
Multicore Assemblies from Three-Component Linear Homo-Copolymer Systems: A Coarse-Grained Modeling Study

 ,
,  and
and
Abstract
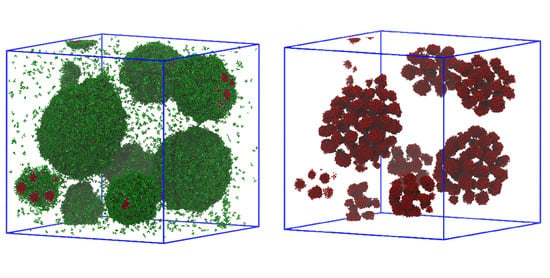
:
1. Introduction
2. Materials and Methods
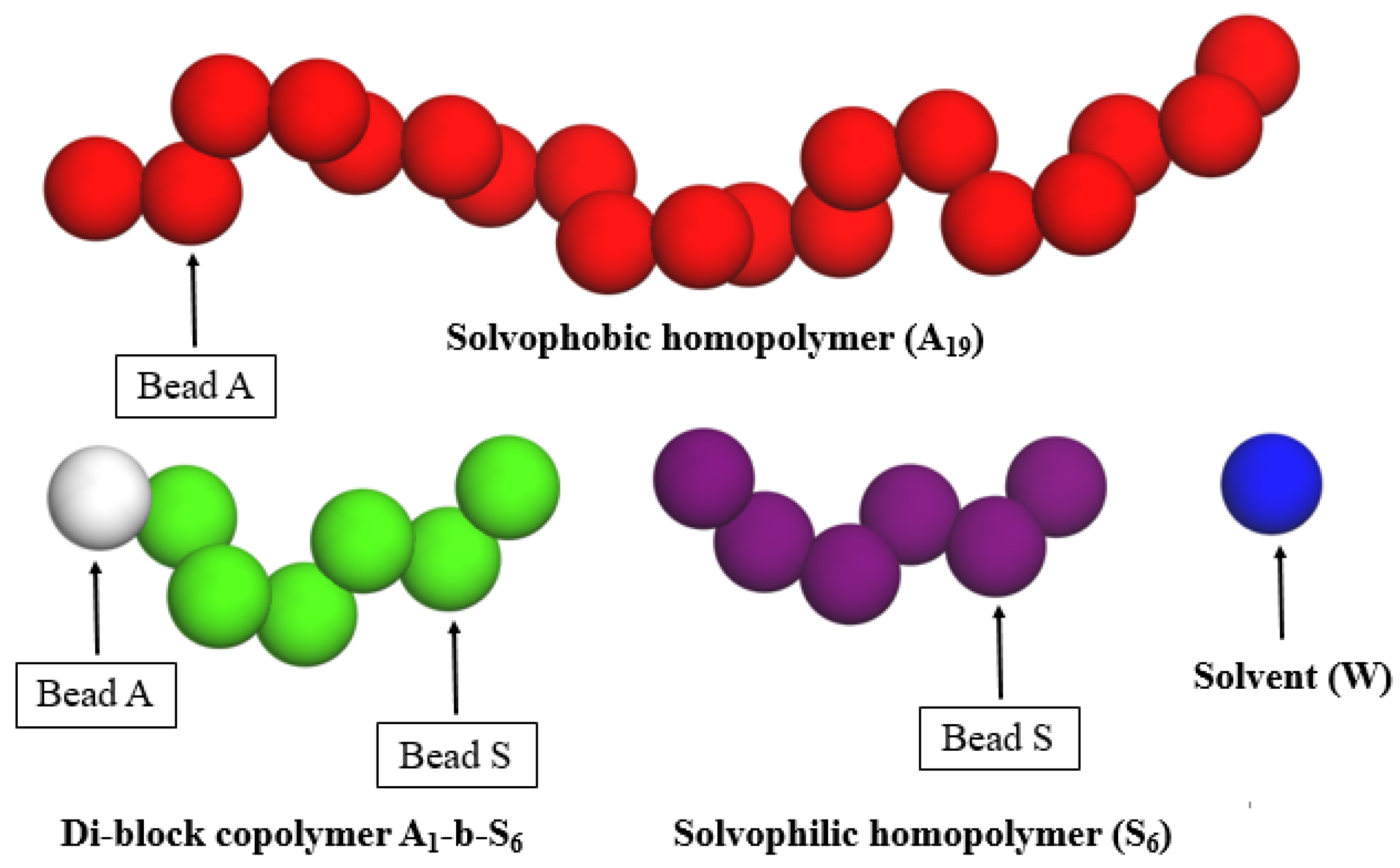
2.1. Simulation Details
2.2. Characterization of Self-Assembled Structures
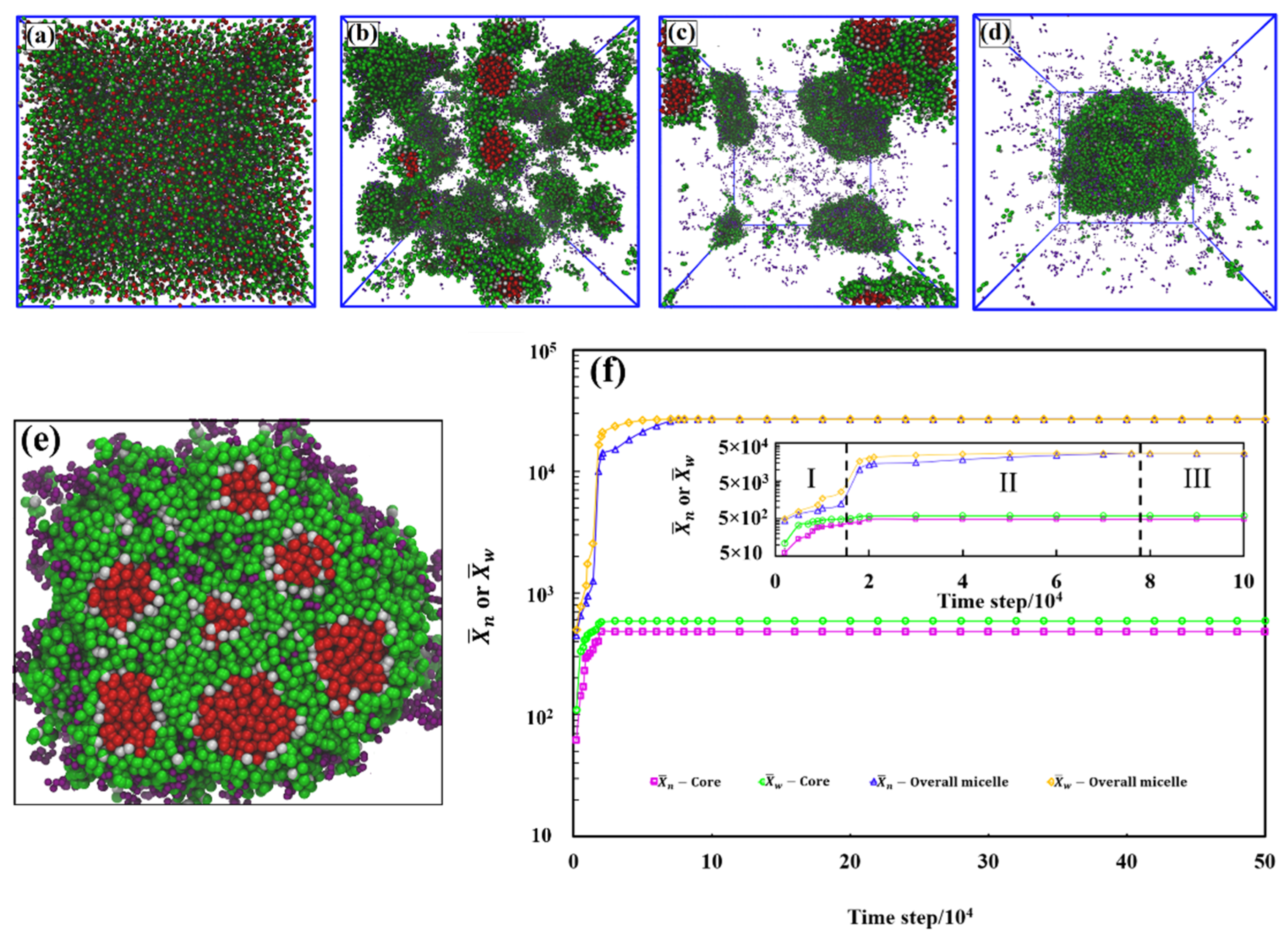
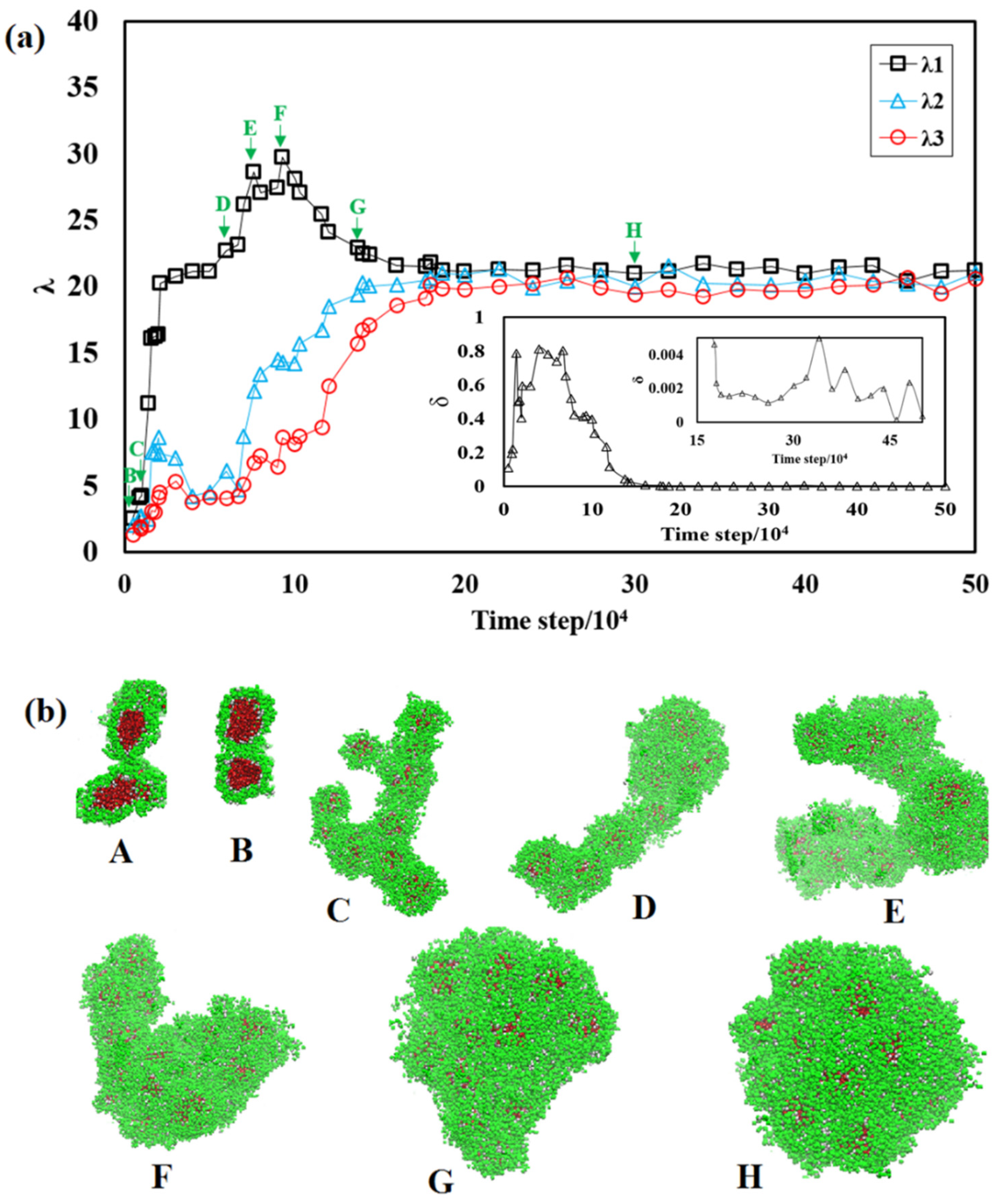
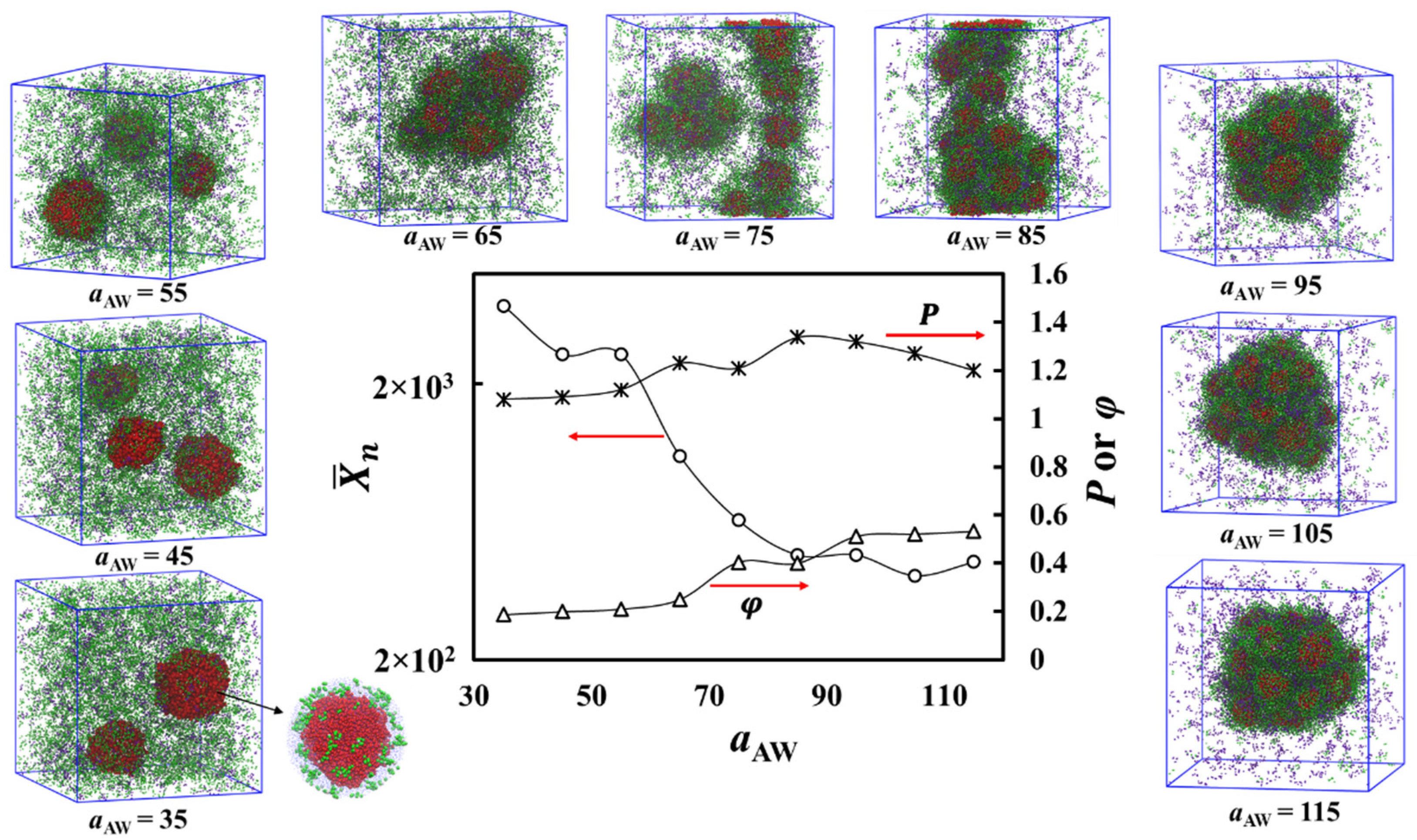
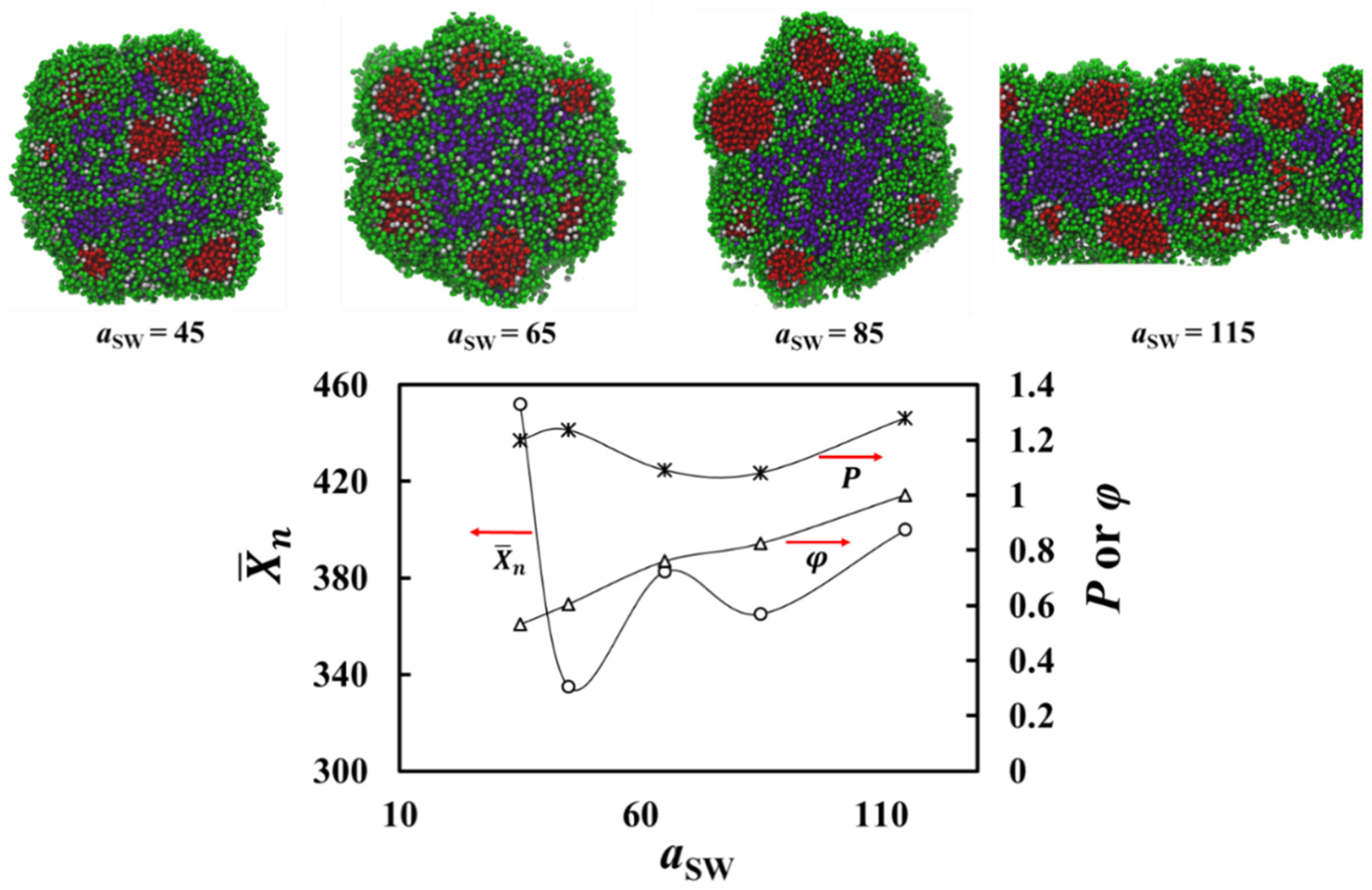
3. Results
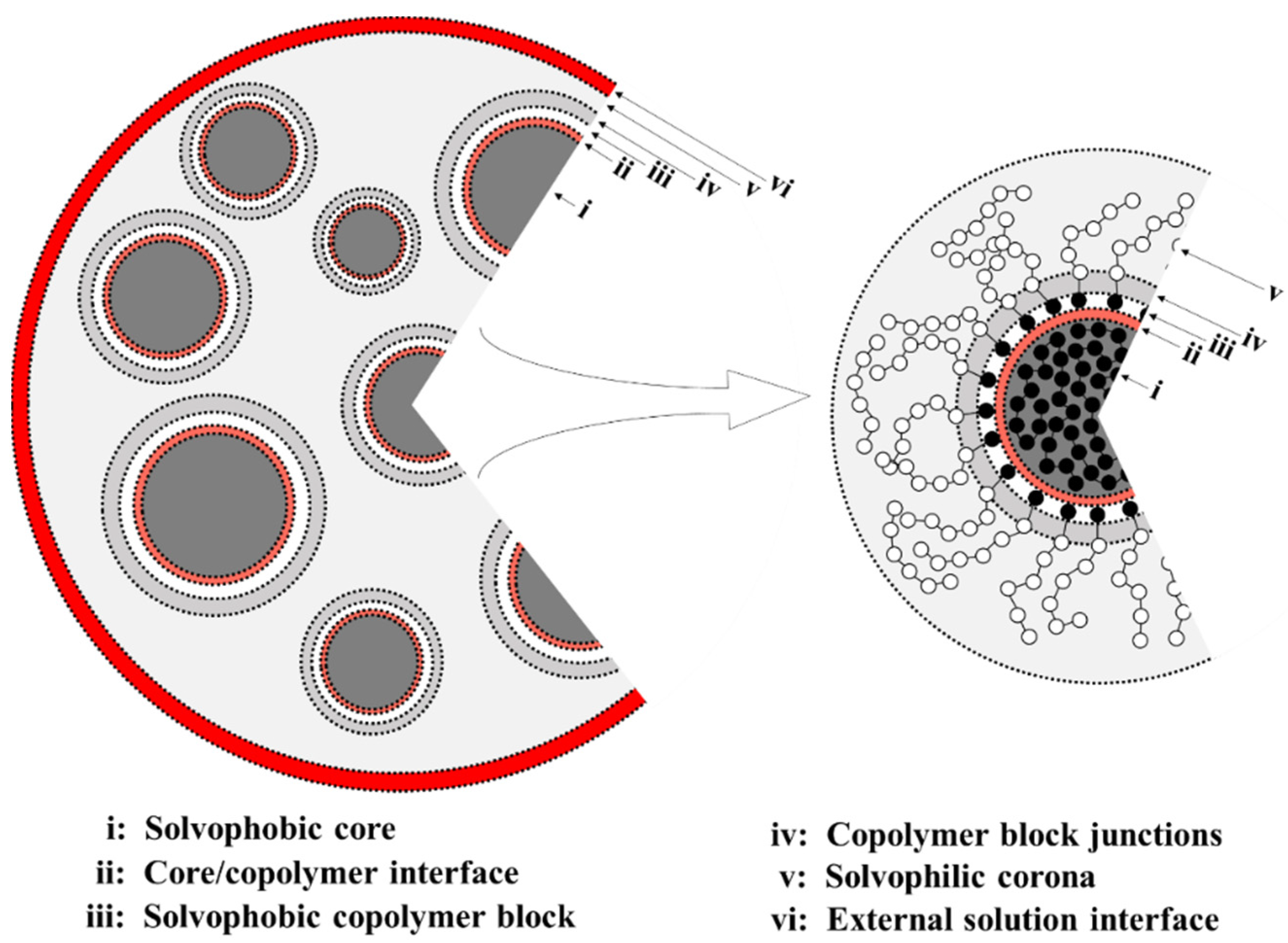
Theoretical Arguments
- (i) Cores filled with solvophobic-free homopolymer A19 with a negative free energy contribution, which dominates here, favoring aggregation of A19 into the core.
- (ii)–(iii) An interface between the core surface and the solvophobic A blocks of the A1-b-S6 copolymers that surround the cores. Here, the free energy has a positive contribution, for the interface between the solvophobic core and the solvent, and a negative contribution, , resulting from removing core-solvent contacts by the A1-b-S6 copolymers.
- (iv)–(v) A corona composed mainly of the solvophilic copolymer blocks and the S6 homopolymer. The free energy contribution is negative and comprises of and . This allows the solvophilic S beads to form the large corona.
- (vi) The external interface between the aggregate and solution mainly composed of solvophilic homopolymers with interface energy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moreno, N.; Nunes, S.P.; Peinemann, K.-V.; Calo, V.M. Topology and Shape Control for Assemblies of Block Copolymer Blends in Solution. Macromology 2015, 48, 8036–8044. [Google Scholar] [CrossRef] [Green Version]
- Callaway, C.P.; Lee, S.M.; Mallard, M.; Clark, B.; Jang, S.S. Effect of Block Length and Side Chain Length Ratios on Determining a Multicompartment Micelle Structure. J. Phys. Chem. B 2019, 123, 4784–4791. [Google Scholar] [CrossRef]
