
Single-Crystal Structure of HP-Sc2TeO6 Prepared by High-Pressure/High-Temperature Synthesis


1
Department for General, Inorganic and Theoretical Chemistry, Leopold-Franzens-University Innsbruck, Innrain 80–82, A-6020 Innsbruck, Austria
2
Institute of Mineralogy and Petrography, Leopold-Franzens-University Innsbruck, Innrain 52, A-6020 Innsbruck, Austria
*
Author to whom correspondence should be addressed.
Crystals 2021, 11(12), 1554; https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11121554
Submission received: 1 December 2021
/
Revised: 10 December 2021
/
Accepted: 11 December 2021
/
Published: 13 December 2021
(This article belongs to the Special Issue Solid State Chemistry: Memorial Issue for Professor Emilio Morán)
Abstract
:The first high-pressure scandium tellurate HP-Sc2TeO6 was synthesized from an NP-Sc2TeO6 normal-pressure precursor at 12 GPa and 1173 K using a multianvil apparatus (1000 t press, Walker-type module). The compound crystallizes in the monoclinic space group P2/c (no. 13) with a = 729.43(3), b = 512.52(2), c = 1095.02(4) pm and β = 103.88(1)°. The structure was refined from X-ray single-crystal diffractometer data: R1 = 0.0261, wR2 = 0.0344, 568 F2 values and 84 variables. HP-Sc2TeO6 is isostructural to Yb2WO6 and is built up from TeO6 octahedra, typical for tellurate(VI) compounds. During synthesis, a reconstructive transition from P321 (normal-pressure modification) to P2/c (high-pressure modification) takes place and the scandium–oxygen distances as well as the coordination number of scandium increase. However, the coordination sphere around the Te6+ cations gets only slightly distorted. High-temperature powder XRD investigations revealed a back-transformation of HP-Sc2TeO6 to the ambient-pressure modification above 973 K.
1. Introduction
Transition metal tellurates show a broad variety of technologically important properties. M3TeO6 (M = Ni, Co, Mn, Cu) compounds exhibit antiferromagnetic ordering at low temperatures combined with magnetoelectric properties. These materials are classified as type-II multiferroics and have recently gained great importance [1,2,3,4,5,6,7,8]. The structural diversity of oxotellurate compounds is related on Te4+ and Te6+ as well as mixed valent Te4+/Te6+ combinations. Oxotellurate(IV) anions are characterized by the presence of the 5s2 lone-electron pair, which causes various oxygen coordination spheres with coordination numbers from three to five and a wide range of possible Te–O bond-lengths. Moreover, stereoactive lone-pair electrons and their directing effects on the crystal structure are responsible for the excellent SHG efficiencies of tellurate crystals [9,10].
Scandium forms both oxotellurates(IV) and oxotellurates(VI), and a total of only three compounds, have been fully described so far. Sc2Te5O13 and Sc2Te3O9 are oxotellurate(IV) representatives. The crystal structure of Sc2Te5O13 [11] is triclinic with space group P (no. 2) and isopuntal to Lu2Te5O13 [12]. In contrast to Sc3+, all other compounds with the formula type M2Te5O13 (M = Y, Dy–Lu) exhibit significantly larger coordination spheres at the M3+ sites [11,12,13]. The second compound, Sc2Te3O9, crystallizes monoclinic (P21/n, no. 14) and is built up from ScO6 octahedra, ScO7-capped octahedra and TeO3 trigonal pyramids [14]. Additional representatives of the composition M2Te3O9 (M = Ga, In, Dy and Lu) form different crystal structure types [15,16,17]. Two additional Sc-Te-O phases published without structural details can be found in the PDF4+ 2021 [18] database, i.e., Sc6TeO12 (PDF# 04-002-9406, [19]) and Sc2Te4O11 (PDF# 00-047-0010).
In oxotellurate(VI) chemistry, the general formula M2TeO6 is common. Three crystal structure types are mainly prevalent: the Na2SiF6-type structure, which crystallizes trigonal in the space group P321 (no. 150) and the two orthorhombic structure types Ta2FeO6 (P42/mnm, no. 136) and Nd2WO6 (P212121, no. 19).
Recently, we focused our work on tellurate compounds at extreme conditions and were able to add now a fourth structure type to the M2TeO6-system. To distinguish the new compound HP-Sc2TeO6 synthesized under high-pressure (HP), the phase synthesized under normal-pressure (NP) will be referred to as NP-Sc2TeO6 [11] in the following. Here we present the synthesis, crystal structure and high-temperature behaviour of HP-Sc2TeO6.
2. Materials and Methods
HP-Sc2TeO6 was synthesized via high-pressure/high-temperature multianvil experiments according to Equation (1). NP-Sc2TeO6 served as precursor and was prepared through a high-temperature solid state route from Sc2O3 (powder, purity 99.99%, ChemPUR, Karlsruhe, Germany) and TeO3 in a 1:1 ratio under air.
To obtain NP-Sc2TeO6, a homogeneous mixture of the reagents was filled in a corundum boat and heated to 1073 K within 6 h. The temperature was kept constant for 36 h. TeO3 was synthesized by dehydration of Te(OH)6 (powder, purity > 99.0%, TCI, Tokyo, Japan) in a corundum boat at 623 K for 36 h in air.
For the high-pressure/high-temperature transformation to HP-Sc2TeO6, the precursor NP-Sc2TeO6 was homogenised and filled in a platinum capsule. The capsule was placed in a crucible made of hexagonal boron nitride and covered by a platelet of the same material (Henze Boron Nitride Products AG, Kempten, Germany). Together with two graphite sleeves acting as resistive heaters, a ZrO2 sleeve for temperature isolation and two molybdenum platelets to ensure conductivity, the crucible was centred in an octahedron of MgO doped with 5% Cr2O3 as the pressure medium. The compression of the specimen was accomplished by a two-stage mechanism of eight inner anvils (first stage) consisting of tungsten carbide cubes with truncated corners (Hawedia, Marklkofen, Germany) and centred octahedral pressure medium. The second stage, the Walker-type module of the 1000 t uniaxial press (both devices from Max Voggenreiter GmbH, Mainleus, Germany), houses the inner anvils with the sample. Detailed information about the multianvil technique and the construction of the various assemblies can be found in numerous references [20,21,22,23].
The pressure was ramped up to 12 GPa in 320 min followed by heating the sample to 1173 K within 10 min. This temperature was kept for the next 10 min and then reduced to 673 K within 20 min. A temperature quench to room temperature completed the heating period and subsequently the pressure was reduced to ambient conditions in 1000 min. Afterwards, the sample was recovered by removing the surrounding assembly materials trough mechanical fragmentation. The sample appeared colourless, polycrystalline and was stable at air.
