277, 390–400. [Google Scholar] [CrossRef]
- Aguilar-Mahecha, A.; Hales, B.F.; Robaire, B. Expression of stress response genes in germ cells during spermatogenesis. Biol. Reprod. 2001, 65, 119–127. [Google Scholar] [CrossRef] [Green Version]
- Aitken, R.J. The amoroso lecture. The human spermatozoon--a cell in crisis? J. Reprod. Fertil. 1999, 115, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Oldereid, N.B.; Thomassen, Y.; Purvis, K. Selenium in human male reproductive organs. Hum. Reprod. 1998, 13, 2172–2176. [Google Scholar] [CrossRef] [Green Version]
- Kemal, D.N.; Morshedi, M.; Oehninger, S. Effects of hydrogen peroxide on DNA and plasma membrane integrity of human spermatozoa. Fertil. Steril. 2000, 74, 1200–1207. [Google Scholar] [CrossRef]
- Aitken, R.J. Free radicals, lipid peroxidation and sperm function. Reprod. Fertil. Dev. 1995, 7, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Aitken, R.J.; Baker, M.A.; Sawyer, D. Oxidative stress in the male germ line and its role in the aetiology of male infertility and genetic disease. Reprod. Biomed. Online 2003, 7, 65–70. [Google Scholar] [CrossRef]
- Agarwal, A.; Nallella, K.P.; Allamaneni, S.S.; Said, T.M. Role of antioxidants in treatment of male infertility: An overview of the literature. Reprod. Biomed. Online 2004, 8, 616–627. [Google Scholar] [CrossRef]
- Cocuzza, M.; Sikka, S.C.; Athayde, K.S.; Agarwal, A. Clinical relevance of oxidative stress and sperm chromatin damage in male infertility: An evidence based analysis. Int. Braz. J. Urol. 2007, 33, 603–621. [Google Scholar] [CrossRef]
- Tai, P.; Ascoli, M. Reactive oxygen species (ROS) play a critical role in the cAMP-induced activation of Ras and the phos-phorylation of ERK1/2 in Leydig cells. Mol. Endocrinol. 2011, 25, 885–893. [Google Scholar] [CrossRef] [Green Version]
- Plante, M.; Lamirande, E.D.; Gagnon, C. Reactive oxygen species released by activated neutrophils, but not by deficient spermatozoa, are sufficient to affect normal sperm motility. Fertil. Steril. 1994, 62, 387–393. [Google Scholar] [CrossRef]
- Agarwal, A.; Saleh, R.; Bedaiwy, M. Role of reactive oxygen species in the pathophysiology of human reproduction. Fertil. Steril. 2003, 79, 829–843. [Google Scholar] [CrossRef] [Green Version]
- Safarnavadeh, T.; Rastegarpanah, M. Antioxidants and infertility treatment, the role of Satureja Khuzestanica: A mini-systematic review. Iran. J. Reprod. Med. 2011, 9, 61–70. [Google Scholar]
- Gao, S.; Li, C.; Chen, L.; Zhou, X. Actions and mechanisms of reactive oxygen species and antioxidative system in semen. Mol. Cell. Toxicol. 2017, 13, 143–154. [Google Scholar] [CrossRef]
- Barati, E.; Nikzad, H.; Karimian, M. Oxidative stress and male infertility: Current knowledge of pathophysiology and role of antioxidant therapy in disease management. Cell. Mol. Life Sci. 2019, 77, 93–113. [Google Scholar] [CrossRef]
- Bardaweel, S.K.; Gul, M.; Alzweiri, M.; Ishaqat, A.; ALSalamat, H.A.; Bashatwah, R.M. Reactive Oxygen Species: The Dual Role in Physiological and Pathological Conditions of the Human Body. Eurasian J. Med. 2018, 50, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Berby, B.; Bichara, C.; Rives-Feraille, A.; Jumeau, F.; Pizio, P.; Sétif, V.; Sibert, L.; Dumont, L.; Rondanino, C.; Rives, N. Oxidative Stress Is Associated with Telomere Interaction Impairment and Chromatin Condensation Defects in Spermatozoa of Infertile Males. Antioxidants 2021, 10, 593. [Google Scholar] [CrossRef] [PubMed]
- Santi, D.; Spaggiari, G.; Simoni, M. Sperm DNA fragmentation index as a promising predictive tool for male infertility diagnosis and treatment management-meta-analyses. Reprod. Biomed. Online 2018, 37, 315–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, A.; Parekh, N.; Selvam, M.K.P.; Henkel, R.; Shah, R.; Homa, S.T.; Ramasamy, R.; Ko, E.; Tremellen, K.; Esteves, S.; et al. Male oxidative stress infertility (MOSI): Proposed terminology and clinical practice guidelines for man-agement of idiopathic male infertility. World J. Mens Health 2019, 37, 296–312. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chian, R.-C. Follicular Development and Oocyte Growth. In Development of In Vitro Maturation for Human Oocytes: Natural and Mild Approaches to Clinical Infertility Treatment; Chian, R.-C., Nargund, G., Huang, J.Y.J., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 37–57. [Google Scholar]
