Syntheses, Characterization, and Application of Tridentate Phenoxyimino-Phenoxy Aluminum Complexes for the Coupling of Terminal Epoxide with CO2: From Binary System to Single Component Catalyst


Abstract
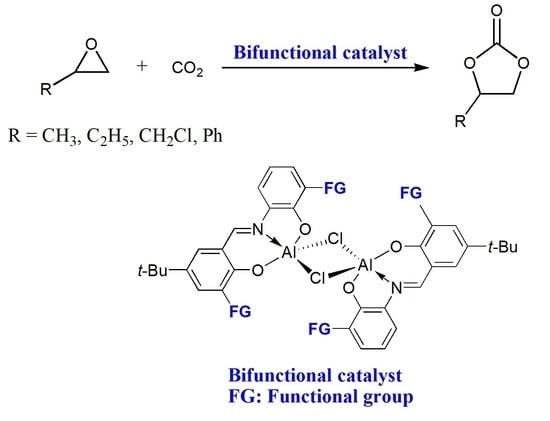
:
1. Introduction
2. Results and Discussion
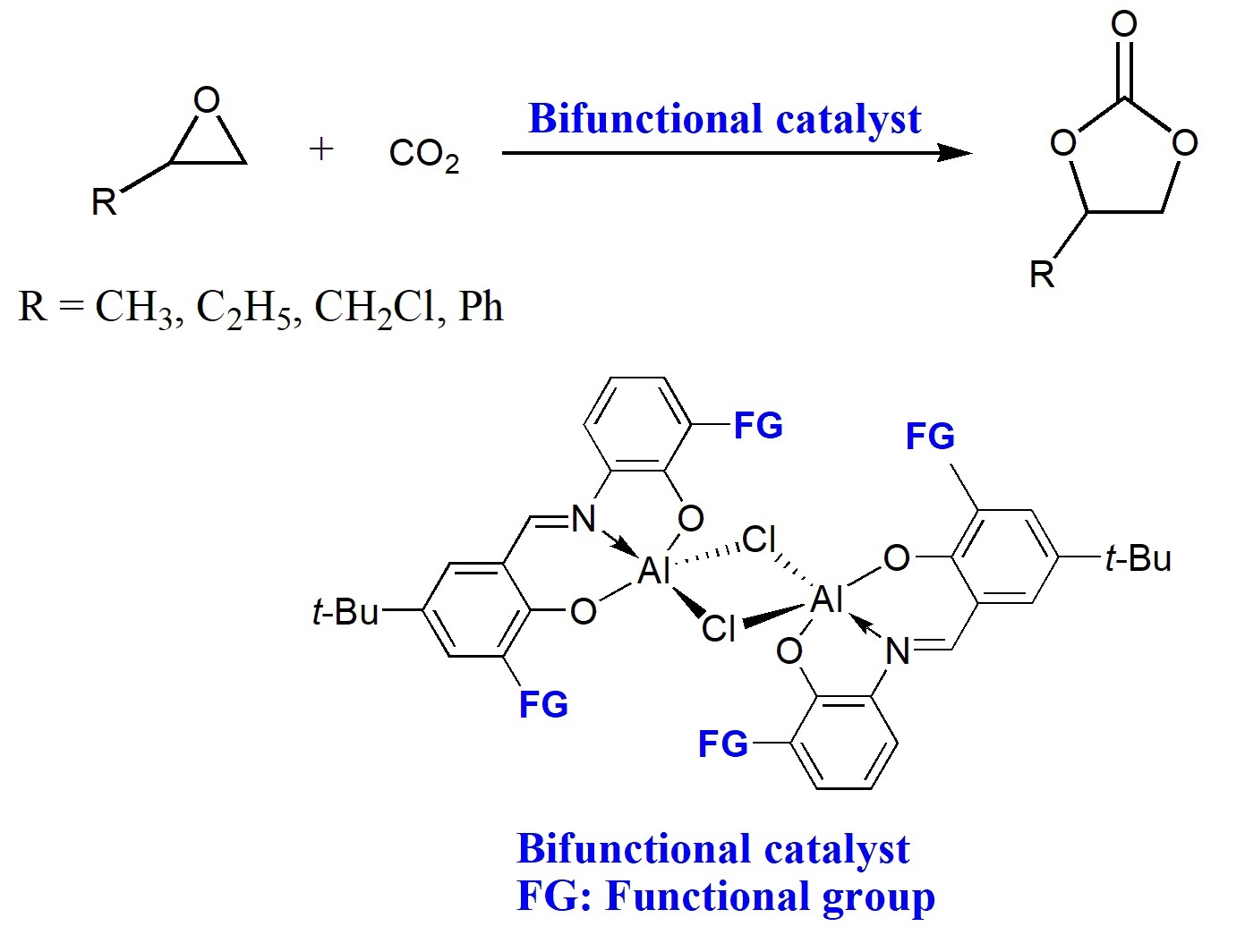
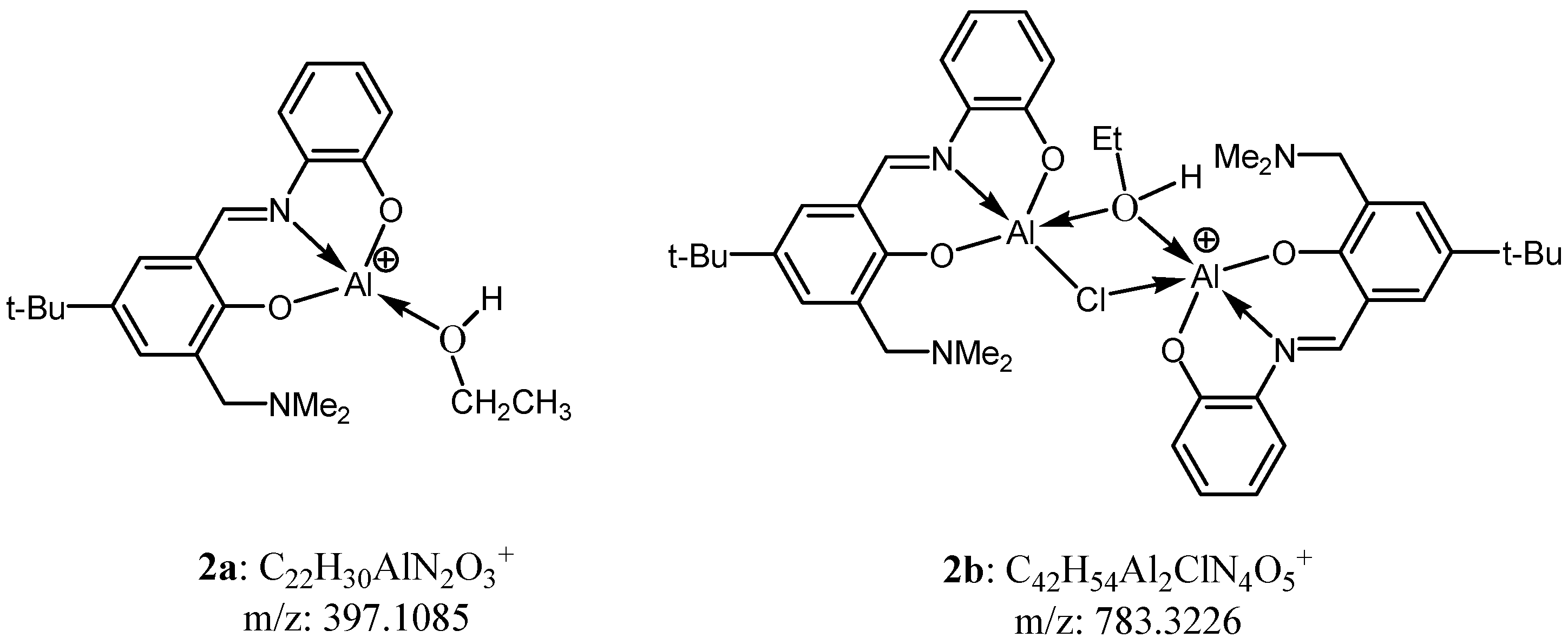
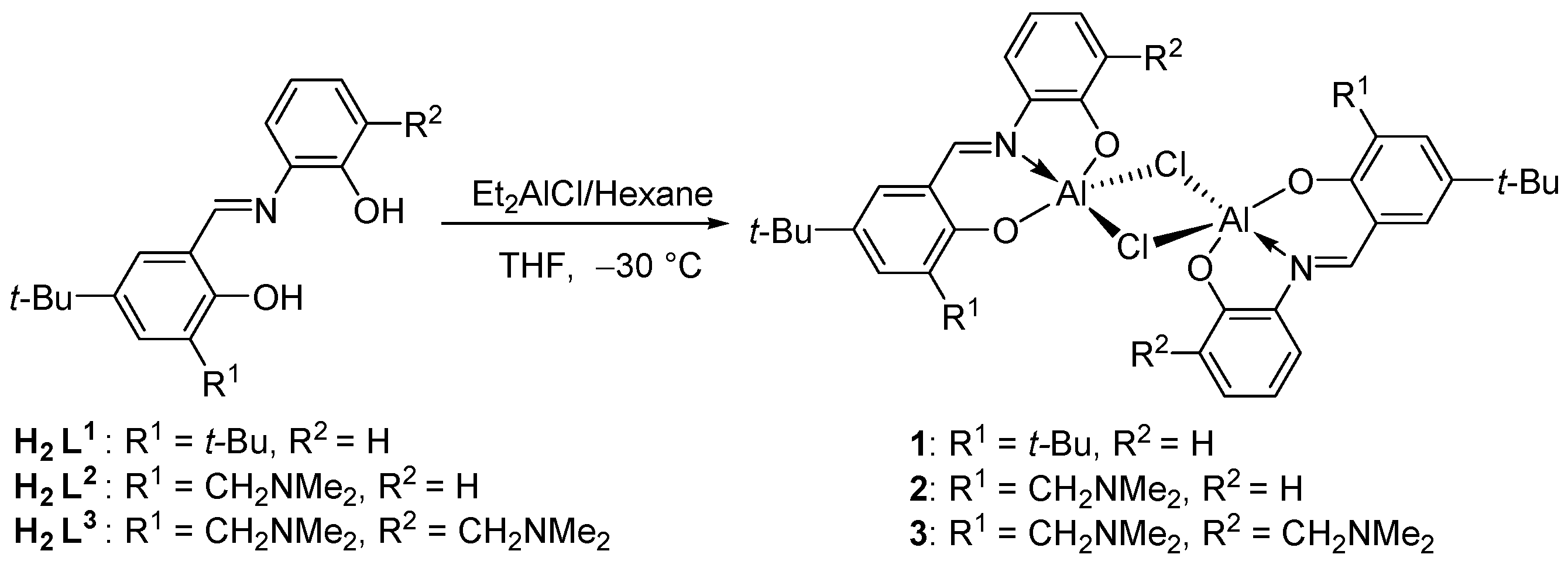
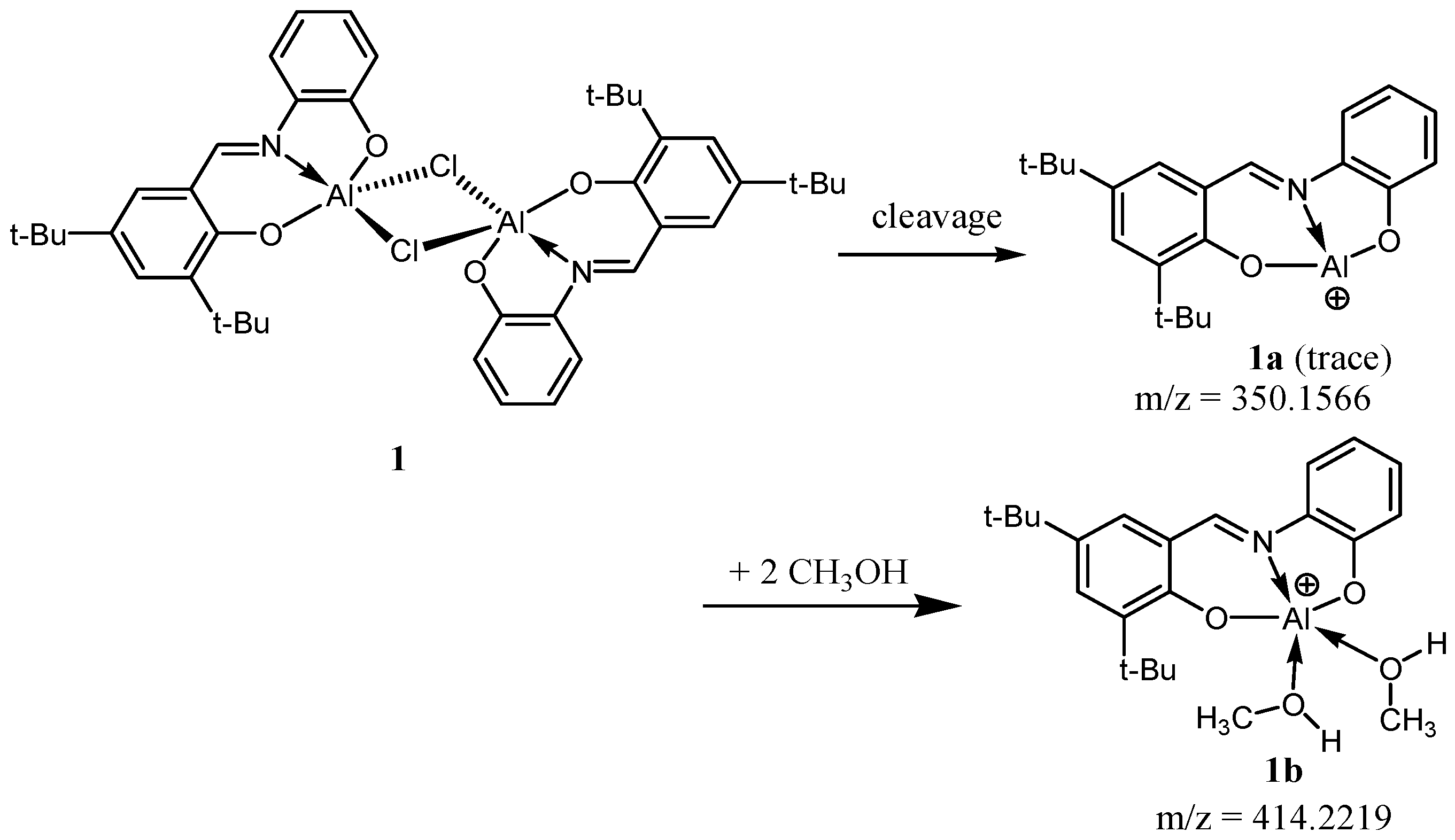
2.1. Syntheses and Characterization of Complexes 1–3
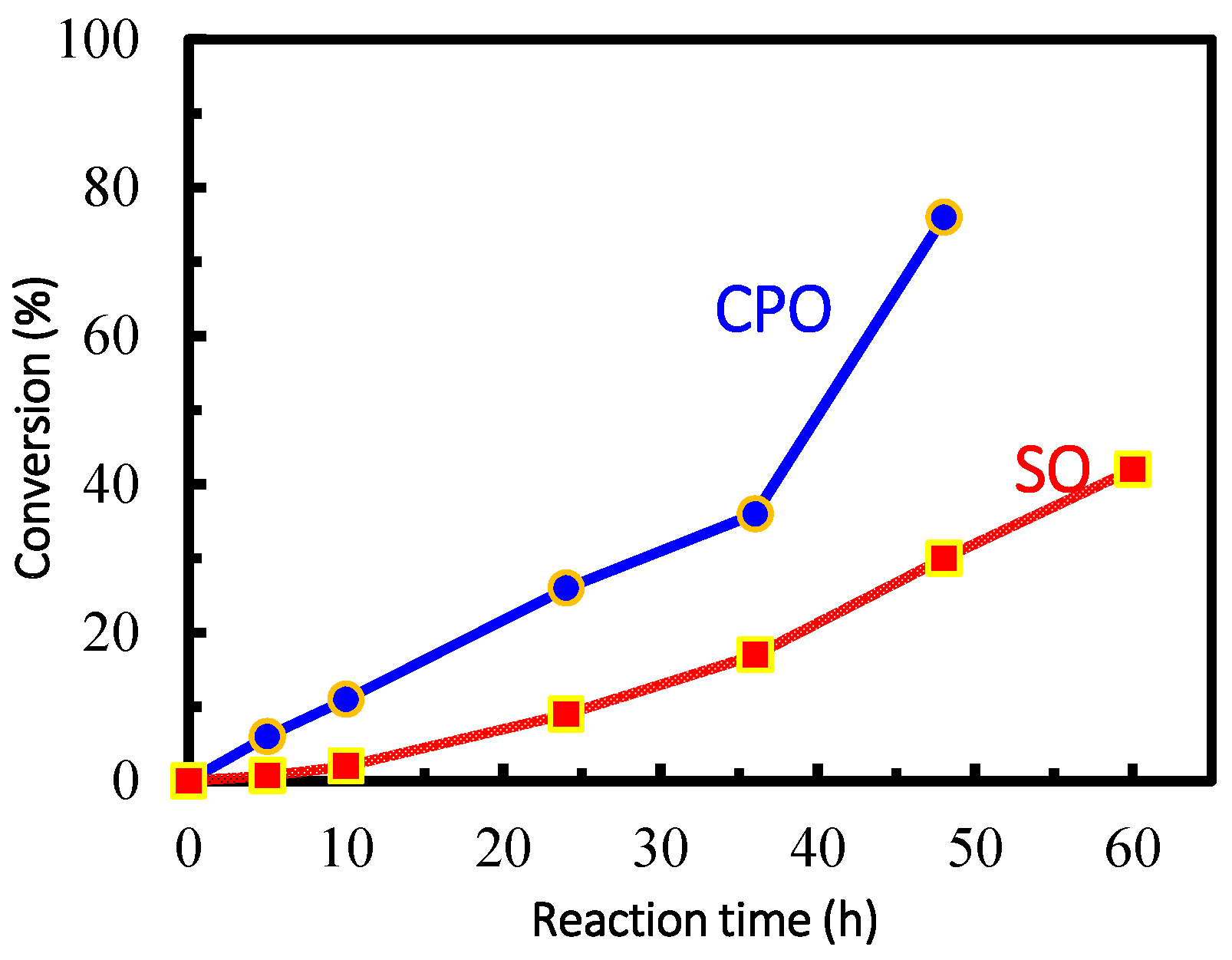
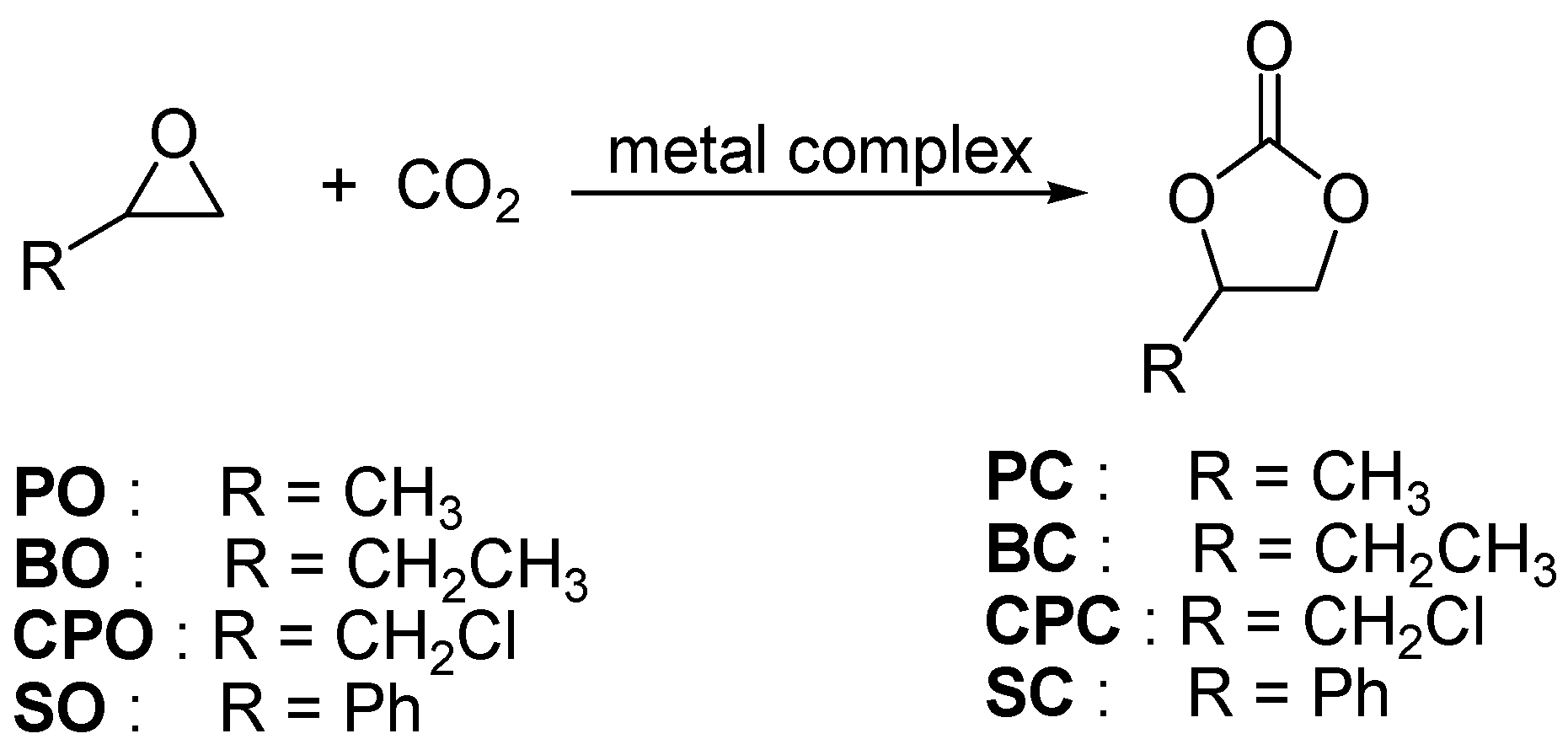
2.2. Catalytic Performance of Complexes 1–3 in the Coupling Reactions of Terminal Epoxide with CO2
3. Materials and Methods
3.1. Coupling Reaction of Epoxide with CO2 by Aluminum Complex 1
3.2. Coupling Reaction of Epoxide with CO2 by Bifunctional Aluminum Complexes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dibenedetto, A.; Angelini, A.; Stufano, P. Use of carbon dioxide as feedstock for chemicals and fuels: Homogeneous and heterogeneous catalysis. J. Chem. Technol. Biotechnol. 2014, 89, 334–353. [Google Scholar] [CrossRef]
- Yu, B.; Diao, Z.-F.; Guo, C.-X.; He, L.-N. Carboxylation of olefins/alkynes with CO2 to industrially relevant acrylic acid derivatives. J. CO2 Util. 2013, 1, 60–68. [Google Scholar] [CrossRef]
- Drees, M.; Cokoja, M.; Kühn, F.E. Recycling CO2? Computational Considerations of the Activation of CO2 with Homogeneous Transition Metal Catalysts. ChemCatChem 2012, 4, 1703–1712. [Google Scholar] [CrossRef]
- Cokoja, M.; Bruckmeier, C.; Rieger, B.; Herrmann, W.A.; Kuehn, F.E. Transformation of Carbon Dioxide with Homogeneous Transition-Metal Catalysts: A Molecular Solution to a Global Challenge? Angew. Chem. Int. Ed. 2011, 50, 8510–8537. [Google Scholar] [CrossRef] [PubMed]
- Omae, I. Aspects of carbon dioxide utilization. Catal. Today 2006, 115, 33–52. [Google Scholar] [CrossRef]
- Cokoja, M.; Wilhelm, M.E.; Anthofer, M.H.; Herrmann, W.A.; Kuehn, F.E. Synthesis of Cyclic Carbonates from Epoxides and Carbon Dioxide by Using Organocatalysts. ChemSusChem 2015, 8, 2436–2454. [Google Scholar] [CrossRef] [PubMed]
- Maeda, C.; Miyazaki, Y.; Ema, T. Recent progress in catalytic conversions of carbon dioxide. Catal. Sci. Technol. 2014, 4, 1482–1497. [Google Scholar] [CrossRef] [Green Version]
- Xu, K. Nonaqueous liquid electrolytes for lithium-based rechargeable batteries. Chem. Rev. 2004, 104, 4303–4417. [Google Scholar] [CrossRef]
- Schaeffner, B.; Schaeffner, F.; Verevkin, S.P.; Boerner, A. Organic Carbonates as Solvents in Synthesis and Catalysis. Chem. Rev. 2010, 110, 4554–4581. [Google Scholar] [CrossRef]
- Shaikh, A.A.G.; Sivaram, S. Organic carbonates. Chem. Rev. 1996, 96, 951–976. [Google Scholar] [CrossRef]