- Zhao, Y.; You, L.-Y.; Lu, Z.-Y.; Sun, C.-C. Dissipative particle dynamics study on the multicompartment micelles self-assembled from the mixture of diblock copolymer poly(ethyl ethylene)-block-poly(ethylene oxide) and homopolymer poly(propylene oxide) in aqueous solution. Polymers 2009, 50, 5333–5340. [Google Scholar] [CrossRef]
- Boucher-Jacobs, C.; Rabnawaz, M.; Katz, J.S.; Even, R.; Guironnet, D. Encapsulation of catalyst in block copolymer micelles for the polymerization of ethylene in aqueous medium. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef]
- Shum, H.C.; Zhao, Y.-J.; Kim, S.-H.; Weitz, D.A. Multicompartment Polymersomes from Double Emulsions. Angew. Chem. Int. Ed. 2011, 50, 1648–1651. [Google Scholar] [CrossRef]
- Cuomo, F.; Ceglie, A.; De Leonardis, A.; Lopez, F. Polymer Capsules for Enzymatic Catalysis in Confined Environments. Catalyst 2018, 9, 1. [Google Scholar] [CrossRef] [Green Version]
- Peters, R.J.R.W.; Marguet, M.; Marais, S.; Fraaije, M.W.; Van Hest, J.C.M.; Lecommandoux, S. Cascade Reactions in Multicompartmentalized Polymersomes. Angew. Chem. Int. Ed. 2014, 53, 146–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larrañaga, A.; Lomora, M.; Sarasua, J.-R.; Palivan, C.; Pandit, A. Polymer capsules as micro-/nanoreactors for therapeutic applications: Current strategies to control membrane permeability. Prog. Mater. Sci. 2017, 90, 325–357. [Google Scholar] [CrossRef]
- Godoy-Gallardo, M.; Labay, C.; Trikalitis, V.D.; Kempen, P.; Larsen, J.B.; Andresen, T.L.; Hosta-Rigau, L. Multicompartment Artificial Organelles Conducting Enzymatic Cascade Reactions inside Cells. ACS Appl. Mater. Interfaces 2017, 9, 15907–15921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durán, M.J.Y.; Godoy-Gallardo, M.; Labay, C.; Urquhart, A.; Andresen, T.L.; Hosta-Rigau, L. Recent advances in compartmentalized synthetic architectures as drug carriers, cell mimics and artificial organelles. Colloids Surf. B Biointerfaces 2017, 152, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Marguet, M.; Bonduelle, C.; Lecommandoux, S. Multicompartmentalized polymeric systems: Towards biomimetic cellular structure and function. Chem. Soc. Rev. 2013, 42, 512–529. [Google Scholar] [CrossRef]