X-ray powder diffraction experiments were conducted on a STOE STADI P diffractometer (STOE & Cie GmbH, Darmstadt, Germany) with (111) curved Ge-monochromatized Mo-Kα1 radiation (λ = 70.93 pm) in transmission geometry. The diffraction intensities were collected by a DECTRIS MYTHEN 1K microstrip detector with 1280 strips. Polycrystalline samples of HP-Sc2TeO6 were ground in an agate mortar and placed between two thin acetate foils.
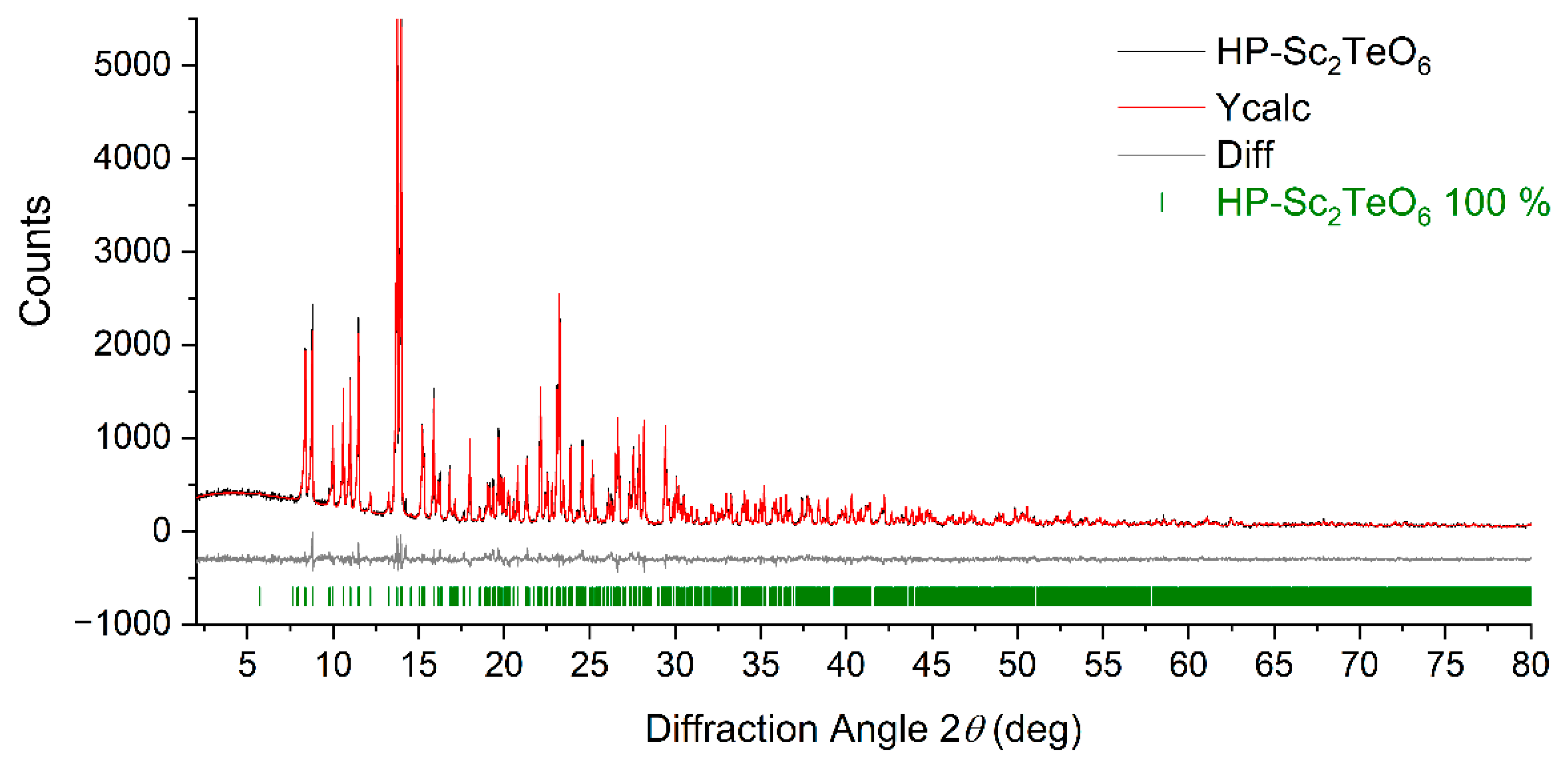
Rietveld refinement was accomplished with the software package DIFFRACplus-TOPAS 4.2 (Bruker AXS, Karlsruhe, Germany). The refinement was based on the single-crystal structure model and the peak shapes were optimized using modified Thompson–Cox–Hastings pseudo-Voigt profiles [24,25]. The lattice parameters derived from the refinement are comparable with those received by single-crystal X-ray diffraction (see Table 1). Moreover, theoretical powder pattern calculated from the single-crystal data fits well to the measured powder pattern, which proves the purity of the HP-Sc2TeO6 specimen (see Figure 1).
A single-crystal was isolated in perfluoropolyalkylether (viscosity 1800 cSt) and mounted on the tip of a loop (MicroMountsTM, MiTeGen, LLC, Ithaca, NY, USA) using a microscope. The data collection was conducted at a Bruker D8 Quest diffractometer (Bruker AXS, Karlsruhe, Germany) equipped with an Incoatec IµS sealed tube microfocus source with multi-layered optic producing monochromatized Mo-Kα radiation (λ = 71.073 pm) and a Photon 100 CMOS detector. The diffraction patterns were indexed and the total number of runs and images was based on the strategy calculation from the program Apex3 [26]. The unit cell refinement, integration and data reduction were performed using Saint [26]. A semi-empirical multi-scan absorption correction was carried out using Sadabs [26].
X-ray powder diffraction patterns at elevated temperatures were recorded using a Rigaku SmartLab-3kW instrument in parallel beam setting and reflection mode (Cu-Kα, λ = 154.18 pm) using a HyPix3000 detector (Rigaku, Tokyo, Japan). The ground sample was placed on a flat Al2O3-sample holder compatible with an Anton Paar HTK1200N (Anton Paar, Graz, Austria) high-temperature chamber used for heating. The patterns were recorded in a range of 15 to 60° 2θ with a step width of 0.01° and a speed of 1.5°/min. The temperature was increased by 20 K/min and held for one minute before each measurement. Patterns were recorded in 100 K steps up to 773 K and subsequently in 50 K steps up to 1373 K, on temperature decrease, a measurement was performed every 100 K.
SmartLab Studio-II (Rigaku Corporation 2014) was used for data collection, temperature control and data analysis such as phase identification and WPPF refinement. For phase identification, the PDF4+ 2021 [18] database was used.