- Lu, X.B.; Feng, X.J.; He, R. Catalytic formation of ethylene carbonate from supercritical carbon dioxide/ethylene oxide mixture with tetradentate Schiff-base complexes as catalyst. Appl. Catal. Gen. 2002, 234, 25–33. [Google Scholar] [CrossRef]
- Fuchs, M.A.; Altesleben, C.; Zevaco, T.A.; Dinjus, E. An Efficient Homogeneous Chloro-Aluminum- N2O2 Catalyst for the Coupling of Epoxides with Carbon Dioxide. Eur. J. Inorg. Chem. 2013, 2013, 4541–4545. [Google Scholar] [CrossRef]
- Clegg, W.; Harrington, R.W.; North, M.; Pasquale, R. Cyclic Carbonate Synthesis Catalysed by Bimetallic Aluminium-Salen Complexes. Chem. Eur. J. 2010, 16, 6828–6843. [Google Scholar] [CrossRef] [PubMed]
- Kasuga, K.; Nagao, S.; Fukumoto, T.; Handa, M. Cycloaddition of carbon dioxide to propylene oxide catalysed by tetra-t-butylphthalocyaninatoaluminium(III) chloride. Polyhedron 1996, 15, 69–72. [Google Scholar] [CrossRef]
- Woo, W.H.; Hyun, K.; Kim, Y.; Ryu, J.Y.; Lee, J.; Kim, M.; Park, M.H.; Kim, Y. Highly Active Salen-Based Aluminum Catalyst for the Coupling of Carbon Dioxide with Epoxides at Ambient Temperature. Eur. J. Inorg. Chem. 2017, 5372–5378. [Google Scholar] [CrossRef]
- Wu, X.; North, M. A Bimetallic Aluminium(Salphen) Complex for the Synthesis of Cyclic Carbonates from Epoxides and Carbon Dioxide. ChemSusChem 2017, 10, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-Y.; Liu, D.-C.; Ko, B.-T. Synthesis, characterization and reactivity of single-site aluminium amides bearing benzotriazole phenoxide ligands: Catalysis for ring-opening polymerization of lactide and carbon dioxide/propylene oxide coupling. Dalton Trans. 2013, 42, 11488–11496. [Google Scholar] [CrossRef]
- Melendez, J.; North, M.; Pasquale, R. Synthesis of cyclic carbonates from atmospheric pressure carbon dioxide using exceptionally active aluminium(salen) complexes as catalysts. Eur. J. Inorg. Chem. 2007, 3323–3326. [Google Scholar] [CrossRef]
- Qin, Y.; Guo, H.; Sheng, X.; Wang, X.; Wang, F. An aluminum porphyrin complex with high activity and selectivity for cyclic carbonate synthesis. Green Chem. 2015, 17, 2853–2858. [Google Scholar] [CrossRef]
- Aida, T.; Inoue, S. Activation of carbon dioxide with aluminum porphyrin and reaction with epoxide. Studies on (tetraphenylporphinato)aluminum alkoxide having a long oxyalkylene chain as the alkoxide group. J. Am. Chem. Soc. 1983, 105, 1304–1309. [Google Scholar] [CrossRef]
- Lu, X.B.; Zhang, Y.J.; Jin, K.; Luo, L.M.; Wang, H. Highly active electrophile-nucleophile catalyst system for the cycloaddition of CO2 to epoxides at ambient temperature. J. Catal. 2004, 227, 537–541. [Google Scholar] [CrossRef]
- Lu, X.B.; Zhang, Y.J.; Liang, B.; Li, X.; Wang, H. Chemical fixation of carbon dioxide to cyclic carbonates under extremely mild conditions with highly active bifunctional catalysts. J. Mol. Catal. Chem. 2004, 210, 31–34. [Google Scholar] [CrossRef]