- Longstreet, A.R.; McQuade, D.T. Organic Reaction Systems: Using Microcapsules and Microreactors to Perform Chemical Synthesis. Acc. Chem. Res. 2012, 46, 327–338. [Google Scholar] [CrossRef]
- Lu, J.; Dimroth, J.; Weck, M. Compartmentalization of Incompatible Catalytic Transformations for Tandem Catalysis. J. Am. Chem. Soc. 2015, 137, 12984–12989. [Google Scholar] [CrossRef]
- Shi, J.; Zhang, L.; Jiang, Z. Facile Construction of Multicompartment Multienzyme System through Layer-by-Layer Self-Assembly and Biomimetic Mineralization. ACS Appl. Mater. Interfaces 2011, 3, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Womble, C.T.; Kuepfert, M.; Weck, M. Multicompartment Polymeric Nanoreactors for Non-Orthogonal Cascade Catalysis. Macromol. Rapid Commun. 2019, 40, e1800580. [Google Scholar] [CrossRef]
- Cho, H.; Lai, T.C.; Tomoda, K.; Kwon, G.S. Polymeric Micelles for Multi-Drug Delivery in Cancer. AAPS PharmSciTech 2014, 16, 10–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Yu, H.; Mu, H.; Li, G.; Shen, Y. Novel multicore niosomes based on double pH-sensitive mixed micelles for Ginsenoside Rh2 delivery. Artif. Cells Nanomed. Biotechnol. 2013, 42, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Hillmyer, M.A.; Lodge, T.P. Evolution of Multicompartment Micelles to Mixed Corona Micelles Using Solvent Mixtures. Langmuir 2008, 24, 12001–12009. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Liu, D.; Zhong, C. Multicompartment micelles and vesicles from π-shaped ABC block copolymers: A dissipative particle dynamics study. Phys. Chem. Chem. Phys. 2007, 9, 5267–5273. [Google Scholar] [CrossRef]
- Liu, D.; Zhong, C. Multicompartment micelles formed from star-dendritic triblock copolymers in selective solvents: A dissipative particle dynamics study. Polymers 2008, 49, 1407–1413. [Google Scholar] [CrossRef]
- Kubowicz, S.; Baussard, J.-F.; Lutz, J.-F.; Thünemann, A.F.; Von Berlepsch, H.; Laschewsky, A. Multicompartment Micelles Formed by Self-Assembly of Linear ABC Triblock Copolymers in Aqueous Medium. Angew. Chem. Int. Ed. 2005, 44, 5262–5265. [Google Scholar] [CrossRef] [PubMed]