- Von Stetina, J.R.; Orr-Weaver, T.L. Developmental Control of Oocyte Maturation and Egg Activation in Metazoan Models. Cold Spring Harb. Perspect. Biol. 2011, 3, a005553. [Google Scholar] [CrossRef]
- Baker, T.G. A quantitative and cytological study of germ cells in human ovaries. Proc. R. Soc. Lond. Ser. B Boil. Sci. 1963, 158, 417–433. [Google Scholar] [CrossRef]
- Gougeon, A.; Chainy, G. Morphometric studies of small follicles in ovaries of women at different ages. Reproduction 1987, 81, 433–442. [Google Scholar] [CrossRef]
- Sánchez, F.; Smitz, J. Molecular control of oogenesis. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2012, 1822, 1896–1912. [Google Scholar] [CrossRef] [Green Version]
- Rimon-Dahari, N.; Terushalmi-Heinemann, L.; Alyagor, L.; Dekel, N. Ovarian Folliculogenesis. Results Probl. Cell Differ. 2016, 58, 167–190. [Google Scholar]
- Baerwald, A.R.; Adams, G.P.; Pierson, R.A. Ovarian antral folliculogenesis during the human menstrual cycle: A review. Hum. Reprod. Update 2011, 18, 73–91. [Google Scholar] [CrossRef] [Green Version]
- Gellersen, B.; Brosens, J. Cyclic Decidualization of the Human Endometrium in Reproductive Health and Failure. Endocr. Rev. 2014, 35, 851–905. [Google Scholar] [CrossRef] [PubMed]
- Messinis, I.E. Ovarian feedback, mechanism of action and possible clinical implications. Hum. Reprod. Update 2006, 12, 557–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farage, M.A.; Neill, S.; MacLean, A.B. Physiological changes associated with the menstrual cycle: A review. Obstet. Gynecol. Surv. 2009, 64, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.W.; Bowling, M. Physiology of the female reproductive axis. Periodontology 2000 2012, 61, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J. Free Radicals and Antioxidant Protection: Mechanisms and Significance in Toxicology and Disease. Hum. Toxicol. 1988, 7, 7–13. [Google Scholar] [CrossRef]
- Sugino, N.; Nakamura, Y.; Takeda, O.; Ishimatsu, M.; Kato, H. Changes in activities of superoxide dismutase and lipid peroxide in corpus luteum during pregnancy in rats. Reproduction 1993, 97, 347–351. [Google Scholar] [CrossRef] [Green Version]
- Devine, P.J.; Perreault, S.D.; Luderer, U. Roles of Reactive Oxygen Species and Antioxidants in Ovarian Toxicity. Biol. Reprod. 2012, 86, 27. [Google Scholar] [CrossRef]
- Shkolnik, K.; Tadmor, A.; Ben-Dor, S.; Nevo, N.; Galiani, D.; Dekel, N. Reactive oxygen species are indispensable in ovulation. Proc. Natl. Acad. Sci. USA 2011, 108, 1462–1467. [Google Scholar] [CrossRef] [Green Version]
- Ruder, E.H.; Hartman, T.J.; Goldman, M.B. Impact of oxidative stress on female fertility. Curr. Opin. Obstet. Gynecol. 2009, 21, 219–222. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Sugino, N.; Fukaya, T.; Sugyama, S.; Uda, T.; Takaya, R.; Yajima, A.; Sasamo, H. Superoxide dismutase in normal cycling human ovaries: Immunohistochemical localization and characterization. Fertil. Steril. 1999, 72, 720–726. [Google Scholar] [CrossRef]
- Shi, L.; Zhang, J.; Lai, Z.; Tian, Y.; Fang, L.; Wu, M.; Xiong, J.; Qin, X.; Luo, A.; Wang, S. Long-Term Moderate Oxidative Stress Decreased Ovarian Reproductive Function by Reducing Follicle Quality and Progesterone Production. PLoS ONE 2016, 11, e0162194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, E.; Soares, A.I.; Costa, F.; Castro, J.P.; Matos, L.; Almeida, H. Antioxidant Supplementation Modulates Age-Related Placental Bed Morphology and Reproductive Outcome in Mice. Biol. Reprod. 2015, 93, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, F.T.; Mendes, S.; Rocha, N.A.; Matos, L.; Rodrigues, A.R.; Almeida, H.; Silva, E. Apocynin Dietary Supplementation Delays Mouse Ovarian Ageing. Oxidative Med. Cell. Longev. 2019, 2019, 5316984. [Google Scholar]
- Cornelli, U.; Belcaro, G.; Cesarone, M.R.; Finco, A. Analysis of oxidative stress during the menstrual cycle. Reprod. Biol. Endocrinol. 2013, 11, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strehlow, K.; Rotter, S.; Wassmann, S.; Adam, O.; Grohé, C.; Laufs, K.; Bohm, M.; Nickenig, G. Modulation of Antioxidant Enzyme Expression and Function by Estrogen. Circ. Res. 2003, 93, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Leng, J.Y.; Gao, F.; Zhao, Z.A.; Deng, W.B.; Liang, X.H.; Zhang, Y.J.; Zhang, Z.R.; Li, M.; Sha, A.G.; et al. Differential expression and anti-oxidant function of glutathione peroxidase 3 in mouse uterus during decidualization. FEBS Lett. 2014, 588, 1580–1589. [Google Scholar] [CrossRef] [Green Version]