- Rintjema, J.; Kleij, A.W. Aluminum-Mediated Formation of Cyclic Carbonates: Benchmarking Catalytic Performance Metrics. ChemSusChem 2017, 10, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Zhao, Z.; Chen, L.; Yuan, D.; Yao, Y. Dinuclear Aluminum Poly(phenolate) Complexes as Efficient Catalysts for Cyclic Carbonate Synthesis. Organometallics 2016, 35, 1707–1712. [Google Scholar] [CrossRef]
- Cozzolino, M.; Press, K.; Mazzeo, M.; Lamberti, M. Carbon Dioxide/Epoxide Reactions Catalyzed by Bimetallic Salalen Aluminum Complexes. ChemCatChem 2016, 8, 455–460. [Google Scholar] [CrossRef]
- Castro-Osma, J.A.; Lara-Sanchez, A.; North, M.; Otero, A.; Villuendas, P. Synthesis of cyclic carbonates using monometallic, and helical bimetallic, aluminium complexes. Catal. Sci. Technol. 2012, 2, 1021–1026. [Google Scholar] [CrossRef]
- Rios Yepes, Y.; Quintero, C.; Osorio Melendez, D.; Daniliuc, C.G.; Martinez, J.; Rojas, R.S. Cyclic Carbonates from CO2 and Epoxides Catalyzed by Tetra- and Pentacoordinate Amidinate Aluminum Complexes. Organometallics 2019, 38, 469–478. [Google Scholar] [CrossRef]
- Whiteoak, C.J.; Kielland, N.; Laserna, V.; Castro-Gomez, F.; Martin, E.; Escudero-Adan, E.C.; Bo, C.; Kleij, A.W. Highly Active Aluminium Catalysts for the Formation of Organic Carbonates from CO2 and Oxiranes. Chem. Eur. J. 2014, 20, 2264–2275. [Google Scholar] [CrossRef]
- Castro-Osma, J.A.; North, M.; Wu, X. Development of a Halide-Free Aluminium-Based Catalyst for the Synthesis of Cyclic Carbonates from Epoxides and Carbon Dioxide. Chem. Eur. J. 2014, 20, 15005–15008. [Google Scholar] [CrossRef]
- Supasitmongkol, S.; Styring, P. A single centre aluminium(III) catalyst and TBAB as an ionic organo-catalyst for the homogeneous catalytic synthesis of styrene carbonate. Catal. Sci. Technol. 2014, 4, 1622–1630. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Han, S.Y.; Kim, J.H.; Kang, Y.Y.; Lee, J.; Kim, Y. Monomeric or Dimeric Aluminum Complexes as Catalysts for Cycloaddition between CO2 and Epoxides. Eur. J. Inorg. Chem. 2015, 2015, 2323–2329. [Google Scholar] [CrossRef]
- Whiteoak, C.J.; Kielland, N.; Laserna, V.; Escudero-Adan, E.C.; Martin, E.; Kleij, A.W. A Powerful Aluminum Catalyst for the Synthesis of Highly Functional Organic Carbonates. J. Am. Chem. Soc. 2013, 135, 1228–1231. [Google Scholar] [CrossRef] [PubMed]
- Ema, T.; Miyazaki, Y.; Koyama, S.; Yano, Y.; Sakai, T. A bifunctional catalyst for carbon dioxide fixation: Cooperative double activation of epoxides for the synthesis of cyclic carbonates. Chem. Commun. 2012, 48, 4489–4491. [Google Scholar] [CrossRef] [PubMed]
- Bai, D.; Duan, S.; Hai, L.; Jing, H. Carbon Dioxide Fixation by Cycloaddition with Epoxides, Catalyzed by Biomimetic Metalloporphyrins. ChemCatChem 2012, 4, 1752–1758. [Google Scholar] [CrossRef]
