
Pressure Induced Disorder-Order Phase Transitions in the Al4Cr Phases

1
State Key Laboratory of Metastable Materials Science and Technology, Yanshan University, Qinhuangdao 066004, China
2
Hebei Key Lab for Optimizing Metal Product Technology and Performance, College of Materials Science and Technology, Yanshan University, Qinhuangdao 066004, China
*
Author to whom correspondence should be addressed.
Crystals 2022, 12(7), 1008; https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12071008
Submission received: 23 June 2022
/
Revised: 13 July 2022
/
Accepted: 19 July 2022
/
Published: 21 July 2022
(This article belongs to the Section Crystalline Metals and Alloys)
Abstract
:An ordered ω-Al4Cr phase synthesized recently by a high-pressure sintering (HPS) approach was calculated to be stable by density function theory (DFT), implying that high pressure can accelerate the disorder-order phase transitions. The structural building units of the ω-Al4Cr phase as well as the non-stoichiometric disordered ε-Al4Cr and μ-Al4Cr phases have been analyzed by the topological “nanocluster” method in order to explore the structural relations among these phases. Both the ε-and μ-Al4Cr phases contain the typical Macky or pseudo-Macky cluster, and their centered positions were all occupied by Cr atoms, which all occupy the high-symmetry Wyckoff positions. The mechanism of the pressure-induced disorder-order phase transitions from the ε-/μ-Al4Cr to the ω-Al4Cr phase has been analyzed. and the related peritectic and eutectoid reactions have been re-evaluated. All results suggest that the stable ω-Al4Cr phase are transformed from the μ-Al4Cr phase by the eutectoid reaction that is accelerated by high-pressure conditions, whereas the ε-Al4Cr phase should form by the peritectic reaction.
All materials are disordered at a finite temperature and the high temperature provides significant entropic stabilization of phases relative to low-temperature ordered structures [1,2,3,4]. Although the influence of pressure on the solidification and phase transformation has not been studied as widely as that of temperature, investigations on the effects of pressure on the material’s thermodynamics has become popular recently [5,6,7]. For example, the pressure-induced fast ordering phase transition (disordered-fcc to ordered-fcc) structures in a CoCrCuFeNiPr high-entropy alloy was found at the pressure ranging from 7.8 GPa to 16.0 GPa [8]. More recently, a pressure-induced inverse order-disorder transition was reported in double perovskites, Y2CoIrO6 and Y2CoRuO6 [9]. There are also different options for computational methods for high-pressure phase transitions [10,11,12,13].
The Al-rich Al-Cr intermetallic phases, ε-Al4Cr, μ-Al4Cr and η-Al11Cr2, exhibit as prototypes of complex metallic alloys related to the formation of icosahedral quasicrystals (IQC). Historically, the ε-Al4Cr phase has been wrongly considered as the η-Al11Cr2 phase by Audier et al. [14] and finally determined by Li et al. [15]. Furthermore, the ε-Al4Cr phase has also been found to be structurally related to the icosahedral quasicrystal by Wen et al. [16]. Different from the orthorhombic ε-Al4Cr phase, the μ-Al4Cr phase has hexagonal symmetry that had been frequently observed [17], and its crystal structure was finally determined and found in close relation with the η-Al11Cr2 phase by Cao et al. [17,18]. However, the ε-Al4Cr and μ-Al4Cr phases are both non-stoichiometric disordered phases. Therefore, it is very interesting that an ordered ω-Al4Cr phase has been discovered by the high-pressure sintering (HPS) approach recently in our group [19].
In the present work, DFT calculations have been carried out in order to explore the stability of the ordered ω-Al4Cr phase. Then, the topological “nanocluster” method was employed to study the structural relations among the ε-, μ- and ω-Al4Cr phases. Finally, the thermodynamic analysis was performed to understand the mechanism of the pressure-induced disorder-order phase transitions.