Semiquantitative analyses by energy-dispersive X-ray spectroscopy (EDX) on several crystals of HP-Sc2TeO6 were made using a scanning electron microscope (JSM-6010LV, Jeol, Freising, Germany) with an energy-dispersive Quantax X-ray detector-system (Bruker AXS GmbH, Billerica, MA, USA) for element identification. The crystals were placed on a carbon plate on an aluminium sample carrier and sputtered with carbon. Each region was measured for 60 s at an excitation energy of 15 kV in high vacuum. Three crystals (see Figure 2) were measured regarding their Sc to Te ratio, and the values from several measurement points were arithmetically averaged. Due to the preparation method and the low sensitivity of light atoms like oxygen only, the ratio of scandium to tellurium was considered and the absolute values are neglected. The measured ratio of Sc/Te = 36 ± 3 at% Sc/18 ± 3 at% Te agrees well with the theoretical ratio of Sc/Te = 2:1 in HP-Sc2TeO6.
3. Results and Discussion
3.1. Structure Refinements
HP-Sc2TeO6 crystallizes monoclinic with the following lattice parameters: a = 729.43(3), b = 512.52(2), c = 1095.02(4) pm and β = 103.88(1)°. The space group P2/c (no. 13) was indicated by the general systematic extinctions of h0l with l ≠ 2n and 00l with l ≠ 2n as well as the special extinction conditions hkl: l ≠ 2n. Using the “Intrinsic Phasing” algorithm implemented in the Apex3 program package [27], the initial positional parameters were determined. Full-matrix least-squares refinements based on F2 yielded the exact atom positions [28,29] and finally, all atoms were refined with anisotropic displacement parameters. The occupation parameters were refined in a separate series of least-squares cycles in order to verify the correct composition. The correctness of the space group was proved with the Addsym [30] routine of the Platon program package [31]. Experimental details, the positional parameters and anisotropic displacement parameters are listed in Table 1, Table 2 and Table 3. CSD 2124916 (HP-Sc2TeO6) contains the ESI data for this paper.
3.2. Crystal Chemistry
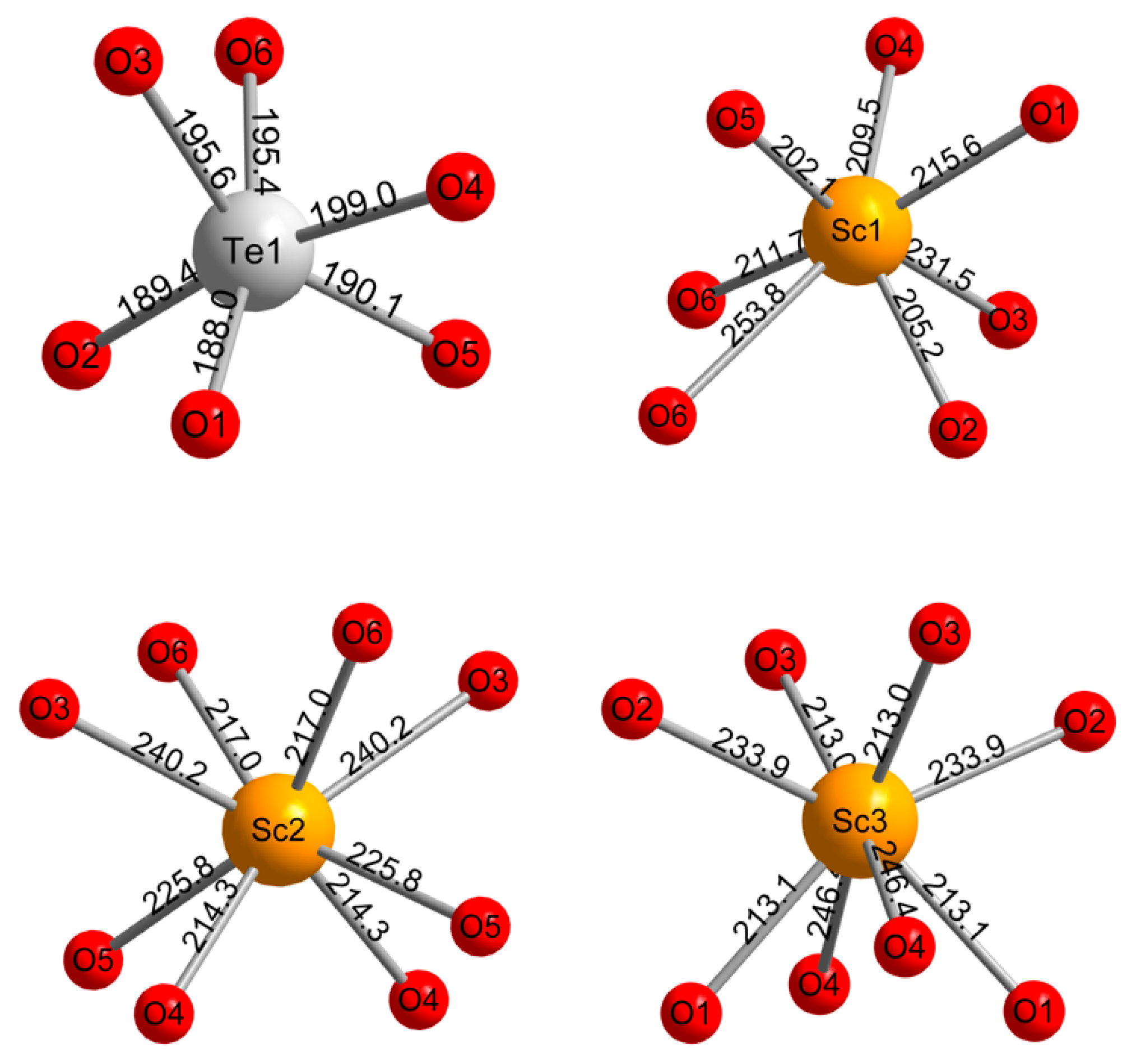
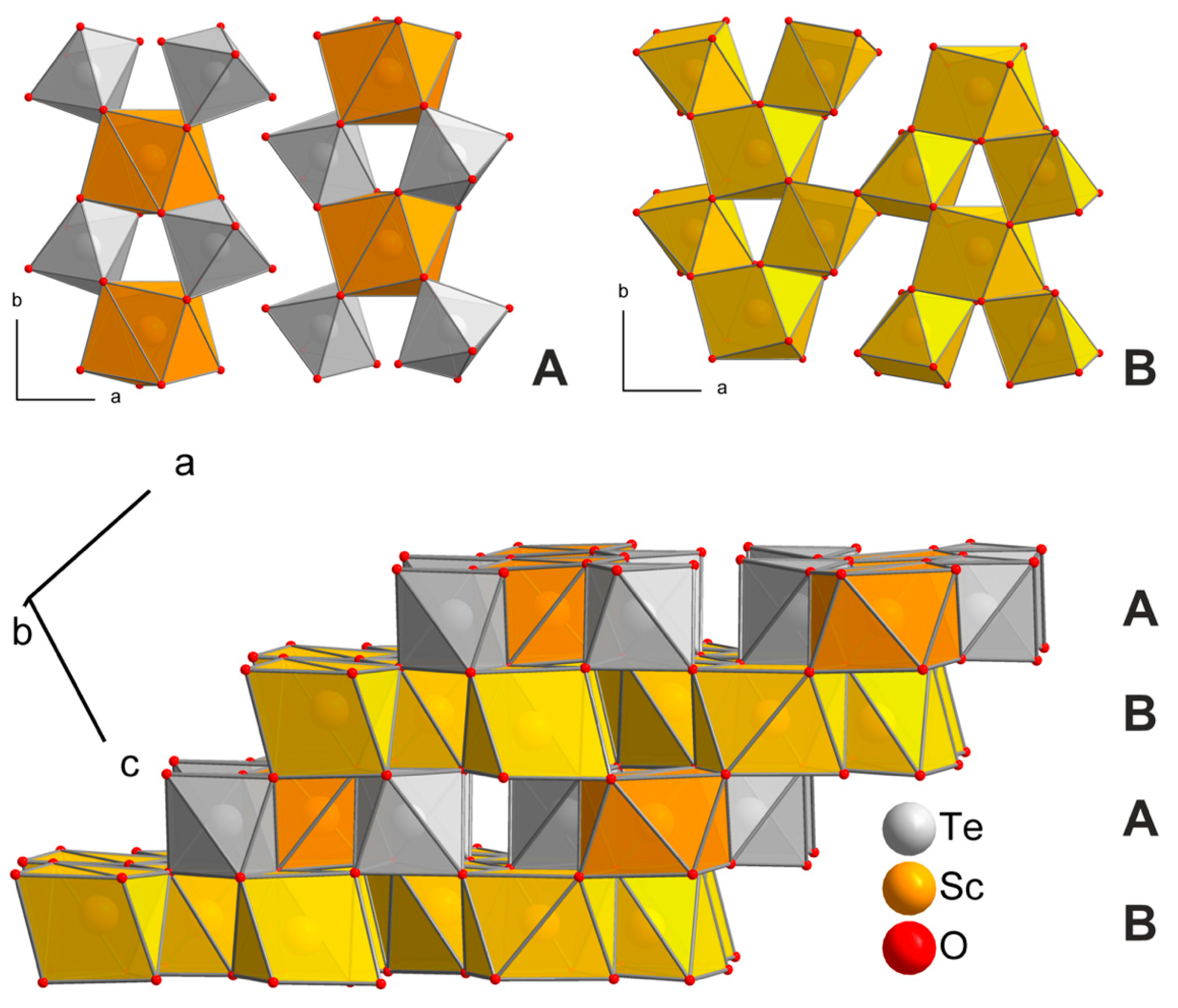
HP-Sc2TeO6 forms at high-pressure/high-temperature conditions of 12 GPa and 1173 K from the normal-pressure precursor compound. It crystallizes in the monoclinic space group P2/c (no. 13) and is isostructural to Yb2WO6 [32]. The Te6+ cations are distorted octahedrally coordinated by six oxygen ions, whereas Sc1 is coordinated by seven oxygen ions as a capped trigonal prism. Sc2- and Sc3-atoms are distorted quadratic antiprismatically coordinated by eight oxygen ions. All coordination geometries with given atom distances are displayed in Figure 3.