- Raghavendra, B.; Shashank, P.V.S.; Pandey, M.K.; Reddy, N.D. CO2/Epoxide Coupling and the ROP of epsilon-Caprolactone: Mg and Al Complexes of gamma-Phosphino-ketiminates as Dual-Purpose Catalysts. Organometallics 2018, 37, 1656–1664. [Google Scholar] [CrossRef]
- Ema, T.; Miyazaki, Y.; Shimonishi, J.; Maeda, C.; Hasegawa, J.-Y. Bifunctional Porphyrin Catalysts for the Synthesis of Cyclic Carbonates from Epoxides and CO2: Structural Optimization and Mechanistic Study. J. Am. Chem. Soc. 2014, 136, 15270–15279. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Fang, C.C.; Rodgers, J.L. Catalytic coupling of carbon dioxide and 2,3-epoxy-1,2,3,4-tetrahydronaphthalene in the presence of a (Salen)(CrCl)-Cl-III derivative. Organometallics 2004, 23, 924–927. [Google Scholar] [CrossRef]
- Castro-Osma, J.A.; North, M.; Wu, X. Synthesis of Cyclic Carbonates Catalysed by Chromium and Aluminium Salphen Complexes. Chem. Eur. J. 2016, 22, 2100–2107. [Google Scholar] [CrossRef]
- Ramidi, P.; Sullivan, S.Z.; Gartia, Y.; Munshi, P.; Griffin, W.O.; Darsey, J.A.; Biswas, A.; Shaikh, A.U.; Ghosh, A. Catalytic Cyclic Carbonate Synthesis Using Epoxide and Carbon Dioxide: Combined Catalytic Effect of Both Cation and Anion of an Ionic Cr-v(O) Amido Macrocyclic Complex. Ind. Eng. Chem. Res. 2011, 50, 7800–7807. [Google Scholar] [CrossRef]
- Cuesta-Aluja, L.; Djoufak, M.; Aghmiz, A.; Rivas, R.; Christ, L.; Masdeu-Bulto, A.M. Novel chromium (III) complexes with N-4-donor ligands as catalysts for the coupling of CO2 and epoxides in supercritical CO2. J. Mol. Catal. Chem. 2014, 381, 161–170. [Google Scholar] [CrossRef]
- Adolph, M.; Zevaco, T.A.; Altesleben, C.; Walter, O.; Dinjus, E. New cobalt, iron and chromium catalysts based on easy-to-handle N-4-chelating ligands for the coupling reaction of epoxides with CO2. Dalton Trans. 2014, 43, 3285–3296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iksi, S.; Aghmiz, A.; Rivas, R.; Dolores Gonzalez, M.; Cuesta-Aluja, L.; Castilla, J.; Orejon, A.; El Guemmout, F.; Masdeu-Bulto, A.M. Chromium complexes with tridentate NN′O Schiff base ligands as catalysts for the coupling of CO2 and epoxides. J. Mol. Catal. Chem. 2014, 383, 143–152. [Google Scholar] [CrossRef]
- Ambrose, K.; Robertson, K.N.; Kozak, C.M. Cobalt amino-bis(phenolate) complexes for coupling and copolymerization of epoxides with carbon dioxide. Dalton Trans. 2019, 48, 6248–6260. [Google Scholar] [CrossRef]
- Lu, X.B.; Liang, B.; Zhang, Y.J.; Tian, Y.Z.; Wang, Y.M.; Bai, C.X.; Wang, H.; Zhang, R. Asymmetric catalysis with CO2: Direct synthesis of optically active propylene carbonate from racemic epoxides. J. Am. Chem. Soc. 2004, 126, 3732–3733. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.; Jin, L.; Jing, H. Bifunctional Chiral Catalyst for the Synthesis of Chiral Cyclic Carbonates from Carbon Dioxide and Epoxides. ChemCatChem 2009, 1, 379–383. [Google Scholar] [CrossRef]