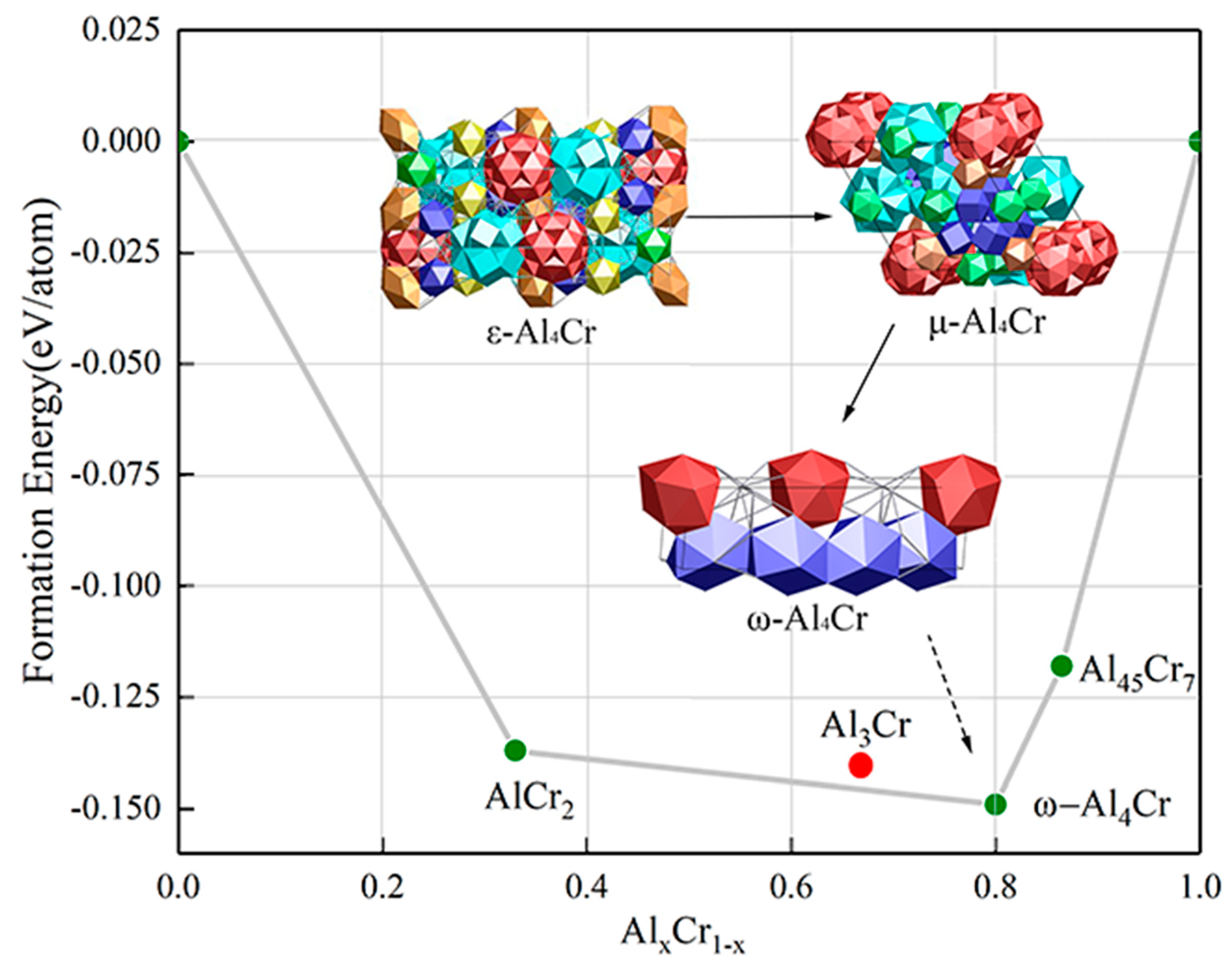
All first-principles calculations were carried out with the DFT calculations method as implanted in the Vienna ab initio simulation package (VASP) [20]. The generalized gradient approximation (GGA) in the Perdew-Burke-Ernzerhof (PBE) form [21] was used to describe the exchange-correlation function, and the projected augmented wave (PAW) method [22] was used to describe the pseudopotentials, i.e., to deal with the core and valence electrons. The k-point samplings in the Brillouin zone were performed using the Monkhorst–Pack scheme [23]. A 5 × 1 × 5 mesh k-point and a 500-eV plane-wave cutoff energy have been tested to satisfy the strict force gradient (EDIFG) self-consistent convergence criterion: 1 × 10−6 eV/atom. From the calculation results shown in Figure 1, one can find that the ω-Al4Cr phase is thermodynamically stable as its formation energy lies on the convhull along with other stable phases, such as AlCr2 and Al45Cr7 (Al3Cr becomes metastable) of the Al-Cr system from the open quantum materials database (OQMD) [24]. Such findings reveal that high pressure can accelerate the disorder-order phase transitions from the ε-/μ-Al4Cr to the ω-Al4Cr phase. Therefore, it is necessary to understand the structural relations between these phases and to decipher the phase transition mechanisms.
The “nanocluster” method in the ToposPro package [25,26,27,28] was adopted to explore the topological features of the titled Al11Cr2 and its related crystal structures. The following criteria needs to be emphasized among all the algorithms used to determine the composition and structure of nanoclusters: (i) the clusters are represented as shell graphs with centers at the most symmetrical positions; (ii) the clusters do not interpenetrate and include all atoms of the structure, making the “nanocluster” method ideal for exploring the building units of intermetallics, including the ε-Al4Cr [26].
The ε-Al4Cr phase is an extremely complex intermetallic compound with 682 atoms in the unit cell. We have re-analyzed the structural building units of the ε-Al4Cr phase by the nanoclustering procedure in order to explore the similarities between the ε-Al4Cr phase and the μ-Al11Cr2 phase, although it has been carried out previously and it captured the Mackay polyhedral [26] that was misled by Li et al. [15].
