
Morphological Effect of Side Chain Length in Sulfonated Poly(arylene ether sulfone)s Polymer Electrolyte Membranes via Molecular Dynamics Simulation

Abstract
:1. Introduction
2. Simulation Methodology
2.1. Atomistic Models and Amorphous Cells Construction
2.2. Simulation Methods
2.3. Model Analysis and Property Calculations
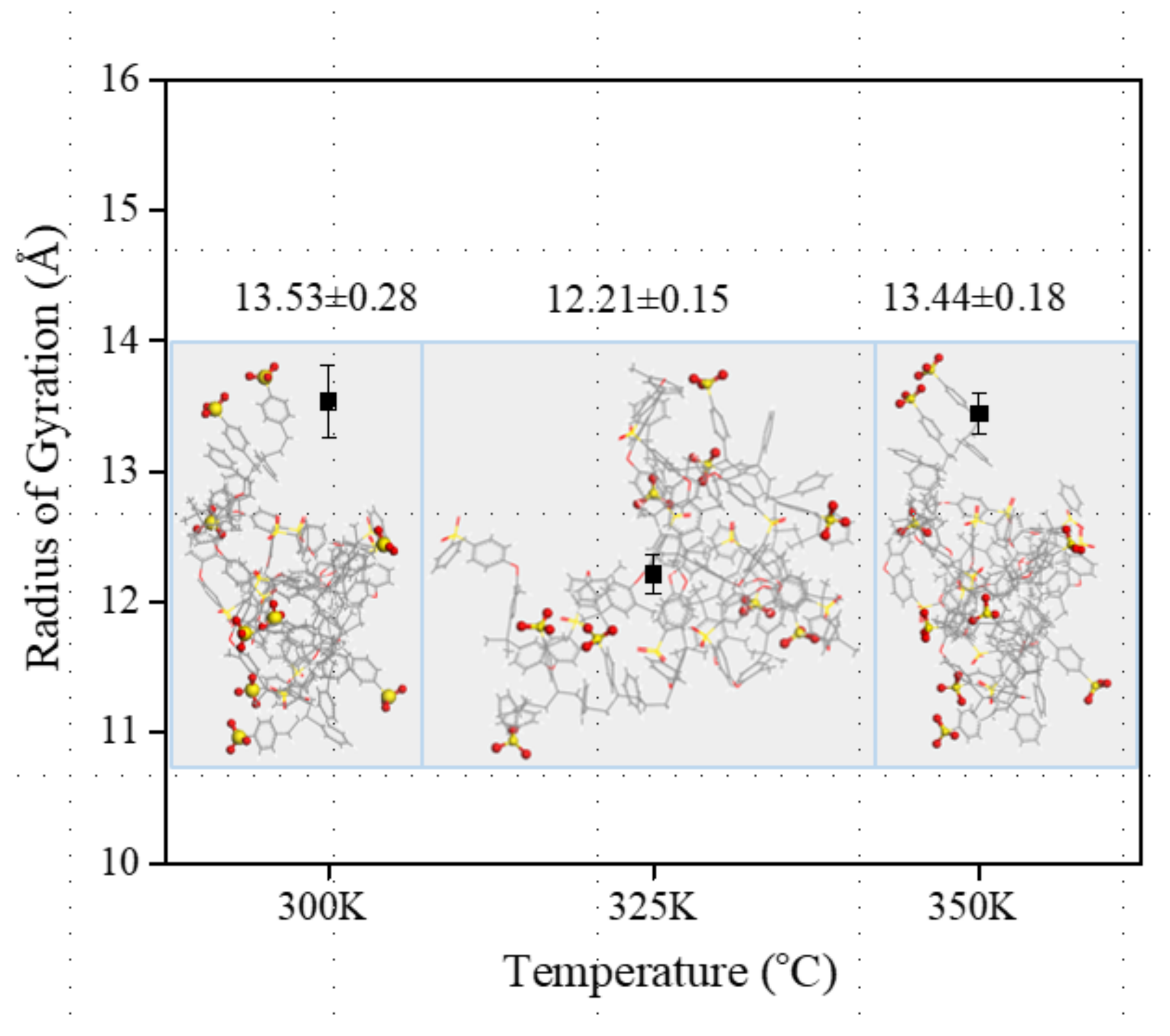
3. Results and Discussions
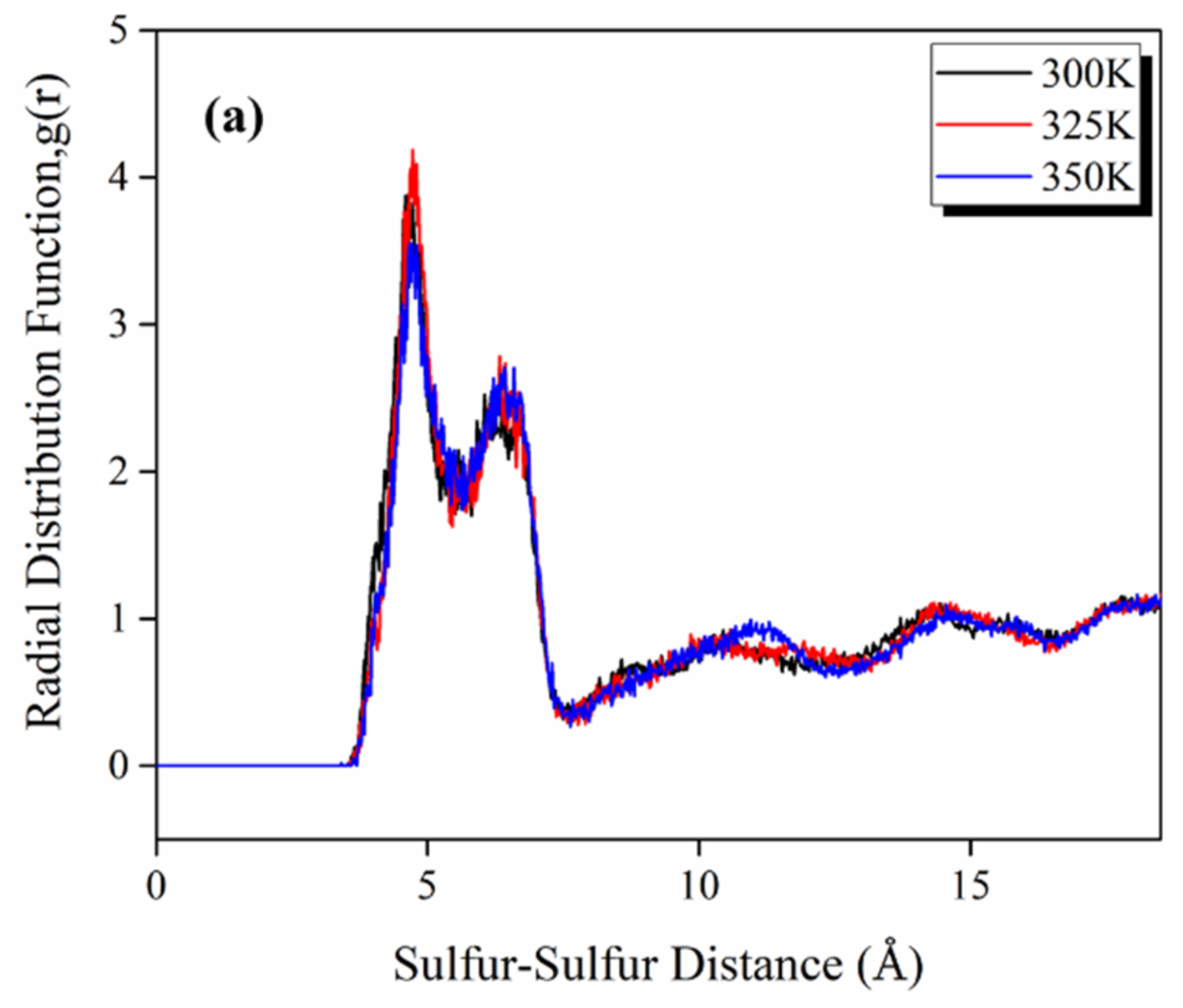
3.1. Amorphous Cell Equilibration
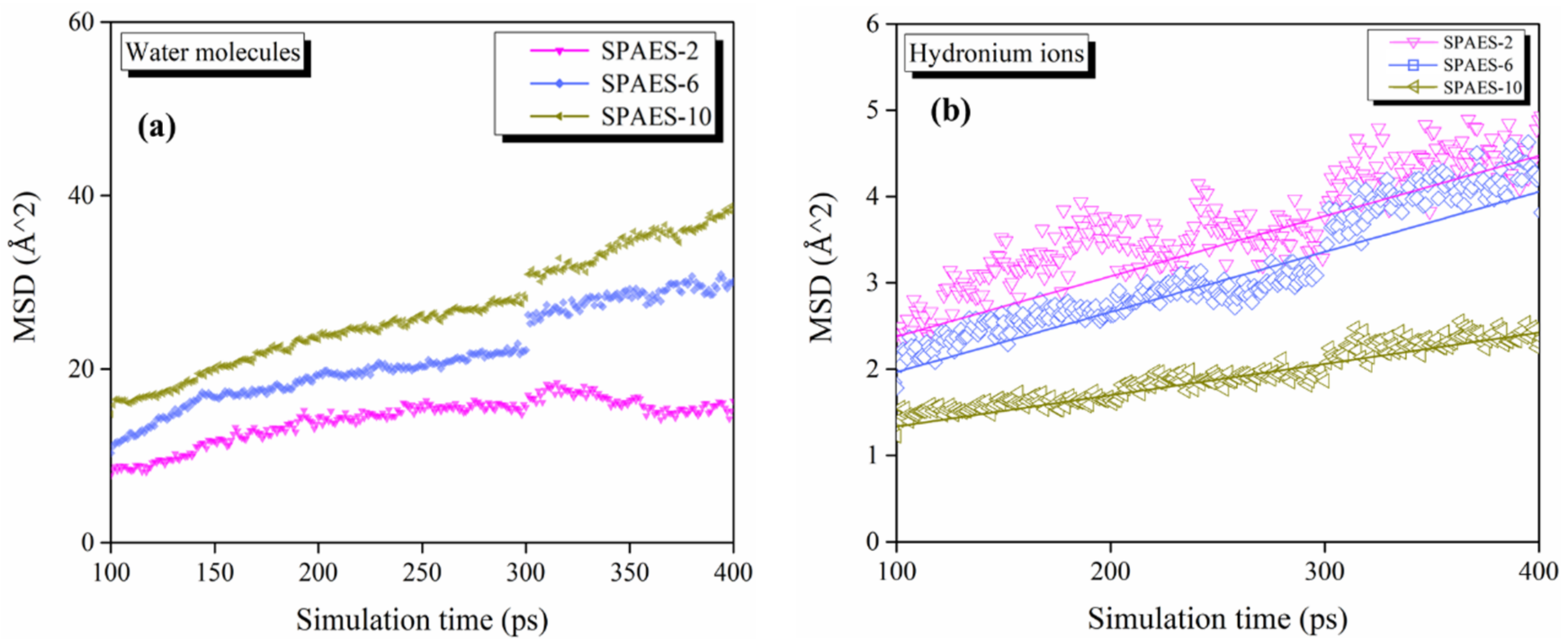
3.2. Numerical Evaluation of Molecular Diffusion