- Reiter, M.; Altenbuchner, P.T.; Kissling, S.; Herdtweck, E.; Rieger, B. Amine-bis(phenolato)cobalt(II) Catalysts for the Formation of Organic Carbonates from Carbon Dioxide and Epoxides. Eur. J. Inorg. Chem. 2015, 2015, 1766–1774. [Google Scholar] [CrossRef]
- Yu, C.-Y.; Chuang, H.-J.; Ko, B.-T. Bimetallic bis(benzotriazole iminophenolate) cobalt, nickel and zinc complexes as versatile catalysts for coupling of carbon dioxide with epoxides and copolymerization of phthalic anhydride with cyclohexene oxide. Catal. Sci. Technol. 2016, 6, 1779–1791. [Google Scholar] [CrossRef]
- Adolph, M.; Zevaco, T.A.; Altesleben, C.; Staudt, S.; Walter, O.; Dinjus, E. New ionic cobalt(III) complexes based on the N, N-bis(2-pyrazinecarboxamide)-1,2-benzene ligand: Application to the formation of organic carbonates from epoxides and CO2. New J. Chem. 2015, 39, 9858–9865. [Google Scholar] [CrossRef]
- Buonerba, A.; De Nisi, A.; Grassi, A.; Milione, S.; Capacchione, C.; Vagin, S.; Rieger, B. Novel iron(III) catalyst for the efficient and selective coupling of carbon dioxide and epoxides to form cyclic carbonates. Catal. Sci. Technol. 2015, 5, 118–123. [Google Scholar] [CrossRef]
- Buchard, A.; Kember, M.R.; Sandeman, K.G.; Williams, C.K. A bimetallic iron(III) catalyst for CO2/epoxide coupling. Chem. Commun. 2011, 47, 212–214. [Google Scholar] [CrossRef] [Green Version]
- Whiteoak, C.J.; Martin, E.; Martinez Belmonte, M.; Benet-Buchholz, J.; Kleij, A.W. An Efficient Iron Catalyst for the Synthesis of Five- and Six-Membered Organic Carbonates under Mild Conditions. Adv. Synth. Catal. 2012, 354, 469–476. [Google Scholar] [CrossRef]
- Della Monica, F.; Buonerba, A.; Capacchione, C. Homogeneous Iron Catalysts in the Reaction of Epoxides with Carbon Dioxide. Adv. Synth. Catal. 2019, 361, 265–282. [Google Scholar] [CrossRef]
- Della Monica, F.; Vummaleti, S.V.C.; Buonerba, A.; De Nisi, A.; Monari, M.; Milione, S.; Grassi, A.; Cavallo, L.; Capacchione, C. Coupling of Carbon Dioxide with Epoxides Efficiently Catalyzed by Thioether-Triphenolate Bimetallic Iron(III) Complexes: Catalyst Structure-Reactivity Relationship and Mechanistic DFT Study. Adv. Synth. Catal. 2016, 358, 3231–3243. [Google Scholar] [CrossRef]
- Zhao, Z.; Qin, J.; Zhang, C.; Wang, Y.; Yuan, D.; Yao, Y. Recyclable Single-Component Rare-Earth Metal Catalysts for Cycloaddition of CO2 and Epoxides at Atmospheric Pressure. Inorg. Chem. 2017, 56, 4568–4575. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, X.; Dai, Z.; Sun, Y.; Tang, N.; Wu, J. Yttrium complex supported by a sterically encumbering N-anchored tris-arylphenoxide ligand: Heteroselective ROP of rac-lactide and CO2/epoxide coupling. Inorg. Chem. Commun. 2015, 56, 69–72. [Google Scholar] [CrossRef]
- Luinstra, G.A.; Haas, G.R.; Molnar, F.; Bernhart, V.; Eberhardt, R.; Rieger, B. On the formation of aliphatic polycarbonates from epoxides with chromium(III) and aluminum(III) metal-salen complexes. Chem. Eur. J. 2005, 11, 6298–6314. [Google Scholar] [CrossRef]