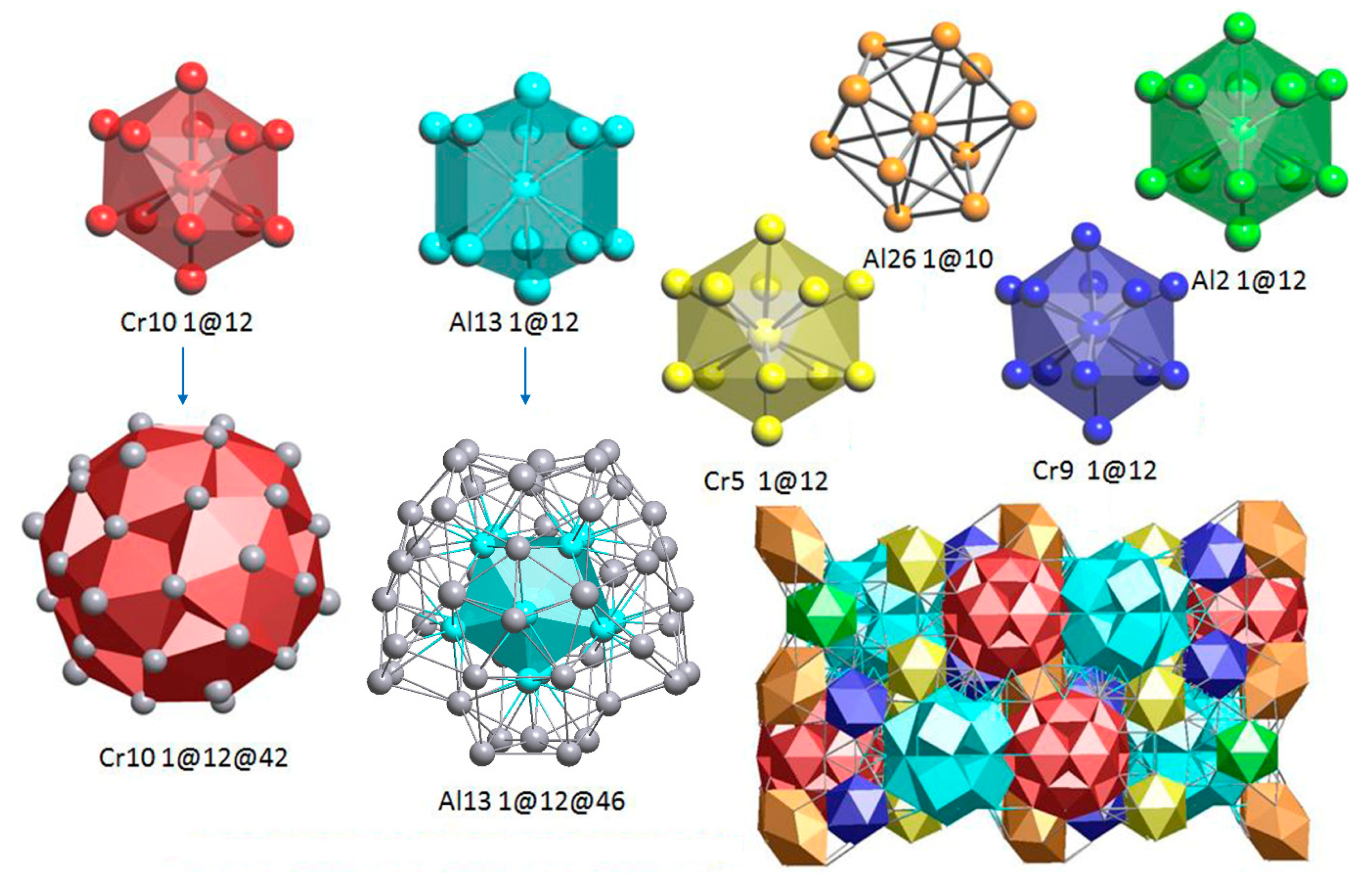
Figure 2 shows the nanocluster model of the ε-Al4Cr phase which is composed of six clusters: Cr10(2)(1@12@42), Al13(2)(1@12@46), Cr5(1)(1@12), Al26(1)(1@10), Cr9(1)(1@12) and Al2(1)(1@12). Firstly, it is confirmed that Mackay clusters centered by Cr10 atoms do exist and are similar to the double-shell cluster Cr11(2) (1@12@38) (not shown here). Secondly, the ε-Al4Cr also contains another double-shell cluster, which is Al13(2)(1@12@46).
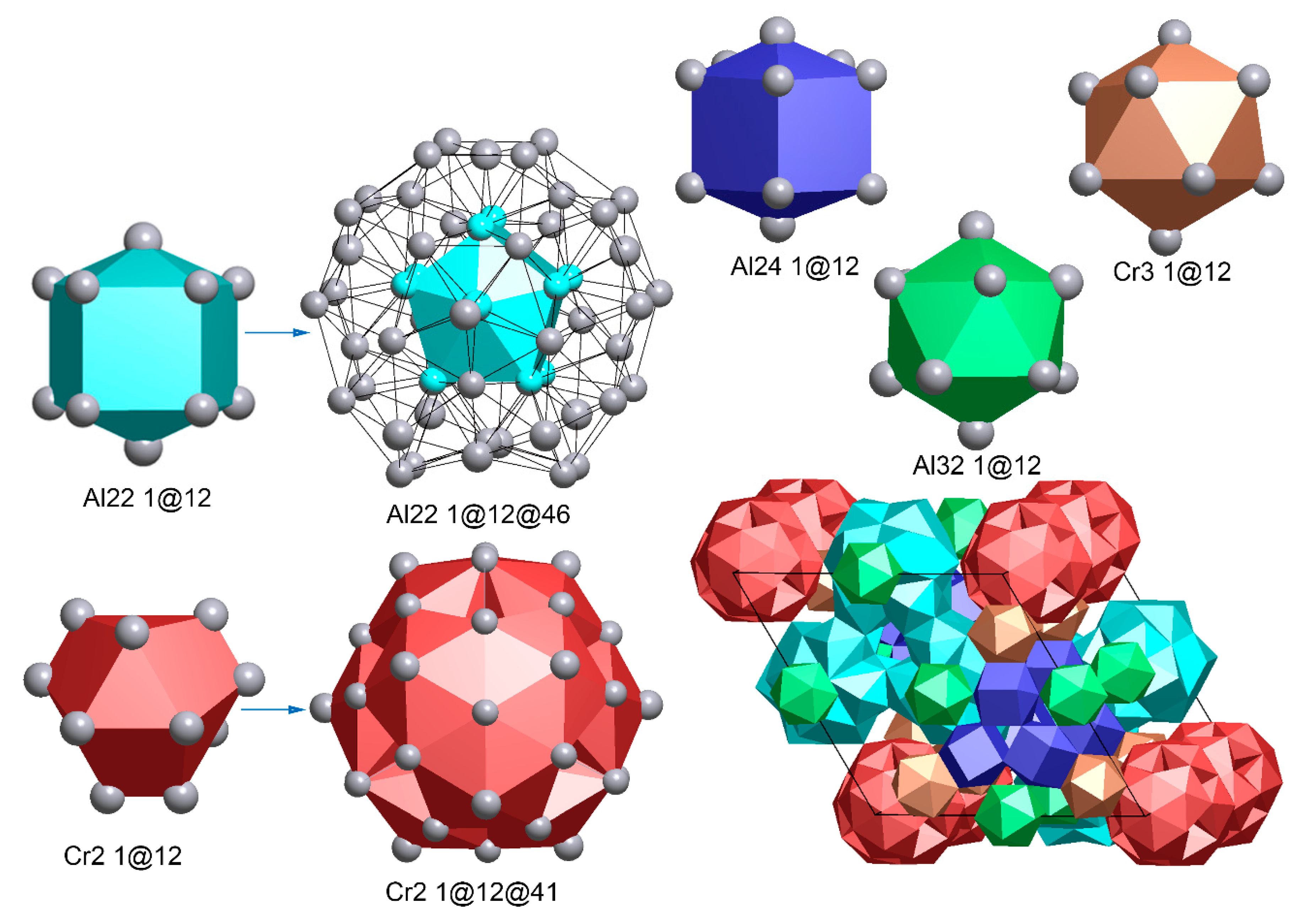
Figure 3 shows one kind of nanocluster model of the μ-Al4Cr phase, which contains six clusters: Cr2(2)(1@12@41), Al22(2)(1@12@46), Al24(1)(1@12), Cr3(1)(1@12), Al32(1)(1@12) and Al31(0). The pseudo-Mackay polyhedral appears in the form of Cr2(2)(1@12@41); the icosahedra first cell was replaced by an irregular 12-coordinated polyhedron although there are only 41 atoms instead of 42, owing to the line-shared instead of vertex-shared pentagonal curved planes. Interestingly, the double-shell clusters, Al22(2)(1@12@46) appear again as Al13(2)(1@12@46) in the ε-Al4Cr. The rest of the clusters include one pentahedron and two icosahedrons, which are very popular in Al-Cr quasicrystal approximants besides one single-atom-type cluster.
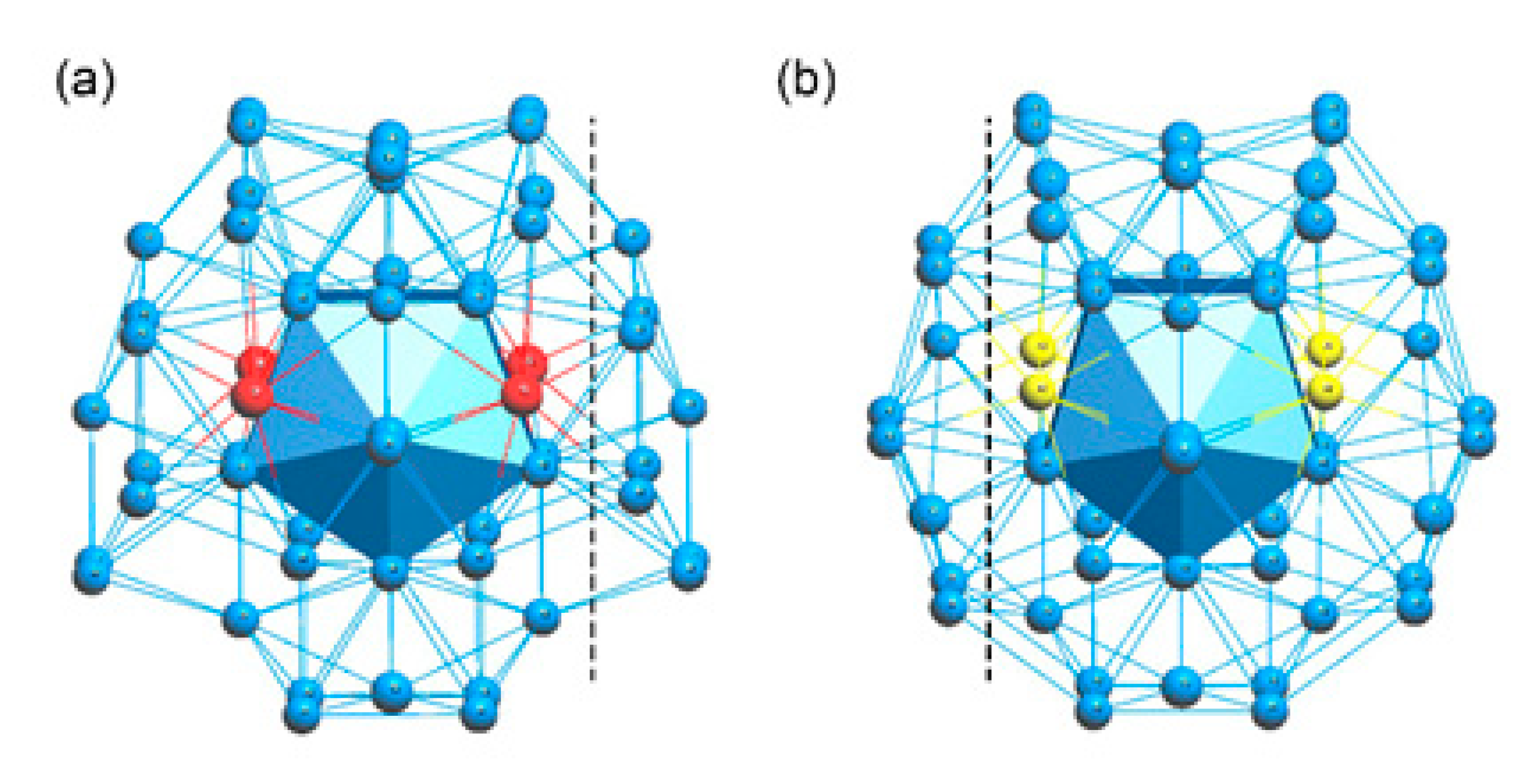
To compare the ε-and μ-Al4Cr phase, both phases contain a 1@12@42 Macky or pseudo-Macky cluster, which is known as one of the most viable structural units for complex metallic alloys and quasicrystals and their approximants. The centered positions inside the Macky or pseudo-Macky cluster were all occupied by Cr atoms and these Cr atoms all occupy the high-symmetry Wyckoff positions (See Table S1 in the Supplementary Materials). Very interestingly, the 1@12@46 double-shell nanocluster also appears in both phases, as shown in Figure 4. Figure 4a,b represents the 1@12@46 nanoclusters found in ε-Al4Cr and μ- Al4Cr, and the marked four red balls are the Cr atoms and the four yellow balls are the Al atoms. For the ω-Al4Cr phase, the building unit cluster is quite simple as it contains only 30 rather than several hundred atoms. There are two types of distinct coordination polyhedrals around two chromium atoms in the ω-Al4Cr phase; the first one connects to eleven aluminum atoms whereas the second one is connected to ten aluminum atoms [19], implying it has a completely different configuration with those of the ε-and μ-Al4Cr phase.