- Duxin, N.; Liu, F.; Vali, H.; Eisenberg, A. Cadmium Sulphide Quantum Dots in Morphologically Tunable Triblock Copolymer Aggregates. J. Am. Chem. Soc. 2005, 127, 10063–10069. [Google Scholar] [CrossRef]
- Thünemann, A.F.; Kubowicz, S.; Von Berlepsch, H.; Möhwald, H. Two-Compartment Micellar Assemblies Obtained via Aqueous Self-Organization of Synthetic Polymer Building Blocks. Langmuir 2006, 22, 2506–2510. [Google Scholar] [CrossRef]
- Berlepsch, H.V.; Böttcher, C.; Skrabania, K.; Laschewsky, A. Complex domain architecture of multicompartment micelles from a linear ABC triblock copolymer revealed by cryogenic electron tomography. Chem. Commun. 2009, 2290–2292. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Kesselman, E.; Talmon, Y.; Hillmyer, M.A.; Lodge, T.P. Multicompartment Micelles from ABC Miktoarm Stars in Water. Science 2004, 306, 98–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iatridi, Z.; Tsitsilianis, C. pH responsive self assemblies from an An-core-(B-b-C)n heteroarm star block terpolymer bearing oppositely charged segments. Chem. Commun. 2011, 47, 5560–5562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, M.; Hashidzume, A.; Sato, T. Unicore−Multicore Transition of the Micelle Formed by an Amphiphilic Alternating Copolymer in Aqueous Media by Changing Molecular Weight. Macromology 2011, 44, 2970–2977. [Google Scholar] [CrossRef]
- Greenall, M.J.; Schuetz, P.; Furzeland, S.; Atkins, D.; Buzza, D.M.A.; Butler, M.F.; McLeish, T.C.B. Controlling the Self-Assembly of Binary Copolymer Mixtures in Solution through Molecular Architecture. Macromology 2011, 44, 5510–5519. [Google Scholar] [CrossRef] [Green Version]
- Schuetz, P.; Greenall, M.J.; Bent, J.; Furzeland, S.; Atkins, D.; Butler, M.F.; McLeish, T.C.B.; Buzza, D.M.A. Controlling the micellar morphology of binary PEO–PCL block copolymers in water–THF through controlled blending. Soft Matter 2010, 7, 749–759. [Google Scholar] [CrossRef] [Green Version]
- Hannon, A.F.; Sunday, D.F.; Bowen, A.; Khaira, G.; Ren, J.; Nealey, P.F.; De Pablo, J.J.; Kline, R.J. Optimizing self-consistent field theory block copolymer models with X-ray metrology. Mol. Syst. Des. Eng. 2018, 3, 376–389. [Google Scholar] [CrossRef]