- Sugino, N.; Takiguchi, S.; Kashida, S.; Karube, A.; Nakamura, Y.; Kato, H. Superoxide dismutase expression in the human corpus luteum during the menstrual cycle and in early pregnancy. Mol. Hum. Reprod. 2000, 6, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Al-Gubory, K.H.; Garrel, C.; Faure, P.; Sugino, N. Roles of antioxidant enzymes in corpus luteum rescue from reactive oxygen species-induced oxidative stress. Reprod. Biomed. Online 2012, 25, 551–560. [Google Scholar] [CrossRef] [Green Version]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Davis, J.M.; Auten, R.L. Maturation of the antioxidant system and the effects on preterm birth. Semin. Fetal Neonatal Med. 2010, 15, 191–195. [Google Scholar] [CrossRef]
- Mier-Cabrera, J.; Jimenez-Zamudio, L.A.; García-Latorre, E.; Cruz-Orozco, O.; Hernández-Guerrero, C. Quantitative and qualitative peritoneal immune profiles, T-cell apoptosis and oxidative stress-associated characteristics in women with minimal and mild endometriosis. BJOG Int. J. Obstet. Gynaecol. 2010, 118, 6–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendes, S.; Timóteo-Ferreira, F.; Soares, A.I.; Rodrigues, A.R.; Silva, A.M.N.; Silveira, S.; Matos, L.; Saraiva, J.; Guedes-Martins, L.; Almeida, H.; et al. Age-related oxidative modifications to uterine albumin impair extravillous trophoblast cells function. Free Radic. Biol. Med. 2020, 152, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Mendes, S.; Timóteo-Ferreira, F.; Almeida, H.; Silva, E. New Insights into the Process of Placentation and the Role of Oxidative Uterine Microenvironment. Oxidative Med. Cell. Longev. 2019, 2019, 9174521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, Q.; Beel, J.; Lillehei, K. A threshold concept for cancer therapy. Med. Hypotheses 2000, 55, 29–35. [Google Scholar] [CrossRef]
- Klaunig, J.E.; Kamendulis, L.M. The Role of Oxidative Stress in Carcinogenesis. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 239–267. [Google Scholar] [CrossRef]
- Storz, P. KRas, ROS and the initiation of pancreatic cancer. Small GTPases 2016, 8, 38–42. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic ap-proach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Raza, M.H.; Siraj, S.; Arshad, A.; Waheed, U.; Aldakheel, F.; Alduraywish, S.; Arshad, M. ROS-modulated therapeutic approaches in cancer treatment. J. Cancer Res. Clin. Oncol. 2017, 143, 1789–1809. [Google Scholar] [CrossRef]
- Wang, J.; Yi, J. Cancer cell killing via ROS: To increase or decrease, that is the question. Cancer Biol. Ther. 2008, 7, 1875–1884. [Google Scholar] [CrossRef]
- Hwang, P.M.; Bunz, F.; Yu, J.; Rago, C.; Chan, T.A.; Murphy, M.P.; Kelso, G.F.; Smith, R.A.; Kinzler, K.W.; Volgelstein, B. Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat. Med. 2001, 7, 1111–1117. [Google Scholar] [CrossRef] [Green Version]
- Fribley, A.; Wang, C.-Y. Proteasome inhibitor induces apoptosis through induction of endoplasmic reticulum stress. Cancer Biol. Ther. 2006, 5, 745–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin Induces a Mitochondrial-ROS Response That Contributes to Cytotoxicity Depending on Mitochondrial Redox Status and Bioenergetic Functions. PLoS ONE 2013, 8, e81162. [Google Scholar] [CrossRef]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharmacogenet. Genomics 2011, 21, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Shan, F.; Shao, Z.; Jiang, S.; Cheng, Z. Erlotinib induces the human non-small-cell lung cancer cells apoptosis via activating ROS-dependent JNK pathways. Cancer Med. 2016, 5, 3166–3175. [Google Scholar] [CrossRef]
- Chang, S.-P.; Shen, S.-C.; Lee, W.-R.; Yang, L.-L.; Chen, Y.-C. Imatinib mesylate induction of ROS-dependent apoptosis in melanoma B16F0 cells. J. Dermatol. Sci. 2011, 62, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Alas, S.; Ng, C.P.; Bonavida, B. Rituximab modifies the cisplatin-mitochondrial signaling pathway, resulting in apoptosis in cisplatin-resistant non-Hodgkin’s lymphoma. Clin. Cancer Res. 2002, 8, 836–845. [Google Scholar]