- North, M.; Quek, S.C.Z.; Pridmore, N.E.; Whitwood, A.C.; Wu, X. Aluminum(salen) Complexes as Catalysts for of Terminal Epoxides via CO2 Coupling. ACS Catal. 2015, 5, 3398–3402. [Google Scholar] [CrossRef]
- North, M.; Young, C. Bimetallic aluminium(acen) complexes as catalysts for the synthesis of cyclic carbonates from carbon dioxide and epoxides. Catal. Sci. Technol. 2011, 1, 93–99. [Google Scholar] [CrossRef]
- Rulev, Y.A.; Gugkaeva, Z.; Maleev, V.I.; North, M.; Belokon, Y.N. Robust bifunctional aluminium-salen catalysts for the preparation of cyclic carbonates from carbon dioxide and epoxides. Beilstein. J. Org. Chem. 2015, 11, 1614–1623. [Google Scholar] [CrossRef] [Green Version]
- Martinez, J.; Castro-Osma, J.A.; Alonso-Moreno, C.; Rodriguez-Dieguez, A.; North, M.; Otero, A.; Lara-Sanchez, A. One-Component Aluminum(heteroscorpionate) Catalysts for the Formation of Cyclic Carbonates from Epoxides and Carbon Dioxide. ChemSusChem 2017, 10, 1175–1185. [Google Scholar] [CrossRef]
- de la Cruz-Martinez, F.; Martinez, J.; Gaona, M.A.; Fernandez-Baeza, J.; Sanchez-Barba, L.F.; Rodriguez, A.M.; Castro-Osma, J.A.; Otero, A.; Lara-Sanchez, A. Bifunctional Aluminum Catalysts for the Chemical Fixation of Carbon Dioxide into Cyclic Carbonates. ACS Sustain. Chem. Eng. 2018, 6, 5322–5332. [Google Scholar] [CrossRef]
- Melendez, J.; North, M.; Villuendas, P. One-component catalysts for cyclic carbonate synthesis. Chem. Commun. 2009, 2577–2579. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Zhou, X.; Chen, S.; Li, Y.; Zhou, L.; Ji, H. Highly efficient synthesis of cyclic carbonates from epoxides catalyzed by salen aluminum complexes with built-in “CO2 capture” capability under mild conditions. Green Chem. 2014, 16, 1496–1506. [Google Scholar] [CrossRef]
- Martinez, J.; de la Cruz-Martinez, F.; Gaona, M.A.; Pinilla-Penalver, E.; Fernandez-Baeza, J.; Rodriguez, A.M.; Castro-Osma, J.A.; Otero, A.; Lara-Sanchez, A. Influence of the Counterion on the Synthesis of Cyclic Carbonates Catalyzed by Bifunctional Aluminum Complexes. Inorg. Chem. 2019, 58, 3396–3408. [Google Scholar] [CrossRef]
- Ren, W.-M.; Liu, Y.; Lu, X.-B. Bifunctional Aluminum Catalyst for CO2 Fixation: Regioselective Ring Opening of Three-Membered Heterocyclic Compounds. J. Org. Chem. 2014, 79, 9771–9777. [Google Scholar] [CrossRef]
- Tian, D.; Liu, B.; Zhang, L.; Wang, X.; Zhang, W.; Han, L.; Park, D.-W. Coupling reaction of carbon dioxide and epoxides efficiently catalyzed by one-component aluminum-salen complex under solvent-free conditions. J. Ind. Eng. Chem. 2012, 18, 1332–1338. [Google Scholar] [CrossRef]
- Tian, D.; Liu, B.; Gan, Q.; Li, H.; Darensbourg, D.J. Formation of Cyclic Carbonates from Carbon Dioxide and Epoxides Coupling Reactions Efficiently Catalyzed by Robust, Recyclable One-Component Aluminum-Salen Complexes. ACS Catal. 2012, 2, 2029–2035. [Google Scholar] [CrossRef]