- Wang, R.; Jiang, Z.; Xue, G. Excluded volume effect on the self-assembly of amphiphilic AB diblock copolymer in dilute solution. Polymers 2011, 52, 2361–2365. [Google Scholar] [CrossRef]
- Daoulas, K.C.; Müller, M.; De Pablo, J.J.; Nealey, P.F.; Smith, G.D. Morphology of multi-component polymer systems: Single chain in mean field simulation studies. Soft Matter 2006, 2, 573–583. [Google Scholar] [CrossRef]
- Greenall, M.J.; Buzza, D.M.A.; McLeish, T. Micelle Formation in Block Copolymer/Homopolymer Blends: Comparison of Self-Consistent Field Theory with Experiment and Scaling Theory. Macromology 2009, 42, 5873–5880. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Lin, J. Discovering multicore micelles: Insights into the self-assembly of linear ABC terpolymers in midblock-selective solvents. Soft Matter 2011, 7, 3383–3391. [Google Scholar] [CrossRef]
- Kim, S.H.; Jo, W.H. A Monte Carlo simulation for the micellization of ABA- and BAB-type triblock copolymers in a selective solvent. II. Effects of the block composition. J. Chem. Phys. 2002, 117, 8565–8572. [Google Scholar] [CrossRef]
- Koga, T. Multicanonical Monte Carlo simulations on intramolecular micelle formation in copolymers. Eur. Phys. J. E 2005, 17, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, N.; Maeda, S.; Ishii, S.; Ohno, K. A Monte Carlo Simulation of the Formation of Micelles in a Ternary System of Water, Oil and Amphiphilic Polymers. Mater. Trans. 2007, 48, 653–657. [Google Scholar] [CrossRef] [Green Version]
- Patti, A. Monte Carlo simulations of self-assembling star-block copolymers in dilute solutions. Colloids Surf. A Physicochem. Eng. Asp. 2010, 361, 81–89. [Google Scholar] [CrossRef]
- Hafezi, M.-J.; Sharif, F. Brownian Dynamics Simulation of Comicellization of Amphiphilic Block Copolymers with Different Tail Lengths. Langmuir 2012, 28, 16243–16253. [Google Scholar] [CrossRef]