- Griffith, O.W.; Meister, A. Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (S-n-butyl homo-cysteine sulfoximine). J. Biol. Chem. 1979, 254, 7558–7560. [Google Scholar] [CrossRef]
- Sheveleva, E.V.; Landowski, T.H.; Samulitis, B.K.; Bartholomeusz, G.; Powis, G.; Dorr, R.T. Imexon Induces an Oxidative Endoplasmic Reticulum Stress Response in Pancreatic Cancer Cells. Mol. Cancer Res. 2012, 10, 392–400. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-J.; Oridate, N.; Lotan, R. Increased level of the p67phox subunit of NADPH oxidase by 4HPR in head and neck squamous carcinoma cells. Int. J. Oncol. 2005, 27, 787–790. [Google Scholar]
- Wondrak, G.T. NQO1-activated phenothiazinium redox cyclers for the targeted bioreductive induction of cancer cell apop-tosis. Free Radic. Biol. Med. 2007, 43, 178–190. [Google Scholar] [CrossRef] [Green Version]
- Pelicano, H.; Feng, L.; Zhou, Y.; Carew, J.S.; Hileman, E.O.; Plunkett, W.; Keating, M.J.; Huang, P. Inhibition of mitochondrial respiration: A novel strategy to enhance drug-induced apoptosis in human leukemia cells by a reactive oxygen species-mediated mechanism. J. Biol. Chem. 2003, 278, 37832–37839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotamraju, S.; Chitambar, C.R.; Kalivendi, S.V.; Joseph, J.; Kalyanaraman, B. Transferrin receptor-dependent iron uptake is responsible for doxorubicin-mediated apoptosis in endo-thelial cells: Role of oxidant-induced iron signaling in apoptosis. J. Biol. Chem. 2002, 277, 17179–17187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostrzewa-Nowak, D.; Paine, M.J.I.; Wolf, C.R.; Tarasiuk, J. The role of bioreductive activation of doxorubicin in cytotoxic activity against leukaemia HL60-sensitive cell line and its multidrug-resistant sublines. Br. J. Cancer 2005, 93, 89–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvorakova, K.; Payne, C.M.; Tome, M.E.; Briehl, M.M.; McClure, T.; Dorr, R.T. Induction of oxidative stress and apoptosis in myeloma cells by the aziridine-containing agent imexon. Biochem. Pharmacol. 2000, 60, 749–758. [Google Scholar] [CrossRef]
- Glasauer, A.; Chandel, N.S. Targeting antioxidants for cancer therapy. Biochem. Pharmacol. 2014, 92, 90–101. [Google Scholar] [CrossRef]
- Fang, J.; Sawa, T.; Akaike, T.; Greish, K.; Maeda, H. Enhancement of chemotherapeutic response of tumor cells by a heme oxygenase inhibitor, pegylated zinc protoporphyrin. Int. J. Cancer 2003, 109, 1–8. [Google Scholar] [CrossRef]
- Smith, P.S.; Zhao, W.; Spitz, D.R.; Robbins, M.E. Inhibiting catalase activity sensitizes 36B10 rat glioma cells to oxidative stress. Free Radic. Biol. Med. 2007, 42, 787–797. [Google Scholar] [CrossRef]
- Kilic, U.; Kilic, E.; Tuzcu, Z.; Ozercan, I.H.; Yilmaz, O.; Sahin, F.; Kazim, S. Melatonin suppresses cisplatin-induced nephrotoxicity via activation of Nrf-2/HO-1 pathway. Nutr. Metab. 2013, 10, 7. [Google Scholar] [CrossRef] [Green Version]
- Surh, Y.-J.; Kundu, J.K.; Na, H.-K. Nrf2 as a Master Redox Switch in Turning on the Cellular Signaling Involved in the Induction of Cytoprotective Genes by Some Chemopreventive Phytochemicals. Planta Med. 2008, 74, 1526–1539. [Google Scholar] [CrossRef] [Green Version]
- Fiedor, J.; Burda, K. Potential Role of Carotenoids as Antioxidants in Human Health and Disease. Nutrients 2014, 6, 466–488. [Google Scholar] [CrossRef] [Green Version]
- Sak, K. Cytotoxicity of dietary flavonoids on different human cancer types. Pharmacogn. Rev. 2014, 8, 122–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavsan, Z.; Kayali, H.A. Flavonoids showed anticancer effects on the ovarian cancer cells: Involvement of reactive oxygen species, apoptosis, cell cycle and invasion. Biomed. Pharmacother. 2019, 116, 109004. [Google Scholar] [CrossRef] [PubMed]
- Joshi, G.; Sultana, R.; Tangpong, J.; Cole, M.P.; Clair, D.K.S.; Vore, M.; Estus, S.; Butterfield, D.A. Free radical mediated oxidative stress and toxic side effects in brain induced by the anti-cancer drug adriamycin: Insight into chemobrain. Free Radic. Res. 2005, 39, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Berthiaume, J.M.; Wallace, K.B. Adriamycin-induced oxidative mitochondrial cardiotoxicity. Cell Biol. Toxicol. 2006, 23, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Qi, Y.; Xu, L.; Tao, X.; Han, X.; Yin, L.; Peng, J. MicroRNA-140-5p aggravates doxorubicin-induced cardiotoxicity by promoting myocardial oxidative stress via targeting Nrf2 and Sirt2. Redox Biol. 2017, 15, 284–296. [Google Scholar] [CrossRef]