Concerning the large unit cell of all these titled complex metallic alloys, density-functional-theory (DFT)-based methods are not plausible as a huge computational cost is required. As a novel tool for the efficient prediction of a material’s properties, machine learning has emerged, and it claims that the machine-learned models for the formation energy of compounds can approach the accuracy of DFT. In the present work, we choose the deep neural network model named ElemNet [29] to estimate the formation energies and the molar fragment descriptor (MFD) [30] to predict the vibrational free energies and entropies, along with the property-labeled material fragments (PLMF) methods [31] to predict the bulk and shear moduli.
Conventional machine learning approaches for predicting a material’s properties involves representing the material composition in the model input format, manual feature engineering, and selection by incorporating the necessary domain knowledge and human intuition by computing the very important chemical and physical attributes of each element, and finally to construct the predictive model. However, “ElemNet” is a deep neural network model which can bypass conventional, manual feature engineering requiring domain knowledge, and it automatically captures the physical and chemical interactions and similarities between different elements to predict the material’s properties, such as the formation enthalpy, with better accuracy and speed. The “ElemNet” model have been trained on the DFT-computed formation enthalpies of 275, 759 compounds with unique elemental compositions from the Open Quantum Materials Database (OQMD) [29].
The calculation of stability at high temperatures is fundamental for the prediction of phase diagrams and chemical reactions. Solving this problem is challenging as high-throughput (HT) phase diagrams have typically been calculated using the ab initio formation enthalpies at 0 K. Legrain et al. show that several machine learning approaches can be applied to the calculation of vibrational entropies and free energies (Svib and Fvib) of solids with satisfactory accuracy and reasonable computational expense. Practically, they have tested a set of descriptors simply based on the chemical formula of compounds and by training a set of 25,705 compounds in the Inorganic Crystal Structure Database (ICSD). They concluded that the descriptors based on the chemical composition and the elemental properties of atoms of the material perform best for small training sets [30].