- Moultos, O.; Gergidis, L.N.; Vlahos, C. Brownian Dynamics Simulations on Self-Assembly Behavior of H-Shaped Copolymers and Terpolymers. Macromology 2010, 43, 6903–6911. [Google Scholar] [CrossRef]
- Georgiadis, C.; Moultos, O.; Gergidis, L.N.; Vlahos, C. Brownian Dynamics Simulations on the Self-Assembly Behavior of AB Hybrid Dendritic−Star Copolymers. Langmuir 2011, 27, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gao, J.; Ustach, V.; Li, C.; Sun, S.; Hu, S.; Faller, R. Tunable Permeability of Cross-Linked Microcapsules from pH-Responsive Amphiphilic Diblock Copolymers: A Dissipative Particle Dynamics Study. Langmuir 2017, 33, 7288–7297. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Sun, S.; Li, C.; Hu, S.; Faller, R. Controllable multicompartment morphologies from cooperative self-assembly of copolymer–copolymer blends. Soft Matter 2017, 13, 5877–5887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Jiang, T.; Lin, J.; Cai, C. Toroid Formation through Self-Assembly of Graft Copolymer and Homopolymer Mixtures: Experimental Studies and Dissipative Particle Dynamics Simulations. Langmuir 2013, 29, 8417–8426. [Google Scholar] [CrossRef]
- Šindelka, K.; Limpouchová, Z.; Lísal, M.; Procházka, K. Dissipative Particle Dynamics Study of Electrostatic Self-Assembly in Aqueous Mixtures of Copolymers Containing One Neutral Water-Soluble Block and One Either Positively or Negatively Charged Polyelectrolyte Block. Macromology 2014, 47, 6121–6134. [Google Scholar] [CrossRef]
- Prhashanna, A.; Khan, S.A.; Chen, S.B. Co-Micellization Behavior in Poloxamers: Dissipative Particle Dynamics Study. J. Phys. Chem. B 2015, 119, 572–582. [Google Scholar] [CrossRef]
- Vuorte, M.; Määttä, J.; Sammalkorpi, M. Simulations Study of Single-Component and Mixed n-Alkyl-PEG Micelles. J. Phys. Chem. B 2018, 122, 4851–4860. [Google Scholar] [CrossRef] [Green Version]
- Murat, M.; Grest, G.S.; Kremer†, K. Statics and Dynamics of Symmetric Diblock Copolymers: A Molecular Dynamics Study. Macromology 1999, 32, 595–609. [Google Scholar] [CrossRef]
- Chakraborty, K.; Shinoda, W.; Loverde, S.M. Molecular simulation of the shape deformation of a polymersome. Soft Matter 2020, 16, 3234–3244. [Google Scholar] [CrossRef]