- Ren, Y.; Jiang, O.; Zeng, H.; Mao, Q.; Jiang, H. Lewis acid-base bifunctional aluminum-salen catalysts: Synthesis of cyclic carbonates from carbon dioxide and epoxides. RSC Adv. 2016, 6, 3243–3249. [Google Scholar] [CrossRef]
- Hong, M.; Kim, Y.; Kim, H.; Cho, H.J.; Baik, M.-H.; Kim, Y. Scorpionate Catalysts for Coupling CO2 and Epoxides to Cyclic Carbonates: A Rational Design Approach for Organocatalysts. J. Org. Chem. 2018, 83, 9370–9380. [Google Scholar] [CrossRef]
- Hanhart, W.; Ingold, C.K. CXXXIX.—The nature of the alternating effect in carbon chains. Part XVIII. Mechanism of exhaustive methylation and its relation to anomalous hydrolysis. J. Chem. Soc. 1927, 997–1020. [Google Scholar] [CrossRef]
- Hughes, E.D.; Ingold, C.K.; Patel, C.S. 135. Influence of poles and polar linkings on the course pursued by elimination reactions. Part XVI. Mechanism of the thermal decomposition of quaternary ammonium compounds. J. Chem. Soc. 1933, 526–530. [Google Scholar] [CrossRef]
- De la Zerda, J.; Neumann, R.; Sasson, Y. Hofmann decomposition of quaternary ammonium salts under phase-transfer catalytic conditions. J. Chem. Soc. Perkin Trans. 2 1986, 823–826. [Google Scholar] [CrossRef]
- Hofman, A.W., IX. Contributions towards the history of the monamines.—No. III. Compound ammonias by inverse substitution. Proc. R. Soc. Lond. 1859, 10, 594–596. [Google Scholar]
- Collie, N.; Schryver, S.B. LIII. The action of heat on the chlorides and hydroxides of mixed quaternary ammonium compounds. J. Chem. Soc. Trans. 1890, 57, 767–782. [Google Scholar] [CrossRef] [Green Version]
- Zaki, A.; Fahim, H. Some quaternary ammonium salts and their decomposition products. J. Chem. Soc. 1942, 270–272. [Google Scholar] [CrossRef]
- Gordon, J.E. Fused Organic Salts. III.1a Chemical Stability of Molten Tetra-n-alkylammonium Salts. Medium Effects on Thermal R4N+X- Decomposition. RBr + I- = RI + Br- Equilibrium Constant in Fused Salt Medium. J. Org. Chem. 1965, 30, 2760–2763. [Google Scholar] [CrossRef]
- Xie, Y.; Zhang, Z.; Jiang, T.; He, J.; Han, B.; Wu, T.; Ding, K. CO2 cycloaddition reactions catalyzed by an ionic liquid grafted onto a highly cross-linked polymer matrix. Angew. Chem. Int. Ed. 2007, 46, 7255–7258. [Google Scholar] [CrossRef]
- North, M.; Pasquale, R. Mechanism of Cyclic Carbonate Synthesis from Epoxides and CO2. Angew. Chem. Int. Ed. 2009, 48, 2946–2948. [Google Scholar] [CrossRef] [Green Version]
- Cho, W.; Shin, M.S.; Hwang, S.; Kim, H.; Kim, M.; Kim, J.G.; Kim, Y. Tertiary amines: A new class of highly efficient organocatalysts for CO2 fixations. J. Ind. Eng. Chem. 2016, 44, 210–215. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Epoxide | P(CO2) (atm) | Temperature (°C) | Time (h) | Conversion 2 (%) |
|---|---|---|---|---|---|