Bulk/shear moduli are both predicted within the property-labeled material fragments (PLMF) scheme. In this approach, data from the ab initio calculated AFLOW repository is combined with the quantitative material’s structure–property relationship models. The prediction only requires the structural information and has comparable accuracy with those of the training data (2829 materials were extracted from the AFLOW repository) [31].
The calculated results of the above mentioned five phases are shown in Table 1. For the ElemNet and the MFD methods, only chemical compositions are necessary, whereas for the PLMF method, the configurations of phases are required. In the present work, the stoichiometric compositions of both the ordered and disordered phases have been calculated and listed, but only the properties of ordered phases are considered and compared. From the calculated stoichiometric values in Table 1, one can find that all disordered phases of ε-Al4Cr and μ-Al4Cr are deficient of Al element compared to the corresponding ordered phases. Such universal deficiency of Al atoms could be understood from the close-packed FCC Al compared to the non-close-packed BCC Cr. The calculated vibrational free energy and entropy of Cr (−3.24 meV/atom) compared to Al (15.29 meV/atom) by the MFD method also suggests that the Cr atoms diffuse more easily than Al atoms. It is found that the bulk modulus values of different Al–Cr alloys are between 105 and 122 GPa; these values are close to the average between the bulk modulus of Cr (182–185 GPa) [32] and Al (73 GPa) [33]. Strikingly, not only stoichiometric, but all the predicted properties including formation energies, vibrational free energies and entropies, and bulk modulus and shear modulus of the ordered phases of ε-Al4Cr and μ-Al4Cr are nearly equivalent (see Table 1). Although, both phases have completely different crystallographic symmetry and have quite different stoichiometric disordered structural models. Such findings encourage us to deduce that the forming conditions of both phases are indistinguishable, and the ε-Al4Cr should also be the precursor in the forming of the η-Al11Cr2 phase by the peritectic reaction as that of μ-Al4Cr phase [34]:
μ-Al4Cr + L(Al)→η-Al11Cr2.