- Gilliam, L.A.; St Clair, D.K. Chemotherapy-Induced Weakness and Fatigue in Skeletal Muscle: The Role of Oxidative Stress. Antioxid. Redox Signal. 2011, 15, 2543–2563. [Google Scholar] [CrossRef] [Green Version]
- Visacri, M.B.; Quintanilha, J.C.; Sousa, V.M.; Amaral, L.S.; Ambrósio, R.F.L.; Calonga, L.; Curi, S.F.B.B.; Leme, M.F.T.; Chone, C.T.; Altemani, J.M.C.; et al. Can acetylcysteine ameliorate cisplatin-induced toxicities and oxidative stress without decreasing antitumor efficacy? A randomized, double-blind, placebo-controlled trial involving patients with head and neck cancer. Cancer Med. 2019, 8, 2020–2030. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Weng, H.; Yang, J.; Wu, C. Protective effect of Liuwei Dihuang Pill on cisplatin-induced reproductive toxicity and genotoxicity in male mice. J. Ethnopharmacol. 2019, 247, 112269. [Google Scholar] [CrossRef]
- Kulhan, N.G.; Kulhan, M.; Turkler, C.; Ata, N.; Kiremitli, T.; Kiremitli, S.; Cimen, F.K.; Suleyman, H.; Toprak, V. Effect of lycopene on oxidative ovary-damage induced by cisplatin in rats. Gen. Physiol. Biophys. 2019, 38, 253–258. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.C.; Meistrich, M.L. Cytotoxic effects of chemotherapeutic drugs on mouse testis cells. Cancer Res. 1979, 39, 3575–3582. [Google Scholar]
- Howell, S.J.; Radford, J.A.; Ryder, W.; Shalet, S.M. Testicular Function After Cytotoxic Chemotherapy: Evidence of Leydig Cell Insufficiency. J. Clin. Oncol. 1999, 17, 1493–1498. [Google Scholar] [CrossRef] [PubMed]
- Gerl, A.; Mühlbayer, D.; Hansmann, G.; Mraz, W.; Hiddemann, W. The impact of chemotherapy on Leydig cell function in long term survivors of germ cell tumors. Cancer 2001, 91, 1297–1303. [Google Scholar] [CrossRef]
- Bedoschi, G.; Navarro, P.A.; Oktay, K. Chemotherapy-induced damage to ovary: Mechanisms and clinical impact. Future Oncol. 2016, 12, 2333–2344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, J.R.; Barton, D.L.; Loprinzi, C.L. Chemotherapy-induced ovarian failure: Manifestations and management. Drug Saf. 2005, 28, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xie, Y.; Yang, D.; Ren, D. Oxidative stress-induced apoptosis in granulosa cells involves JNK, p53 and Puma. Oncotarget 2017, 8, 25310–25322. [Google Scholar] [CrossRef] [Green Version]
- Arnon, J.; Meirow, D.; Lewis-Roness, H.; Ornoy, A. Genetic and teratogenic effects of cancer treatments on gametes and embryos. Hum. Reprod. Update 2001, 7, 394–403. [Google Scholar] [CrossRef]
- Hales, B.F.; Barton, T.S.; Robaire, B. Impact of Paternal Exposure to Chemotherapy on Offspring in the Rat. J. Natl. Cancer Inst. Monogr. 2005, 28–31. [Google Scholar] [CrossRef] [Green Version]
- Signorello, L.B.; Mulvihill, J.J.; Green, D.M.; Munro, H.M.; Stovall, M.; Weathers, R.E.; Mertens, A.C.; Whitton, J.A.; Robison, L.L.; Boice, J.D. Congenital Anomalies in the Children of Cancer Survivors: A Report From the Childhood Cancer Survivor Study. J. Clin. Oncol. 2012, 30, 239–245. [Google Scholar] [CrossRef]
- Yimit, A.; Adebali, O.; Sancar, A.; Jiang, Y. Differential damage and repair of DNA-adducts induced by anti-cancer drug cisplatin across mouse organs. Nat. Commun. 2019, 10, 309. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Schumaker, L.M.; Egorin, M.J.; Zuhowski, E.G.; Guo, Z.; Cullen, K.J. Cisplatin preferentially binds mitochondrial DNA and voltage-dependent anion channel protein in the mito-chondrial membrane of head and neck squamous cell carcinoma: Possible role in apoptosis. Clin. Cancer Res. 2006, 12, 5817–5825. [Google Scholar] [CrossRef] [Green Version]
- Türk, G.; Ateşşahin, A.; Sönmez, M.; Ceribasi, A.O.; Yüce, A. Improvement of cisplatin-induced injuries to sperm quality, the oxidant-antioxidant system, and the histologic structure of the rat testis by ellagic acid. Fertil. Steril. 2008, 89, 1474–1481. [Google Scholar] [CrossRef] [Green Version]
- Fouad, A.A.; Qutub, H.O.; Fouad, A.E.A.; Audeh, A.M.; Al-Melhim, W.N. Epigallocatechin-3-gallate counters cisplatin toxicity of rat testes. Pharm. Biol. 2017, 55, 1710–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.-E.; Lai, Y.-H.; Yang, K.-C.; Lin, S.-J.; Chen, C.-L.; Tsai, P.-S. Counteracting Cisplatin-Induced Testicular Damages by Natural Polyphenol Constituent Honokiol. Antioxidants 2020, 9, 723. [Google Scholar] [CrossRef] [PubMed]