- Patel, S.; Lavasanifar, A.; Choi, P. Application of Molecular Dynamics Simulation To Predict the Compatability between Water-Insoluble Drugs and Self-Associating Poly(ethylene oxide)-b-poly(ε-caprolactone) Block Copolymers. Biomacromolecules 2008, 9, 3014–3023. [Google Scholar] [CrossRef]
- Wengenmayr, M.; Dockhorn, R.; Sommer, J.-U. Multicore Unimolecular Structure Formation in Single Dendritic–Linear Copolymers under Selective Solvent Conditions. Macromology 2016, 49, 9215–9227. [Google Scholar] [CrossRef]
- Nie, S.Y.; Sun, Y.; Lin, W.J.; Wu, W.S.; Guo, X.D.; Qian, Y.; Zhang, L.J. Dissipative Particle Dynamics Studies of Doxorubicin-Loaded Micelles Assembled from Four-Arm Star Triblock Polymers 4AS-PCL-b-PDEAEMA-b-PPEGMA and their pH-Release Mechanism. J. Phys. Chem. B 2013, 117, 13688–13697. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, X.; Zhang, L.; He, L. Vesicles from the self-assembly of coil–rod–coil triblock copolymers in selective solvents. Polymers 2014, 55, 2921–2927. [Google Scholar] [CrossRef]
- Chen, H.; Ruckenstein, E. Self-assembly of π-shaped copolymers. Soft Matter 2012, 8, 1327–1333. [Google Scholar] [CrossRef]
- Chen, H.; Ruckenstein, E. Formation and Degradation of Multicomponent Multicore Micelles: Insights from Dissipative Particle Dynamics Simulations. Langmuir 2013, 29, 5428–5434. [Google Scholar] [CrossRef]
- Hoogerbrugge, P.J.; Koelman, J.M.V.A. Simulating Microscopic Hydrodynamic Phenomena with Dissipative Particle Dynamics. EPL Europhys. Lett. 1992, 19, 155–160. [Google Scholar] [CrossRef]
- Groot, R.D.; Warren, P.B. Dissipative particle dynamics: Bridging the gap between atomistic and mesoscopic simulation. J. Chem. Phys. 1997, 107, 4423–4435. [Google Scholar] [CrossRef]
- Wang, Y.; Li, B.; Zhou, Y.; Lu, Z.; Yan, D. Dissipative particle dynamics simulation study on the mechanisms of self-assembly of large multimolecular micelles from amphiphilic dendritic multiarm copolymers. Soft Matter 2013, 9, 3293–3304. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Dormidontova, E.E. Kinetics of Diblock Copolymer Micellization by Dissipative Particle Dynamics. Macromology 2010, 43, 3521–3531. [Google Scholar] [CrossRef]