The structural building units of the ε-Al4Cr and μ-Al4Cr discussed before also support that they are nearly indistinguishable as both phases have identical or slightly different clusters. It was also mentioned that the following eutectoid reaction has been observed: η-Al11Cr2 → μ-Al4Cr + θ-Al7Cr (Al45Cr7) [34]. It needs to be noted that the situation should be more complicated as both the η-Al11Cr2 and μ-Al4Cr phase are non-stoichiometric compounds (see Table 1). The Al:Cr ratio equals to 5.16 in the η-Al11Cr2 phase. The Al:Cr ratio equals to 4.2 for both the ε-Al4Cr and μ-Al4Cr phase, and the only exception is the ω-Al4Cr whose Al:Cr ratio is exactly equal to 4. In addition. the stable ω-Al4Cr should be the final product of the eutectoid reaction as it has the lowest formation energy (see Figure 1).
In the present work, high pressure affects the solidification in synergy with high temperature, which makes the situation more complex. It is essential to explain the pressure-induced disorder-order phase transitions (disordered ε-Al4Cr or/and μ-Al4Cr to the ordered ω-Al4Cr) firstly. The well-known thermodynamic potential difference between ordered and disordered phases at certain temperature T and pressure P is [4]:
where ∆G means the thermodynamic potential difference between that of disordered phase Gdis and ordered phase Gord; ΔE and ΔS are the internal energy and entropy difference between the ordered and disordered phases. If , then the ordered phase is favorable. For simplicity, we take the disordered μ-Al4Cr to compare with the ordered ω-Al4Cr phase as there are only two vacancy positions in the former phase. As the first term is generally satisfied owing to the local distortion, it will enhance the internal energy of the disordered phase. Therefore, will be satisfied even when the sum of the second and third terms equal to zero. Taking the HPS conditions, 5GPa and 1169 K, into consideration, the second term and the third term equals to −3.4 eV and 7.3 eV, respectively. Therefore, is satisfied at HPS conditions in synthesizing the ω-Al4Cr phase, and the ordered ω-Al4Cr phase is more stable than the disordered μ-Al4Cr phase. It can also expect that the critical pressure to ignite the disorder to order phase transition is about 2.3 GPa from the same thermodynamic potential difference calculations. Comparing the ε-Al4Cr and μ-Al4Cr, it is obvious that the former is more disordered as it contains more vacancy positions (7 vs. 2), besides the additional ten co-occupied positions, than the latter. The increasing of entropy caused by the vacancy positions was calculated to be about 118kB, which corresponds to 10.8 eV at 1058 K (the eutectoid reaction temperature). If there exists a phase transition from the ε-Al4Cr to the μ-Al4Cr phase directly, the external pressure required should be around 22.5 GPa. In other words, high pressure (5 GPa) will probably affect the solidification by reducing the diffusion coefficients of the involved atoms, but it has little effect on a possible phase transition from the ε-Al4Cr to the μ-Al4Cr phase. In the following, the peritectic and eutectoid reactions based on the above discussions are reconsidered. It is most likely that the ε-Al4Cr phase appears firstly by the peritectic reaction, and then the μ-Al4Cr phase was produced by the eutectoid reaction: (1) there are more disordered atoms in the ε-Al4Cr and the η-Al11Cr2 phases than that of the μ-Al4Cr phase which has only two vacancy positions, thus the formation energies of the ε-Al4Cr and the η-Al11Cr2 phases should be much closer; (2) the solidification is controlled by the diffusion of the involved atoms, especially the sluggish Al atoms, thus the Al-deficiency ε-Al4Cr phase (the Al:Cr ratio 3.93 and 4.16 for the disordered ε-Al4Cr and the μ-Al4Cr phase, respectively) should be formed firstly; (3) the transformation from the μ-Al4Cr phase to the ω-Al4Cr phase is easily understood from the pressure-induced disorder-order transition mechanism.