- García, M.M.S.; Acquier, A.; Suarez, G.; Gomez, N.V.; Gorostizaga, A.; Mendez, C.F.; Paz, C. Cisplatin inhibits testosterone synthesis by a mechanism that includes the action of reactive oxygen species (ROS) at the level of P450scc. Chem. Interact. 2012, 199, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Afsar, T.; Razak, S.; Khan, M.R.; Almajwal, A. Acacia hydaspica ethyl acetate extract protects against cisplatin-induced DNA damage, oxidative stress and testicular injuries in adult male rats. BMC Cancer 2017, 17, 883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aksu, E.H.; Kandemir, F.M.; Özkaraca, M.; Ömür, A.D.; Küçükler, S.; Çomaklı, S. Rutin ameliorates cisplatin-induced reproductive damage via suppression of oxidative stress and apoptosis in adult male rats. Andrologia 2016, 49, e12593. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Iwase, A.; Murase, T.; Bayasula; Ishida, C.; Kato, N.; Nakamura, T.; Osuka, S.; Takikawa, S.; Goto, M.; et al. Protective effects of mangafodipir against chemotherapy-induced ovarian damage in mice. Reprod. Biol. Endocrinol. 2018, 16, 106. [Google Scholar] [CrossRef]
- Gouveia, B.B.; Barberino, R.D.S.; Silva, R.L.D.S.; Lins, T.L.B.G.; Guimarães, V.D.S.; Monte, A.P.O.D.; Palheta, R.C.; De Matos, M.H.T. Involvement of PTEN and FOXO3a Proteins in the Protective Activity of Protocatechuic Acid Against Cisplatin-Induced Ovarian Toxicity in Mice. Reprod. Sci. 2020, 28, 865–876. [Google Scholar] [CrossRef]
- Li, X.; Yang, S.; Lv, X.; Sun, H.; Weng, J.; Liang, Y.; Zhou, D. The mechanism of mesna in protection from cisplatin-induced ovarian damage in female rats. J. Gynecol. Oncol. 2013, 24, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Özcan, P.; Fıçıcıoğlu, C.; Yıldırım, Ö.K.; Özkan, F.; Akkaya, H.; Aslan, İ. Protective effect of resveratrol against oxidative damage to ovarian reserve in female Sprague-Dawley rats. Reprod. Biomed. Online 2015, 31, 410. [Google Scholar]
- Altuner, D.; Gulaboglu, M.; Yapca, O.E.; Cetin, N. The Effect of Mirtazapine on Cisplatin-Induced Oxidative Damage and Infertility in Rat Ovaries. Sci. World J. 2013, 2013, 627240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Maddawy, Z.K.; Abd El Naby, W.S.H. Protective effects of zinc oxide nanoparticles against doxorubicin induced testicular toxicity and DNA damage in male rats. Toxicol. Res. 2019, 8, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Aksu, E.H.; Kandemir, F.M.; Yıldırım, S.; Küçükler, S.; Dörtbudak, M.B.; Çağlayan, C.; Benzer, F. Palliative effect of curcumin on doxorubicin-induced testicular damage in male rats. J. Biochem. Mol. Toxicol. 2019, 33, e22384. [Google Scholar] [CrossRef] [PubMed]
- Kabel, A.M. Zinc/alogliptin combination attenuates testicular toxicity induced by doxorubicin in rats: Role of oxidative stress, apoptosis and TGF-β1/NF-κB signaling. Biomed. Pharm. 2018, 97, 439–449. [Google Scholar] [CrossRef]
- Cabral, R.E.L.; Mendes, T.B.; Vendramini, V.; Miraglia, S.M. Carnitine partially improves oxidative stress, acrosome integrity, and reproductive competence in doxo-rubicin-treated rats. Andrology 2018, 6, 236–246. [Google Scholar] [CrossRef] [Green Version]
- Badkoobeh, P.; Parivar, K.; Kalantar, S.M.; Hosseini, S.D.; Salabat, A. Effect of nano-zinc oxide on doxorubicin- induced oxidative stress and sperm disorders in adult male Wistar rats. Iran. J. Reprod. Med. 2013, 11, 355–364. [Google Scholar]
- Yang, C.-C.; Chen, Y.-T.; Chen, C.-H.; Chiang, J.Y.; Zhen, Y.-Y.; Yip, H.-K. Assessment of doxorubicin-induced mouse testicular damage by the novel second-harmonic generation microscopy. Am. J. Transl. Res. 2017, 9, 5275–5288. [Google Scholar]
- Erdem, G.E.; Tektemur, N.K.; Tektemur, A. Alpha-lipoic acid may ameliorate testicular damage by targeting dox-induced altered antioxidant parameters, mitofusin-2 and apoptotic gene expression. Andrology 2021, 53, e13990. [Google Scholar] [CrossRef]
- Abdelaziz, M.H.; El-Din, E.Y.S.; El-Dakdoky, M.; Ahmed, T.A. The impact of mesenchymal stem cells on doxorubicin-induced testicular toxicity and progeny outcome of male prepubertal rats. Birth Defects Res. 2019, 111, 906–919. [Google Scholar] [CrossRef]