The crystal structure is built up by two alternating layers A and B (Figure 4, top). In layer A, square antiprisms of Sc2O8 share edges with four octahedra of TeO6, which in turn are connected to two Sc2O8 units. Thus, the polyhedra form unconnected bands along the crystallographic b-axis. Layer B consists of Sc3O8 and Sc1O7 units, which are similarly connected to each other as in layer A. The resulting bands are edge connected via the Sc1O7 polyhedra, unlike in layer A. Both layers are linked via corners and edges stacked along [10] (Figure 4, bottom). The Te–O distances vary between 188.0 and 199.0 pm, which is in good agreement with other tellurates(VI) [33]. The fourfold linked oxygen atom O4 exhibits the longest Te–O distance. The Sc–O distances in HP-Sc2TeO6 are longer than in Sc2O3 (208 to 216 pm), which agrees well with the pressure coordination rule [34,35]. A comparison with the normal-pressure modification NP-Sc2TeO6 is given later on in the section. Sc1 has six shorter (202.1 to 231.5 pm) and one longer (253.8 pm) distance to oxygen atoms. The long distance between Sc1 and O6 is responsible for the connection of the bands in layer B. The quadratic antiprismatically coordinated atoms Sc2 and Sc3 show similar distances to oxygen atoms of 214.3 to 240.2 pm and 213.0 to 246.4 pm, respectively.
NP-Sc2TeO6 [11] crystallizes in the Na2SiF6-type structure (trigonal, P321, no. 150). The structural differences between the high-pressure and normal-pressure modifications of Sc2TeO6 are confirmed by changes in molar volume, coordination numbers and atom distances. The molar volume decreases from the normal-pressure phase to the high-pressure phase from 63.73 cm3⋅mol−1 to 59.83 cm3⋅mol−1. The coordination of tellurium remains unchanged, but the distortion is enhanced in the high-pressure phase. On the one hand, this is indicated by different Te–O distances; on the other hand, the distortion is seen in the angles of the octahedra. The axial octahedra angles range from 154° to 169° (ideal: 180°) and the equatorial angles from 78° to 107° (ideal: 90°). In any case, the average Te–O distances do not change significantly between the normal-pressure (191 pm) and high-pressure (193 pm) modifications. In contrast to the coordination of the Te atoms, the coordination number of all three scandium atoms increases from six in the normal-pressure modification to seven and eight in the high-pressure phase. Therefore, Sc–O distances increase from NP-Sc2TeO6 (208.4 to 216.4 pm) to HP-Sc2TeO6 (202.2 to 253.8 pm), with most distances longer than 208 pm.
3.3. Theoretical Calculations
For better evidence of the crystal structure of HP-Sc2TeO6 Maple (Madelung Part of Lattice Energy), Chardi (Charge Distribution) as well as BL/BS (bond length/bond strength) calculations have been performed [36,37,38,39,40,41]. The calculated bond-valence sums and charge distributions agree well with the formal charges Te6+, Sc3+ and O2− acquired by X-ray structure analysis (see Table 4).
The Mapleter value of HP-Sc2TeO6 (42,018 kJ/mol) fits to the Maplebin value (42,159 kJ/mol) with a deviation of 0.3% or 141 kJ/mol, which is within the limits of the concept. Maplebin is calculated by the sum of MapleSc2O3 (16,435 kJ/mol) and MapleTeO3 (25,724 kJ/mol) in stoichiometric ratio.