In summary, the mechanism of the pressure-induced disorder-order phase transitions in the Al4Cr system have been analyzed. The thermodynamic analysis results suggest that the stable ω-Al4Cr phase is transformed from the μ-Al4Cr phase by the eutectoid reaction accelerated by high-pressure conditions. The ε-Al4Cr phase should form by the peritectic reaction, although its structural building units are nearly indistinguishable from that of the μ-Al4Cr phase. The modified reaction process can also be understood easily by the diffusion-controlled solidification and phase transition. Such findings could be utilized to tune the final products with different phases and different mechanical properties on the one hand, and on the other hand would promote the understanding of the process of solidification including the forming mechanism of the quasicrystal phases in the Al-Cr binary system.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/cryst12071008/s1, Table S1: The Wyckoff positions of the centered atoms inside the Macky or pseudo-Macky cluster for the ε-and μ-Al4Cr phase along with the η-Al11Cr2 phase
Author Contributions
Conceptualization, C.F.; investigation, X.G. and C.F.; writing—original draft preparation, C.F. and X.G.; writing—review and editing, X.G., C.F. and B.W.; project administration, C.F.; funding acquisition, C.F. and B.W. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by: The National Natural Science Foundation of China (grant No. 52173231/51771165), the Natural Science Foundation of Hebei Province (grant No. E2022203182) and Hebei Province Innovation Ability Promotion Project (22567609H).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Simonov, A.; Goodwin, A.L. Designing disorder into crystalline materials. Nat. Rev. Chem. 2020, 4, 657–673. [Google Scholar] [CrossRef]
- Redfem, S.A.T. Order-Disorder Phase Transitions. Rev. Miner. Geochem. 2000, 39, 105–133. [Google Scholar]
- Skakunova, K.D.; Rychkov, D.A. Low Temperature and High-Pressure Study of Bending L-Leucinium Hydrogen Maleate Crystals. Crystals 2021, 11, 1575. [Google Scholar] [CrossRef]
- Errandonea, D. Pressure-Induced Phase Transformations. Crystals 2020, 10, 595. [Google Scholar] [CrossRef]
- Baty, S.; Burakovsky, L.; Errandonea, D. Ab Initio Phase Diagram of Copper. Crystals 2021, 11, 537. [Google Scholar] [CrossRef]
- Anzellini, S.; Burakovsky, L.; Turnbull, R.; Bandiello, E.; Errandonea, D. P–V–T Equation of State of Iridium Up to 80 GPa and 3100 K. Crystals 2021, 11, 452. [Google Scholar] [CrossRef]
- Baty, S.; Burakovsky, L.; Preston, D. Topological Equivalence of the Phase Diagrams of Molybdenum and Tungsten. Crystals 2020, 10, 20. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Fan, J.; Zhang, L.; Zhang, M.; Cui, P.; Dong, W.; Yu, P.; Li, Y.; Liaw, P.K.; Li, G. Pressure-induced ordering phase transition in high-entropy alloy. Intermetallics 2018, 103, 63–66. [Google Scholar] [CrossRef]
- Deng, Z.; Kang, C.J.; Croft, M.; Li, W.M.; Shen, X.; Zhao, J.F.; Yu, R.C.; Jin, C.Q.; Kotliar, G.; Liu, S.Z.; et al. A Pressure-Induced Inverse Order–Disorder Transition in Double Perovskites. Angew. Chem. 2020, 132, 2–9. [Google Scholar]
- Fedorov, A.Y.; Rychkov, D.A. Comparison of different computational approaches for unveiling the high-pressure behavior of organic crystals at molecular level. Case study of tolazamide polymorphs. J. Struct. Chem. 2020, 61, 1356–1366. [Google Scholar] [CrossRef]
- Colmenero, F. Organic acids under pressure: Elastic properties, negative mechanical phenomena and pressure induced phase transitions in the lactic, maleic, succinic and citric acids. Mater. Adv. 2020, 1, 1399–1426. [Google Scholar] [CrossRef]
- Korabel’nikov, D.V.; Zhuravlev, Y.N. Semi-empirical and ab initio calculations for crystals under pressure at fixed temperatures: The case of guanidinium perchlorate. RSC Adv. 2020, 10, 42204–42211. [Google Scholar] [CrossRef] [PubMed]
- Rychkov, D.A. A Short Review of Current Computational Concepts for High-Pressure Phase Transition Studies in Molecular Crystals. Crystals 2020, 10, 81. [Google Scholar] [CrossRef] [Green Version]
- Audier, M.; Durand-Charre, M.; Laclau, E.; Klein, H. Phase equilibria in the Al-Cr system. J. Alloys Compd. 1995, 220, 225–230. [Google Scholar] [CrossRef]
- Li, X.Z.; Sugiyama, K.; Hiraga, K.; Sato, A.; Yamamoto, A.; Sui, H.X.; Kuo, K.H. Crystal structure of orthorhombic ε-Al4Cr. Zeitschrift Für Krist. Cryst. Mater. 1997, 212, 628–633. [Google Scholar] [CrossRef]
- Wen, K.Y.; Chen, Y.L.; Kuo, K.H. Crystallographic AI4Cr Crystalline Relationships of the and Quasicrystalline Phases. Metall. Trans. a-Phys. Metall. Mater. Sci. 1992, 23, 2437–2445. [Google Scholar] [CrossRef]
- Cao, B.; Kuo, K. Crystal structure of the monoclinic η-Al11Cr2. J. Alloys Compd. 2008, 458, 238–247. [Google Scholar] [CrossRef]
- Cao, B. On the structure of the hexagonal μ-Al 4 Cr and its relation to the monoclinic η-Al 11 Cr 2. J. Alloys Compd. 2017, 698, 605–610. [Google Scholar] [CrossRef]