| 1 | PO | 10 | 80 | 10 | 7 |
| 2 | PO | 20 | 80 | 10 | 8 |
| 3 | PO | 10 | 100 | 10 | 68 |
| 4 | PO | 20 | 100 | 10 | 94 |
| 5 | BO | 10 | 80 | 10 | 7 |
| 6 | BO | 20 | 100 | 10 | 49 |
| 7 | CPO | 10 | 80 | 10 | 12 |
| 8 | CPO | 20 | 80 | 10 | 26 |
| 9 | CPO | 10 | 100 | 5 | 36 |
| 10 | CPO | 10 | 100 | 10 | 52 |
| 11 | CPO | 10 | 100 | 24 | 90 |
| 12 | CPO | 20 | 100 | 5 | 40 |
| 13 | CPO | 20 | 100 | 10 | 62 |
| 14 | SO | 10 | 100 | 5 | 5 |
| 15 | SO | 10 | 100 | 10 | 11 |
| 16 | SO | 10 | 100 | 18 | 32 |
| 17 | SO | 10 | 100 | 24 | 75 |
| 18 | SO | 20 | 100 | 5 | 7 |
| 19 | SO | 20 | 100 | 10 | 15 |
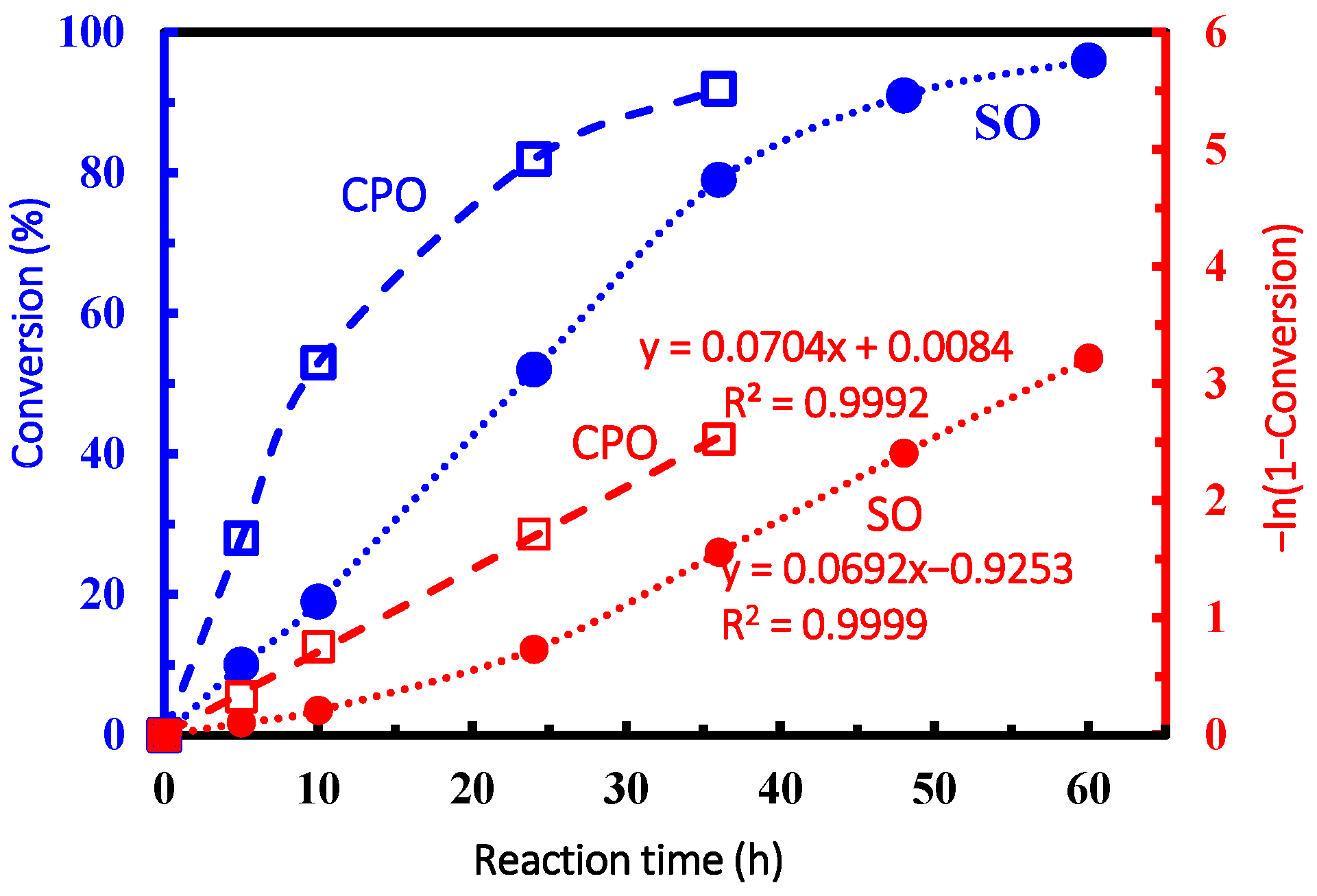
| Entry | Epoxide | Time (h) | Conversion (%) |
|---|---|---|---|
| 1 | CPO | 5 | 6 |
| 2 | CPO | 10 | 11 |
| 3 | CPO | 24 | 28 |
| 4 | CPO | 36 | 35 |
| 5 | CPO | 48 | 68 |
| 6 | SO | 5 | 0.7 |
| 7 | SO | 10 | 2 |
| 8 | SO | 24 | 9 |
| 9 | SO | 36 | 17 |
| 10 | SO | 48 | 30 |
| 11 | SO | 60 | 42 |
| Entry | Epoxide | P(CO2) (atm) | Temperature (°C) | Time (h) | Conversion (%) |
|---|---|---|---|---|---|
| 1 | PO | 20 | 80 | 10 | 14 |
| 2 | PO | 20 | 100 | 5 | 64 |
| 3 | PO | 20 | 100 | 10 | 99 |
| 4 | BO | 10 | 80 | 10 | 5 |
| 5 | BO | 10 | 100 | 10 | 76 |
| 6 | BO | 20 | 100 | 10 | 95 |
| 7 | CPO | 20 | 100 | 5 | 92 |
| 8 | CPO | 20 | 100 | 10 | 98 |
| 9 | SO | 20 | 100 | 10 | 50 |
| 10 | SO | 20 | 100 | 24 | 99 |
| Entry | Epoxide | P(CO2) (atm) | Time (h) | Conversion (%) |
|---|---|---|---|---|
| 1 | CPO | 2 | 5 | 85 |
| 2 | CPO | 2 | 10 | 98 |
| 3 | CPO | 1 | 5 | 28 |
| 4 | CPO | 1 | 10 | 55 |
| 5 | CPO | 1 | 24 | 83 |
| 6 | CPO | 1 | 36 | 91 |
| 7 | SO | 2 | 10 | 26 |
| 8 | SO | 1 | 5 | 10 |
| 9 | SO | 1 | 10 | 19 |
| 10 | SO | 1 | 24 | 50 |
| 11 | SO | 1 | 36 | 82 |
| 12 | SO | 1 | 48 | 92 |
| 13 | SO | 1 | 60 | 96 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Wang, T.; Xiang, P.; Du, Q.; Han, S. Syntheses, Characterization, and Application of Tridentate Phenoxyimino-Phenoxy Aluminum Complexes for the Coupling of Terminal Epoxide with CO2: From Binary System to Single Component Catalyst. Catalysts 2021, 11, 145. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11020145
Zhang Z, Wang T, Xiang P, Du Q, Han S. Syntheses, Characterization, and Application of Tridentate Phenoxyimino-Phenoxy Aluminum Complexes for the Coupling of Terminal Epoxide with CO2: From Binary System to Single Component Catalyst. Catalysts. 2021; 11(2):145. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11020145
Chicago/Turabian StyleZhang, Zhichao, Tianming Wang, Peng Xiang, Qinqin Du, and Shuang Han. 2021. "Syntheses, Characterization, and Application of Tridentate Phenoxyimino-Phenoxy Aluminum Complexes for the Coupling of Terminal Epoxide with CO2: From Binary System to Single Component Catalyst" Catalysts 11, no. 2: 145. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11020145