3.4. Temperature-Dependent X-ray Powder Diffractometry
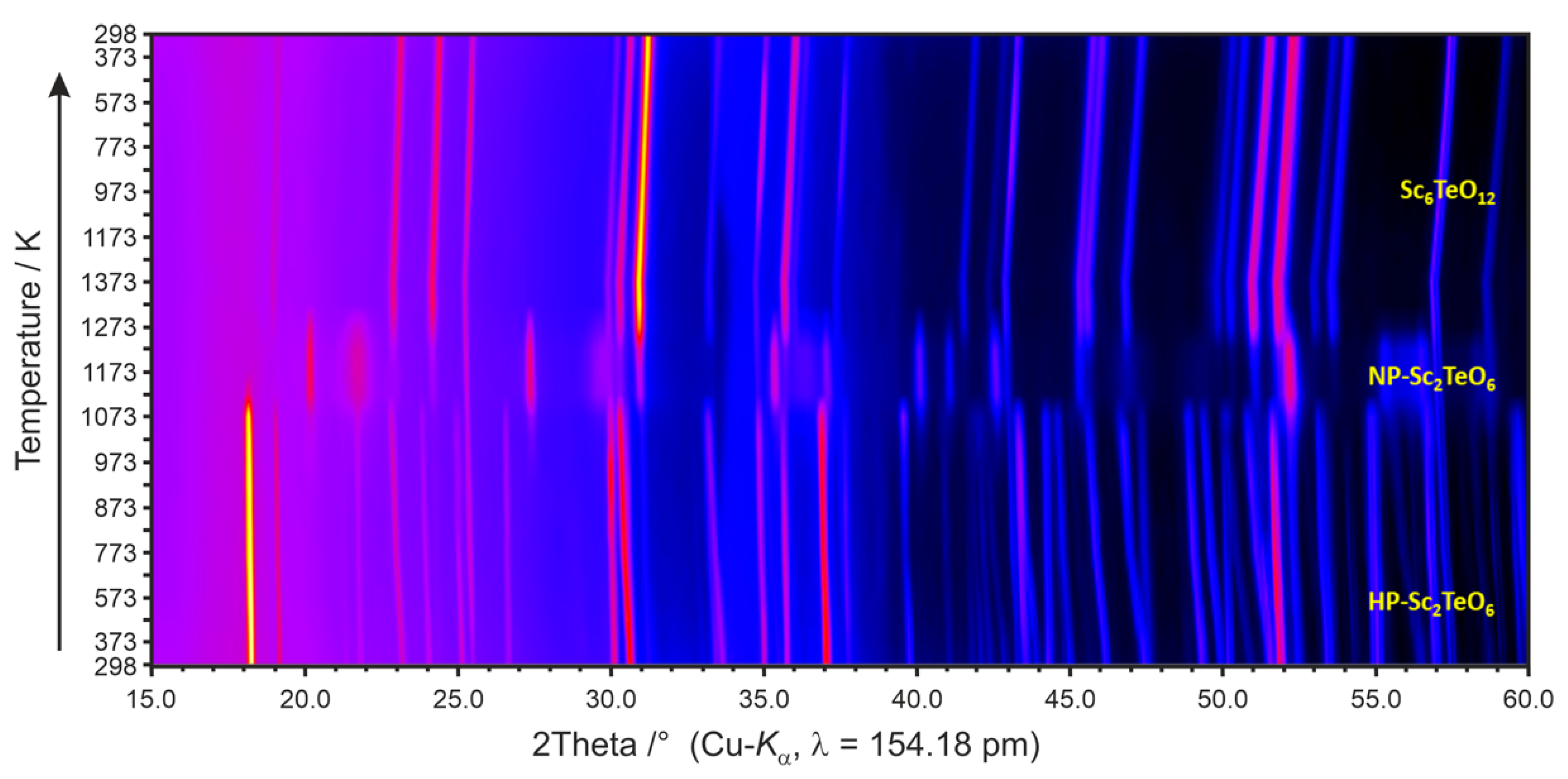
To investigate the high-temperature behaviour of HP-Sc2TeO6, temperature-dependent X-ray powder diffractometry was performed (see Figure 5). At the starting temperature of 298 K, all measured Bragg reflections could be indexed with HP-Sc2TeO6 and sample holder material Al2O3. In comparison to the X-ray powder diffraction experiments performed in transmission geometry (see Figure 1), the patterns recorded in reflection geometry showed a strong preferred orientation for (10), presumably due to the sample preparation. At 298 K, the lattice parameters of HP-Sc2TeO6 were determined as a = 728.89(3), b = 512.11(2), c = 1094.67(4) pm, β = 103.95(2)°. With increasing temperature lattice parameters expanded to a = 731.9(1), b = 514.39(7), c = 1105.7(1) pm, β = 103.28(2)° at 1073 K. Bragg reflections of HP-Sc2TeO6 decreased in intensity at 1073 K, were still present at 1123 K but absent at 1173 K. At 973 K, Bragg reflections of a new phase appeared with very little intensity and increased in intensity at 1123 K. They dominated the pattern from 1123 to 1223 K and decreased at 1273 K, at 1323 K they were not observed any more. This phase, observed overall from 973 to 1273 K, has been identified as NP-Sc2TeO6 [11]. However, whole powder pattern fitting did not yield satisfying results due to very different peak shapes: some Bragg reflections, especially (101), (201), (211) and some further reflections of lower intensity, were significantly broadened compared to (110), (111), (300) and (302). The reason for this could be (1) the formation of an unidentified HT-phase overlapping with NP-Sc2TeO6 or (2) anisotropic peak broadening of NP-Sc2TeO6 due to crystal shape anisotropy.
Another phase started to appear from at least 1123 K, it was identified as Sc6TeO12, R isostructural to Y2UO12 [18,19]. Besides signals from the Al2O3 sample holder, Sc6TeO12 was the only phase present at 1323 and 1373 K and remained stable on temperature decrease to 298 K. Lattice parameters were determined as a = 933.53(2), c = 884.31(5) pm at 1373 K and a = 924.47(2), c = 873.40(2) pm at 298 K.
4. Conclusions
The structural elucidation of the first scandium tellurate (HP-Sc2TeO6) synthesized under high-pressure/high-temperature conditions of 12 GPa and 1173 K revealed a Yb2WO6-type structure. The normal-pressure modification of Sc2TeO6 adopts the Na2SiF6-type structure. From a crystal symmetry point of view, there exists no direct group subgroup relationship between the two space groups P321 and P2/c. Thus, a reconstructive conversion of the precursor material NP-Sc2TeO6 to HP-Sc2TeO6 can be assumed. High-temperature powder X-ray diffractometry characterized the back-transformation of the metastable high-pressure modification to the at ambient conditions thermodynamically stable NP-Sc2TeO6 modification. Rietveld refinements confirmed the single-crystal structure model and the purity of the product.