- Fan, C.Z.; Liu, C. Crystal structure of ω-Al4Cr. IUCrData 2018, 3, x180216. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B (Condens. Matter) 1996, 54, 11169–11181. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B Condens. Matter Mater. Phys. 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Kirklin, S.; Saal, J.E.; Meredig, B.; Thompson, A.; Doak, J.W.; Aykol, M.; Rühl, S.; Wolverton, C.M. The Open Quantum Materials Database (OQMD): Assessing the accuracy of DFT formation energies. NPJ Comput. Mater. 2015, 1, 15010. [Google Scholar] [CrossRef] [Green Version]
- Pankova, A.A.; Blatov, V.A.; Ilyushin, G.D.; Proserpio, D.M. Ilyushin, γ-Brass polyhedral core in intermetallics: The nanocluster model. Inorg. Chem. 2013, 52, 13094–13107. [Google Scholar] [CrossRef]
- Akhmetshina, T.; Blatov, V.A. A fascinating building unit: Mackay cluster in intermetallics. Struct. Chem. 2016, 28, 133–140. [Google Scholar] [CrossRef]
- Akhmetshina, T.G.; Blatov, V.A. Topological methods for complex intermetallics. Zeitschrift für Kristallographie—Cryst. Mater. 2017, 232, 497–506. [Google Scholar] [CrossRef]
- Akhmetshina, T.G.; Blatov, V.A.; Proserpio, D.M.; Shevchenko, A.P. Topology of intermetallic structures: From statistics to rational design. Acc. Chem. Res. 2018, 51, 21–30. [Google Scholar] [CrossRef]
- Jha, D.; Ward, L.; Paul, A.; Liao, W.K.; Choudhary, A.; Wolverton, C.; Agrawal, A. ElemNet: Deep Learning the Chemistry of Materials From Only Elemental Composition. Sci. Rep. 2018, 8, 17593. [Google Scholar] [CrossRef]
- Legrain, F.; Carrete, J.; van Roekeghem, A.; Curtarolo, S.; Mingo, N. How Chemical Composition Alone Can Predict Vibrational Free Energies and Entropies of Solids. Chem. Mater. 2017, 29, 6220–6227. [Google Scholar] [CrossRef] [Green Version]
- Isayev, O.; Oses, C.; Toher, C.; Gossett, E.; Curtarolo, S.; Tropsha, A. Universal fragment descriptors for predicting properties of inorganic crystals. Nat. Commun. 2017, 8, 15679. [Google Scholar] [CrossRef] [PubMed]
- Anzellini, S.; Errandonea, D.; Burakovsky, L.; Proctor, J.E.; Turnbull, R.; Beavers, C.M. Characterization of the high-pressure and high-temperature phase diagram and equation of state of chromium. Sci. Rep. 2022, 12, 6727. [Google Scholar] [CrossRef] [PubMed]
- Dewaele, A.; Loubeyre, P.; Mezouar, M. Equations of state of six metals above 94GPa. Phys. Rev. B 2004, 70, 094112. [Google Scholar] [CrossRef] [Green Version]
- Cao, B.B. Structures of Quasicrystal Approximants in Al-Cr Alloys. Ph.D. Thesis, Dalian University Technology, Dalian, China, 2007. (In Chinese). [Google Scholar]
Figure 1.
Convhull of the Al-Cr system by first-principles calculations with the proposed phase transition sequences for the ε-, μ- and ω-Al4Cr phases in the present work.
Figure 1.
Convhull of the Al-Cr system by first-principles calculations with the proposed phase transition sequences for the ε-, μ- and ω-Al4Cr phases in the present work.

Figure 2.
The entire unit cell of ε-Al4Cr structure composed of six cluster units.

Figure 3.
The entire unit cell of μ-Al4Cr structure composed of six cluster units.

Figure 4.
The 1@12@46 nanoclusters found in (a) ε- Al4Cr and (b) μ- Al4Cr.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Formation Energy (eV/atom), vibrational free energy and entropy, bulk modulus and shear modulus of related phases in the Al-Cr system by the deep neural network model.
Table 1.
Formation Energy (eV/atom), vibrational free energy and entropy, bulk modulus and shear modulus of related phases in the Al-Cr system by the deep neural network model.
| Phases | Stoichio-Metric | Al:Cr Ratio | Ef a | Fvib/Svib b | B (GPa) c | G (GPa) c |
|---|---|---|---|---|---|---|
| ε-Al4Cr | 472:112 (449.1:114.3) | 4.21 (3.93) | −0.167 | 12.43 | 110.70 | 79.18 |
| μ-Al4Cr | 464:110 (457.6:110) | 4.22 (4.16) | −0.167 | 12.43 | 106.01 | 76.97 |
| η-Al11Cr2 | 516:100 (516:100) | 5.16 (5.16) | −0.15 | 12.90 | 105.77 | 62.76 |
| ω-Al4Cr | 24:6 | 4.0 | −0.167 | 12.29 | 121.65 | 81.56 |
| θ-Al45Cr7 | 90:14 | 6.43 | −0.13 | 13.34 | 109.23 | 56.82 |
a Reference [29]; b Reference [30]; c Reference [31]. For the method introduced in Reference [20], the calculations were carried out on the website: http://info.eecs.northwestern.edu/FEpredictor (only the stoichiometric of compounds is required as input, accessed on 13 July 2022). For the method introduced in References [30,31], the calculations were carried out on the website: https://aflow.org/aflow-ml/, (only the structural information in the VASP format is required as input, accessed on 13 July 2022).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fan, C.; Geng, X.; Wen, B. Pressure Induced Disorder-Order Phase Transitions in the Al4Cr Phases. Crystals 2022, 12, 1008. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12071008
AMA Style
Fan C, Geng X, Wen B. Pressure Induced Disorder-Order Phase Transitions in the Al4Cr Phases. Crystals. 2022; 12(7):1008. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12071008
Chicago/Turabian StyleFan, Changzeng, Xu Geng, and Bin Wen. 2022. "Pressure Induced Disorder-Order Phase Transitions in the Al4Cr Phases" Crystals 12, no. 7: 1008. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12071008
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.