- Niringiyumukiza, J.D.; Cai, H.; Chen, L.; Li, Y.; Wang, L.; Zhang, M.; Xu, X.; Xiang, W. Protective properties of glycogen synthase kinase-3 inhibition against doxorubicin-induced oxi-dative damage to mouse ovarian reserve. Biomed. Pharmacother. 2019, 116, 108963. [Google Scholar] [CrossRef]
- Han, J.; Wang, H.; Zhang, T.; Chen, Z.; Zhao, T.; Lin, L.; Xia, G.; Wang, C. Resveratrol attenuates doxorubicin-induced meiotic failure through inhibiting oxidative stress and apoptosis in mouse oocytes. Aging 2020, 12, 7717–7728. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.F.; Young, F. Gamma Tocopherol Reduced Chemotherapeutic-Induced ROS in an Ovarian Granulosa Cell Line, But Not in Breast Cancer Cell Lines In Vitro. Antioxidants 2020, 9, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbri, R.; Macciocca, M.; Vicenti, R.; Caprara, G.; Piccinni, M.; Paradisi, R.; Terzano, P.; Papi, A.; Seracchioli, R. Epigallocatechin-3-gallate inhibits doxorubicin-induced inflammation on human ovarian tissue. Biosci. Rep. 2019, 39, BSR20181424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, K.; Kietzmann, T. Reactive oxygen species and fibrosis: Further evidence of a significant liaison. Cell Tissue Res. 2016, 365, 591–605. [Google Scholar] [CrossRef] [Green Version]
- Holland, B.C.; Shetty, Z.; Alanee, S. The effect of targeted therapy for genitourinary malignancies on sexual function and fer-tility. Curr. Urol. Rep. 2017, 18, 65. [Google Scholar] [CrossRef]
- Lorenzi, E.; Simonelli, M.; Santoro, A. Infertility risk and teratogenicity of molecularly targeted anticancer therapy: A chal-lenging issue. Crit. Rev. Oncol. Hematol. 2016, 107, 1–13. [Google Scholar] [CrossRef]
- Lioni, M.; Noma, K.; Snyder, A.; Klein-Szanto, A.; Diehl, J.A.; Rustgi, A.K.; Herlyn, M.; Smalley, K.S.M. Bortezomib induces apoptosis in esophageal squamous cell carcinoma cells through activation of the p38 mi-togen-activated protein kinase pathway. Mol. Cancer Ther. 2008, 7, 2866–2875. [Google Scholar] [CrossRef] [Green Version]
- Hou, M.; Eriksson, E.; Svechnikov, K.; Jahnukainen, K.; Söder, O.; Meinhardt, A.; Sävendahl, L. Bortezomib treatment causes long-term testicular dysfunction in young male mice. Mol. Cancer 2014, 13, 155. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Fu, J.; Zhang, S.; Zhao, J.; Xie, N.; Cai, G. The proteasome inhibitor bortezomib induces testicular toxicity by upregulation of oxidative stress, AMP-activated protein kinase (AMPK) activation and deregulation of germ cell development in adult murine testis. Toxicol. Appl. Pharmacol. 2015, 285, 98–109. [Google Scholar] [CrossRef]
- D’Souza, U.J. Toxic effects of 5-Fluorouracil on sperm count in wistar rats. Malays. J. Med. Sci. 2003, 10, 43–45. [Google Scholar]
- Focaccetti, C.; Bruno, A.; Magnani, E.; Bartolini, D.; Principi, E.; Dallaglio, K.; Bucci, E.O.; Finzi, G.; Sessa, F.; Noonan, D.M.; et al. Effects of 5-Fluorouracil on Morphology, Cell Cycle, Proliferation, Apoptosis, Autophagy and ROS Production in Endothelial Cells and Cardiomyocytes. PLoS ONE 2015, 10, e0115686. [Google Scholar] [CrossRef] [PubMed]
- Fideles, L.S.; Miranda, J.A.L.; Martins, C.S.; Barbosa, M.L.L.; Pimenta, H.B.; Pimentel, P.V.S.; Teixeira, C.S.; Scafuri, M.A.S.; de Osterno Façanha, S.; Barreto, J.E.F.; et al. Role of Rutin in 5-Fluorouracil-Induced Intestinal Mucositis: Prevention of Histological Damage and Re-duction of Inflammation and Oxidative Stress. Molecules 2020, 25, 2786. [Google Scholar] [CrossRef] [PubMed]
- Bomfin, L.E.; Braga, C.M.; Oliveira, T.A.; Martins, C.S.; Foschetti, D.A.; Santos, A.A.; Costa, D.V.; Leitão, R.F.; Brito, G.A. 5-Fluorouracil induces inflammation and oxidative stress in the major salivary glands affecting salivary flow and saliva composition. Biochem. Pharmacol. 2017, 145, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Orcutt, K.P.; Parsons, A.D.; Sibenaller, Z.A.; Scarbrough, P.M.; Zhu, Y.; Sobhakumari, A.; Wilke, W.W.; Kalen, A.L.; Goswami, P.; Miller, F.J.; et al. Erlotinib-Mediated Inhibition of EGFR Signaling Induces Metabolic Oxidative Stress through NOX4. Cancer Res. 2011, 71, 3932–3940. [Google Scholar] [CrossRef] [Green Version]
- Chang, X.; Zhou, L.; Chen, X.; Xu, B.; Cheng, Y.; Sun, S.; Fang, M.; Xiang, Y. Impact of Imatinib on the Fertility of Male Patients with Chronic Myelogenous Leukaemia in the Chronic Phase. Target. Oncol. 2017, 12, 827–832. [Google Scholar] [CrossRef]