Author Contributions
Synthesis of polycrystalline samples, X-ray phase analysis, X-ray diffraction analysis, theoretical calculations, visualization, writing—original draft preparation, R.Z.; EDX analysis, M.T.; HT-PDXRD analysis, writing—original draft preparation, visualization, C.H.; X-ray diffraction analysis, visualization, conceptualization, writing—original draft preparation, writing—review and editing, supervision, G.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Deposition Number CSD 2124916 (for HP-Sc2TeO6) contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures (accessed on 11 December 2021).
Acknowledgments
We thank H. Huppertz for continuous support and usage of all the facilities of the Institute of General, Inorganic and Theoretical Chemistry, University of Innsbruck.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lass, J.; Andersen, C.R.; Leerberg, H.K.; Birkemose, S.; Toth, S.; Stuhr, U.; Bartkowiak, M.; Niedermayer, C.; Lu, Z.; Toft-Petersen, R.; et al. Field-induced magnetic incommensurability in multiferroic Ni3TeO6. Phys. Rev. B Condens. Matter 2020, 101, 054415. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-H.; Wang, C.-W.; Zhao, Y.; Li, W.-H.; Lynn, J.W.; Harris, A.B.; Rule, K.; Yang, H.-D.; Berger, H. Complex magnetic incommensurability and electronic charge transfer through the ferroelectric transition in multiferroic Co3TeO6. Sci. Rep. 2017, 7, 6437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selb, E.; Buttlar, T.; Janka, O.; Tribus, M.; Ebbinghaus, S.G.; Heymann, G. Multianvil high-pressure/high-temperature synthesis and characterization of magnetoelectric HP-Co3TeO6. J. Mater. Chem. C 2021, 9, 5486–5496. [Google Scholar] [CrossRef]
- Liu, L.; Skogby, H.; Ivanov, S.; Weil, M.; Mathieu, R.; Lazor, P. Bandgap engineering in Mn3TeO6: Giant irreversible bandgap reduction triggered by pressure. Chem. Commun. 2019, 55, 12000–12003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathieu, R.; Ivanov, S.A.; Nordblad, P.; Weil, M. Enhancement of antiferromagnetic interaction and transition temperature in M3TeO6 systems (M = Mn, Co, Ni, Cu). Eur. Phys. J. B 2013, 86, 361. [Google Scholar] [CrossRef] [Green Version]
- Solana-Madruga, E.; Aguilar-Maldonado, C.; Ritter, C.; Huvé, M.; Mentré, O.; Attfield, J.P.; Arévalo-López, Á.M. Complex magnetism in Ni3TeO6-type Co3TeO6 and high-pressure polymorphs of Mn3−xCoxTeO6 solid solutions. Chem. Commun. 2021, 57, 2511–2514. [Google Scholar] [CrossRef]
- Arévalo-López, Á.M.; Solana-Madruga, E.; Aguilar-Maldonado, C.; Ritter, C.; Mentré, O.; Attfield, J.P. Magnetic frustration in the high-pressure Mn2MnTeO6 (Mn3TeO6-II) double perovskite. Chem. Commun. 2019, 55, 14470–14473. [Google Scholar] [CrossRef]
- Kim, J.W.; Artyukhin, S.; Mun, E.D.; Jaime, M.; Harrison, N.; Hansen, A.; Yang, J.J.; Oh, Y.S.; Vanderbilt, D.; Zapf, V.S.; et al. Successive Magnetic-Field-Induced Transitions and Colossal Magnetoelectric Effect in Ni3TeO6. Phys. Rev. Lett. 2015, 115, 137201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ra, H.-S.; Ok, K.M.; Halasyamani, P.S. Combining Second-Order Jahn−Teller Distorted Cations to Create Highly Efficient SHG Materials: Synthesis, Characterization, and NLO Properties of BaTeM2O9 (M = Mo6+ or W6+). J. Am. Chem. Soc. 2003, 125, 7764–7765. [Google Scholar] [CrossRef] [PubMed]
- Halasyamani, P.S.; Zhang, W. Viewpoint: Inorganic Materials for UV and Deep-UV Nonlinear-Optical Applications. Inorg. Chem. 2017, 56, 12077–12085. [Google Scholar] [CrossRef] [PubMed]
- Höss, P.; Schleid, T. Sc2Te5O13 und Sc2TeO6: Die ersten Oxotellurate des Scandiums. Z. Anorg. Allg. Chem. 2007, 633, 1391–1396. [Google Scholar] [CrossRef]
- Meier, S.F.; Schleid, T. Oxotellurate(IV) der Lanthanide: II. Die isotype Reihe M2Te5O13 (M = Dy − Lu) / Oxotellurates(IV) of Lanthanides: II. The Isotypic Series M2Te5O13 (M = Dy − Lu). Z. Für. Nat. B Chem. Sci. 2005, 60, 720–726. [Google Scholar] [CrossRef]
- Höss, P.; Osvet, A.; Meister, F.; Batentschuk, M.; Winnacker, A.; Schleid, T. Synthesis, crystal structures and luminescence properties of the Eu3+-doped yttrium oxotellurates(IV) Y2Te4O11 and Y2Te5O13. J. Solid State Chem. 2008, 181, 2783–2788. [Google Scholar] [CrossRef]
- Song, S.Y.; Lee, D.W.; Ok, K.M. Rich Structural Chemistry in Scandium Selenium/Tellurium Oxides: Mixed-Valent Selenite–Selenates, Sc2(SeO3)2(SeO4) and Sc2(TeO3)(SeO3)(SeO4), and Ternary Tellurite, Sc2(TeO3)3. Inorg. Chem. 2014, 53, 7040–7046. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Xu, X.; Mao, J.-G. A Series of New Ternary and Quaternary Compounds in the LiI–GaIII–TeIV–O System. Inorg. Chem. 2010, 49, 11573–11580. [Google Scholar] [CrossRef] [PubMed]
- Höss, P.; Meier, S.F.; Schleid, T. Lu2Te3O9: The First Example of the Triclinic C-Type Lanthanoid(III) Oxotellurates(IV) with the Composition M2Te3O9. Z. Anorg. Allg. Chem. 2013, 639, 2548–2553. [Google Scholar] [CrossRef]
- Meier, S.F.; Höss, P.; Schleid, T. Dy2Te3O9: Der erste Vertreter von Lanthanoid(III)-Oxotelluraten der Zusammensetzung M2Te3O9. Z. Anorg. Allg. Chem. 2009, 635, 768–775. [Google Scholar] [CrossRef]
- Gates-Rector, S.; Blanton, T. The Powder Diffraction File: A quality materials characterization database. Powder Diffr. 2019, 34, 352–360. [Google Scholar] [CrossRef] [Green Version]
- Blasse, G. Lanthanide tellurates Ln6TeO12. J. Inorg. Nucl. Chem. 1969, 31, 3335–3336. [Google Scholar] [CrossRef]
- Walker, D. Lubrication, gasketing, and precision in multianvil experiments. Am. Mineral. 1991, 76, 1092–1100. [Google Scholar]
- Walker, D.; Carpenter, M.A.; Hitch, C.M. Some simplifications to multianvil devices for high pressure experiments. Am. Mineral. 1990, 75, 1020–1028. [Google Scholar]
- Huppertz, H.; Heymann, G.; Schwarz, U.; Schwarz, M.R. High-Pressure Methods in Solid-State Chemistry. In Handbook of Solid State Chemistry; Dronskowski, R., Kikkawa, S., Stein, A., Eds.; Wiley-VCH: Weinheim, Germany, 2017; Volume 2, pp. 23–48. [Google Scholar]
- Huppertz, H. Multianvil high-pressure/high-temperature synthesis in solid state chemistry. Z. Kristallogr. 2004, 219, 330–338. [Google Scholar] [CrossRef]
- Thompson, P.; Cox, D.E.; Hastings, J.B. Rietveld refinement of Debye-Scherrer synchrotron X-ray data from Al2O3. J. Appl. Crystallogr. 1987, 20, 79–83. [Google Scholar] [CrossRef] [Green Version]
- Young, R.A.; Desai, P. Crystallite Size and Microstrain Indicators in Rietveld Refinement. Arch. Nauki. Mater. 1989, 10, 71–90. [Google Scholar]
- APEX3 (v. 2017.3-0), CELL_NOW (v. 2008/4), SAINT (v. 8.38A), TWINABS (v. 2012/1), and SADABS (v. 2016/2); Bruker AXS GmbH: Karlsruhe, Germany, 2016.