- Marrink, S.; Tieleman, D.P.; Mark, A.E. Molecular Dynamics Simulation of the Kinetics of Spontaneous Micelle Formation. J. Phys. Chem. B 2000, 104, 12165–12173. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.S.; Yang, Y.Q.; Guo, X.D.; Sun, Y.; Qian, Y.; Zhang, L.J. Mesoscopic simulations on the aggregation behavior of pH-responsive polymeric micelles for drug delivery. J. Colloid Interface Sci. 2011, 363, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Tanford, C. Theory of micelle formation in aqueous solutions. J. Phys. Chem. 1974, 78, 2469–2479. [Google Scholar] [CrossRef]
- Nagarajan, R.; Ruckenstein, E. Aggregation of amphiphiles as micelles or vesicles in aqueous media. J. Colloid Interface Sci. 1979, 71, 580–604. [Google Scholar] [CrossRef]
- Nagarajan, R.; Ruckenstein, E. Theory of surfactant self-assembly: A predictive molecular thermodynamic approach. Langmuir 1991, 7, 2934–2969. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| i/j | A | S | W |
|---|---|---|---|
| A | 25 | 72 | 115 |
| S | 72 | 25 | 30 |
| W | 115 | 30 | 25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Javan Nikkhah, S.; Turunen, E.; Lepo, A.; Ala-Nissila, T.; Sammalkorpi, M. Multicore Assemblies from Three-Component Linear Homo-Copolymer Systems: A Coarse-Grained Modeling Study. Polymers 2021, 13, 2193. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13132193
Javan Nikkhah S, Turunen E, Lepo A, Ala-Nissila T, Sammalkorpi M. Multicore Assemblies from Three-Component Linear Homo-Copolymer Systems: A Coarse-Grained Modeling Study. Polymers. 2021; 13(13):2193. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13132193
Chicago/Turabian StyleJavan Nikkhah, Sousa, Elsi Turunen, Anneli Lepo, Tapio Ala-Nissila, and Maria Sammalkorpi. 2021. "Multicore Assemblies from Three-Component Linear Homo-Copolymer Systems: A Coarse-Grained Modeling Study" Polymers 13, no. 13: 2193. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13132193