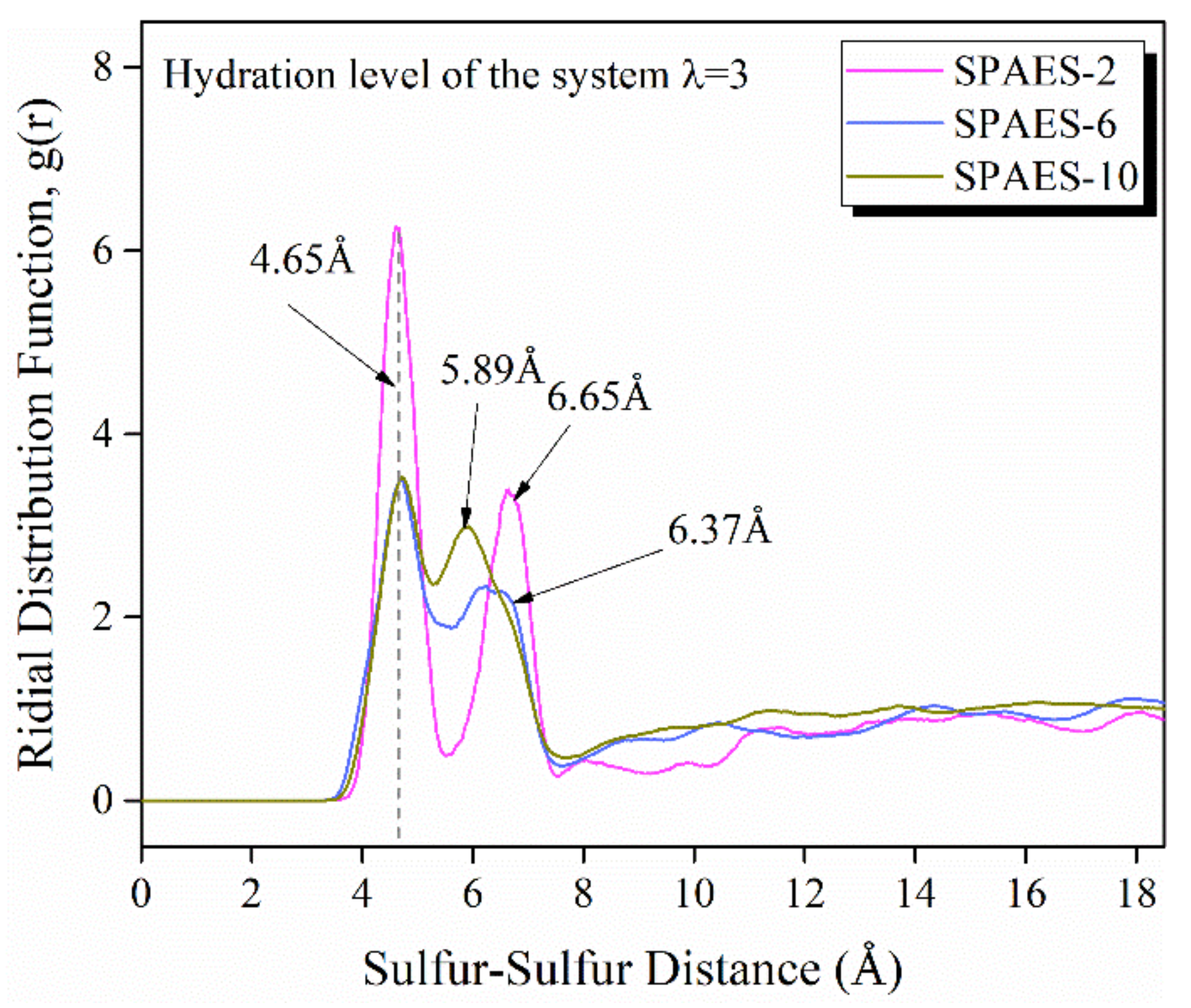
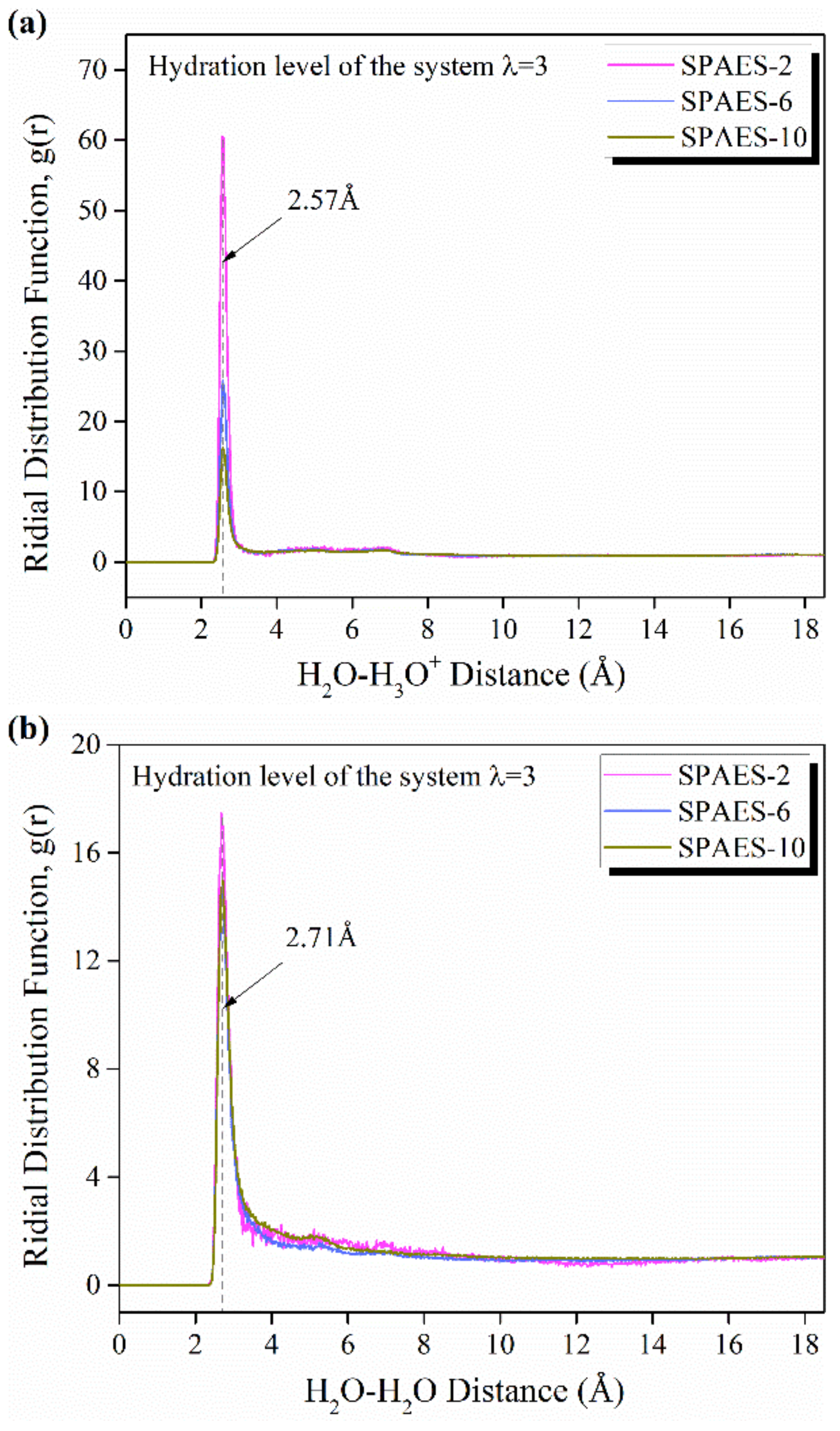
3.3. Morphological Assessment
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Gao, Y.; Robertson, G.P.; Guiver, M.D.; Mikhailenko, S.D.; Li, X.; Kaliaguine, S. Synthesis of poly(arylene ether ether ketone ketone) copolymers containing pendant sulfonic acid groups bonded to naphthalene as proton exchange membrane materials. Macromolecules 2004, 37, 6748–6754. [Google Scholar] [CrossRef]