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. ShelXL—Crystal Structure Refinement—Multi-CPU Version; 2017/1; University of Göttingen: Göttingen, Germany, 2017. [Google Scholar]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Le Page, Y. MISSYM1.1—A flexible new release. J. Appl. Crystallogr. 1988, 21, 983–984. [Google Scholar] [CrossRef]
- Spek, A. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Efremov, V.A.; Tyulin, A.V.; Trunov, V.K.; Kudin, O.V.; Yanovskij, V.K.; Voronkova, V.I. Crystal structure of monoclinic Y2WO6 and Yb2WO6. Kristallografiya 1984, 28, 904–909. [Google Scholar]
- Becker, R.; Johnsson, M.; Berger, H. A new synthetic cobalt tellurate: Co3TeO6. Acta Crystallogr. C 2006, 62, i67–i69. [Google Scholar] [CrossRef]
- Geller, S.; Romo, P.; Remeika, J.P. Refinement of the structure of scandium sesquioxide. Z. Kristallogr. 1967, 124, 136–142. [Google Scholar] [CrossRef]
- Prewitt, C.T.; Downs, R.T. High-pressure crystal chemistry. Rev. Mineral. Geochem. 1998, 37, 283–317. [Google Scholar]
- Hoppe, R.; Voigt, S.; Glaum, H.; Kissel, J.; Müller, H.P.; Bernet, K. A new route to charge distributions in ionic solids. J. Less-Common Met. 1989, 156, 105–122. [Google Scholar] [CrossRef]
- Pauling, L. Atomic Radii and Interatomic Distances in Metals. J. Am. Chem. Soc. 1947, 69, 542–553. [Google Scholar] [CrossRef]
- Brown, I.D.; Altermatt, D. Bond-valence parameters obtained from a systematic analysis of the Inorganic Crystal Structure Database. Acta Crystallogr. B 1985, 41, 244–247. [Google Scholar] [CrossRef] [Green Version]
- Brese, N.E.; O’Keeffe, M. Bond-valence parameters for solids. Acta Crystallogr. B 1991, 47, 192–197. [Google Scholar] [CrossRef]
- Hübenthal, R. MAPLE; v.4; Universität Gießen: Gießen, Germany, 1993. [Google Scholar]
- Nespolo, M.; Guillot, B. CHARDI2015: Charge distribution analysis of non-molecular structures. J. Appl. Crystallogr. 2016, 49, 317–321. [Google Scholar] [CrossRef]
Figure 1.
XRPD pattern (Mo-Kα1 radiation) and Rietveld refinement of X-ray pure HP-Sc2TeO6 displayed with an offset of 750 counts (Rexp = 6.98, Rwp = 7.56, Rp = 5.67 and GooF = 1.08).
Figure 1.
XRPD pattern (Mo-Kα1 radiation) and Rietveld refinement of X-ray pure HP-Sc2TeO6 displayed with an offset of 750 counts (Rexp = 6.98, Rwp = 7.56, Rp = 5.67 and GooF = 1.08).

Figure 2.
Crystals of HP-Sc2TeO6 selected for elemental analysis by energy-dispersive X-ray spectroscopy (EDX).
Figure 2.
Crystals of HP-Sc2TeO6 selected for elemental analysis by energy-dispersive X-ray spectroscopy (EDX).

Figure 3.
Coordination spheres and interatomic distances (pm) of Te1 (top left), Sc1 (top right), Sc2 and Sc3 (bottom left and right). Standard deviations are smaller than 0.2 pm.
Figure 3.
Coordination spheres and interatomic distances (pm) of Te1 (top left), Sc1 (top right), Sc2 and Sc3 (bottom left and right). Standard deviations are smaller than 0.2 pm.

Figure 4.
The crystal structure of HP-Sc2TeO6 is shown with view along the b axis (bottom). Layer (A) (top left) and layer (B) (top right) are displayed along the crystallographic c axis.
Figure 4.
The crystal structure of HP-Sc2TeO6 is shown with view along the b axis (bottom). Layer (A) (top left) and layer (B) (top right) are displayed along the crystallographic c axis.

Figure 5.
Temperature-dependent X-ray powder diffractogram displaying the back-transformation of HP-Sc2TeO6 to the ambient pressure modification NP-Sc2TeO6.
Figure 5.