- Bouitbir, J.; Panajatovic, M.V.; Frechard, T.; Roos, N.J.; Krähenbühl, S. Imatinib and Dasatinib Provoke Mitochondrial Dysfunction Leading to Oxidative Stress in C2C12 Myotubes and Human RD Cells. Front. Pharmacol. 2020, 11, 1106. [Google Scholar] [CrossRef]
- Salem, W.; Li, K.; Krapp, C.; Ingles, S.A.; Bartolomei, M.S.; Chung, K.; Paulson, R.J.; Nowak, R.A.; McGinnis, L.K. Imatinib treatments have long-term impact on placentation and embryo survival. Sci. Rep. 2019, 9, 2535. [Google Scholar] [CrossRef] [Green Version]
- Das, G.; Damotte, V.; Gelfand, J.M.; Bevan, C.; Cree, B.A.C.; Do, L.; Green, A.J.; Hauser, S.L.; Bove, R. Rituximab before and during pregnancy: A systematic review, and a case series in MS and NMOSD. Neurol. Neuroimmunol. Neuroinflamm. 2018, 5, e453. [Google Scholar] [CrossRef] [Green Version]
- Pendergraft, W.F.; McGrath, M.M.; Murphy, A.P.; Murphy, P.; Laliberte, K.A.; Greene, M.F.; Niles, J.L. Fetal outcomes after rituximab exposure in women with autoimmune vasculitis. Ann. Rheum. Dis. 2013, 72, 2051–2053. [Google Scholar] [CrossRef] [Green Version]
- Al-Mogairen, S.M. Safety of rituximab on testicles, a double-blindedcontrolled trial in mice. J. Nat. Sci. Med. 2020, 3, 66. [Google Scholar] [CrossRef]
- Salvi, A.; Patki, G.; Khan, E.; Asghar, M.; Salim, S. Protective Effect of Tempol on Buthionine Sulfoximine-Induced Mitochondrial Impairment in Hippocampal Derived HT22 Cells. Oxidative Med. Cell. Longev. 2016, 2016, 5059043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tatsunami, R.; Oba, T.; Tampo, Y. Buthionine sulfoximine promotes methylglyoxal-induced apoptotic cell death and oxidative stress in en-dothelial cells. Biol. Pharm. Bull. 2010, 33, 556–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Mechanism of Action | Role in Redox System | Ref |
|---|---|---|---|
| Direct ROS generation | |||
| 5-fluorouracil | Thymidylate synthase inhibitor | p53-dependent ROS | [121] |
| Bortezomib | Proteasome inhibitor | ER stress-induced ROS | [122] |
| Cisplatin | nDNA adducts generation | mtDNA and ETC damage | [123] |
| Doxorubicin | nDNA intercalation; topoisomerase-II-mediated nDNA repair disruption | Generation of free radical through iron chelation | [124] |
| Erlotinib | EGFR tyrosine kinase inhibition | Loss of MM potential | [125] |
| Imatinib | Bcr-Abl tyrosine kinase inhibition | Loss of MM potential | [126] |
| Rituximab | Anti-CD20 | Bcl-2 inhibition | [127] |
| Antioxidant process inhibition | |||
| Buthionine sulfoximine | - | GSH synthesis inhibitor | [128] |
| Imexon | Ribonucleotide reductase inhibitor | GSH activity disruption via thiol binding | [129] |
| Name | Fertility Effect | Ref | ROS-Known Effect | Ref |
|---|---|---|---|---|
| 5-fluorouracil | Decreased sperm count (rat) | [191] | Inflammation, autophagy, apoptosis, and senescence induction | [192,193,194] |
| Erlotinib | - | Increase radical’s production through NOX4 | [195] | |
| Imatinib | Reduces sperm count and density (human) | [196] | Reduces MMP and complex I activity of ETC, leading to mitochondrial OS | [197] |
| Decrease vasculature of placenta (mouse) | [198] | |||
| Diminishes primordial follicles (mouse) | [198] | |||
| Rituximab | No mentionable effects (human and mouse) | [199,200,201] | - | |
| Buthionine sulfoximine | - | Mitochondrial impairment | [202,203] | |
| Imexon | - | GSH depletion and induction of ER stress | [129,135] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendes, S.; Sá, R.; Magalhães, M.; Marques, F.; Sousa, M.; Silva, E. The Role of ROS as a Double-Edged Sword in (In)Fertility: The Impact of Cancer Treatment. Cancers 2022, 14, 1585. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14061585
Mendes S, Sá R, Magalhães M, Marques F, Sousa M, Silva E. The Role of ROS as a Double-Edged Sword in (In)Fertility: The Impact of Cancer Treatment. Cancers. 2022; 14(6):1585. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14061585
Chicago/Turabian StyleMendes, Sara, Rosália Sá, Manuel Magalhães, Franklim Marques, Mário Sousa, and Elisabete Silva. 2022. "The Role of ROS as a Double-Edged Sword in (In)Fertility: The Impact of Cancer Treatment" Cancers 14, no. 6: 1585. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14061585