Temperature-dependent X-ray powder diffractogram displaying the back-transformation of HP-Sc2TeO6 to the ambient pressure modification NP-Sc2TeO6.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Crystal data, data collection and structure refinement results for HP-Sc2TeO6.
| Empirical formula | HP-Sc2TeO6 |
| Molar mass, g∙mol−1 | 313.52 |
| Crystal system | monoclinic |
| Space group | P2/c |
| Cell formula units | 4 |
| Powder diffractometer | STOE STADI P |
| Radiation | Mo-Kα1 (λ = 70.93 pm) |
| Powder data: | |
| a, pm | 729.224(7) |
| b, pm | 512.576(5) |
| c, pm | 1095.04(2) |
| β, deg | 103.898(1) |
| V, Å3 | 397.323(7) |
| Single-crystal diffractometer | Bruker D8 Quest |
| Radiation | Mo-Kα (λ = 71.073 pm) |
| Single-crystal data: | |
| a, pm | 729.43(3) |
| b, pm | 512.52(2) |
| c, pm | 1095.02(4) |
| β, deg | 103.88(1) |
| V, Å3 | 397.42(3) |
| Calculated density, g∙cm−3 | 5.24 |
| Crystal size, mm3 | 0.02 × 0.02 × 0.02 |
| Temperature, K | 299(2) |
| Absorption coefficient, mm−1 | 10.54 |
| F(000), e | 568 |
| Detector distance, mm | 34 |
| θ range, deg | 2.88–39.42 |
| Range in hkl | −12 ≤ h ≤ 13, ±9, ±19 |
| Total no. reflections | 13924 |
| Data/ref. parameters | 2379/84 |
| Reflections with I > 2σ(I) | 2077 |
| Rint/Rσ | 0.0363, 0.0254 |
| Goodness-of-fit on F2 | 1.055 |
| Absorption correction | multi-scan |
| R1/wR2 for I > 2σ(I) | 0.0191/0.0331 |
| R1/wR2 for all data | 0.0261/0.0344 |
| Extinction coefficient | 0.0026(2) |
| Transmission max./min. | 0.748/0.646 |
| Largest diff. peak/hole/e∙Å−3 | 0.905/−0.962 |
Table 2.
Atomic coordinates, Wyckoff positions, site occupancy (s. o. f.) and equivalent isotropic displacement parameters Ueq (Å2) 1 for HP-Sc2TeO6 (space group P2/c; no. 13). Standard deviations in parentheses.
Table 2.
Atomic coordinates, Wyckoff positions, site occupancy (s. o. f.) and equivalent isotropic displacement parameters Ueq (Å2) 1 for HP-Sc2TeO6 (space group P2/c; no. 13). Standard deviations in parentheses.
| Atom | Wyck. | x | y | z | SOF | Ueq |
|---|---|---|---|---|---|---|
| Te1 | 4g | 0.22848(2) | 0.26270(2) | 0.61484(2) | 1 | 0.00223(3) |
| Sc1 | 4g | 0.71575(4) | 0.19306(6) | 0.58249(3) | 1 | 0.00365(5) |
| Sc2 | 2f | 1/2 | 0.24868(8) | 1/4 | 1 | 0.00377(6) |
| Sc3 | 2e | 0 | 0.30665(8) | 1/4 | 1 | 0.00442(7) |
| O1 | 4g | 0.0135(2) | 0.6152(2) | 0.3823(2) | 1 | 0.0050(2) |
| O2 | 4g | 0.1418(2) | 0.1363(2) | 0.4484(1) | 1 | 0.0056(2) |
| O3 | 4g | 0.2041(2) | 0.0254(2) | 0.2271(1) | 1 | 0.0044(2) |
| O4 | 4g | 0.3021(2) | 0.5402(2) | 0.2758(1) | 1 | 0.0046(2) |
| O5 | 4g | 0.3462(2) | 0.4355(2) | 0.0674(1) | 1 | 0.0054(2) |
| O6 | 4g | 0.4541(2) | 0.0606(2) | 0.6107(2) | 1 | 0.0050(2) |
1 Ueq is defined as one third of the trace of the orthogonalized Uij tensor.
Table 3.
Anisotropic displacement parameters Uij (Å2) 1 for HP-Sc2TeO6 (space group P2/c; no. 13). Standard deviations in parentheses.
Table 3.
Anisotropic displacement parameters Uij (Å2) 1 for HP-Sc2TeO6 (space group P2/c; no. 13). Standard deviations in parentheses.
| Atom | U11 | U22 | U33 | U23 | U13 | U12 |
|---|---|---|---|---|---|---|
| Te1 | 0.00242(4) | 0.00213(4) | 0.00227(4) | −0.00001(3) | 0.00082(3) | 0.00006(3) |
| Sc1 | 0.0040(1) | 0.0037(1) | 0.0036(1) | 0.00032(8) | 0.00160(8) | 0.00024(8) |
| Sc2 | 0.0041(1) | 0.0030(2) | 0.0041(1) | 0 | 0.0006(1) | 0 |
| Sc3 | 0.0047(2) | 0.0034(2) | 0.0055(2) | 0 | 0.0019(1) | 0 |
| O1 | 0.0031(4) | 0.0062(5) | 0.0060(4) | −0.0009(4) | 0.0017(4) | 0.0006(4) |
| O2 | 0.0076(5) | 0.0054(5) | 0.0030(4) | −0.0018(3) | −0.0004(4) | 0.0007(4) |
| O3 | 0.0059(4) | 0.0031(4) | 0.0043(4) | −0.0013(3) | 0.0013(4) | 0.0006(4) |
| O4 | 0.0068(5) | 0.0043(4) | 0.0027(4) | 0.0010(3) | 0.0012(4) | 0.0017(4) |
| O5 | 0.0061(5) | 0.0049(5) | 0.0051(4) | −0.0013(4) | 0.0010(4) | 0.0015(4) |
| O6 | 0.0029(4) | 0.0044(5) | 0.0081(5) | 0.0013(4) | 0.0023(4) | 0.0014(3) |
1 The anisotropic displacement factor exponent takes the form: −2π2[(ha*)2U11 + ⋯ + 2hka*b*U12].
Table 4.
Charge distribution calculated with BL/BS (ΣV) and Chardi (ΣQ).
| Te1 | Sc1 | Sc2 | Sc3 | O1 | O2 | O3 | O4 | O5 | O6 | |
|---|---|---|---|---|---|---|---|---|---|---|
| ΣV | +5.83 | +3.08 | +2.86 | +2.78 | −2.01 | −1.91 | −1.72 | −1.98 | −2.00 | −1.96 |
| ΣQ | +5.99 | +2.99 | +3.03 | +3.01 | −2.12 | −1.98 | −1.89 | −1.99 | −2.08 | −1.93 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ziegler, R.; Tribus, M.; Hejny, C.; Heymann, G. Single-Crystal Structure of HP-Sc2TeO6 Prepared by High-Pressure/High-Temperature Synthesis. Crystals 2021, 11, 1554. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11121554
AMA Style
Ziegler R, Tribus M, Hejny C, Heymann G. Single-Crystal Structure of HP-Sc2TeO6 Prepared by High-Pressure/High-Temperature Synthesis. Crystals. 2021; 11(12):1554. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11121554
Chicago/Turabian StyleZiegler, Raimund, Martina Tribus, Clivia Hejny, and Gunter Heymann. 2021. "Single-Crystal Structure of HP-Sc2TeO6 Prepared by High-Pressure/High-Temperature Synthesis" Crystals 11, no. 12: 1554. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11121554
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.