- Chikashige, Y.; Chikyu, Y.; Miyatake, K.; Watanabe, M. Poly(arylene ether) ionomers containing sulfofluorenyl groups for fuel cell applications. Macromolecules 2005, 38, 7121–7126. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Wu, L.; Xu, T.W. Synthesis and properties of side-chain-type sulfonated poly(phenylene oxide) for proton exchange membranes. J. Membr. Sci. 2011, 373, 160–166. [Google Scholar] [CrossRef]
- Matsumoto, K.; Higashihara, T.; Ueda, M. Locally and densely sulfonated poly(ether sulfone)s as proton exchange membrane. Macromolecules 2009, 42, 1161–1166. [Google Scholar] [CrossRef]
- Feng, S.G.; Shang, Y.M.; Wang, Y.W.; Liu, G.S.; Xie, X.F.; Dong, W.Q.; Xu, J.M.; Mathur, V.K. Synthesis and crosslinking of hydroxyl-functionalized sulfonated poly(ether ether ketone) copolymer as candidates for proton exchange membranes. J. Membr. Sci. 2010, 352, 14–21. [Google Scholar] [CrossRef]
- Awang, N.; Ismail, A.F.; Jaafar, J.; Matsuura, T.; Junoh, H.; Othman, M.H.D.; Rahman, M.A. Functionalization of polymeric materials as a high performance membrane for direct methanol fuel cell: A review. React. Funct. Polym. 2015, 86, 248–258. [Google Scholar] [CrossRef]
- Zhang, X.; Sheng, L.; Hayakawa, T.; Ueda, M.; Higashihara, T. Polymer electrolyte membranes based on poly(phenylene ether)s with sulfonic acid via long alkyl side chains. J. Mater. Chem. A 2013, 1, 11389–11396. [Google Scholar] [CrossRef]
- Zhong, S.L.; Liu, C.H.; Na, H. Preparation and properties of UV irradiation-induced crosslinked sulfonated poly(ether ether ketone) proton exchange membranes. J. Membr. Sci. 2009, 326, 400–407. [Google Scholar] [CrossRef]
- Kim, D.J.; Choi, D.H.; Park, C.H.; Nam, S.Y. Characterization of the sulfonated PEEK/sulfonated nanoparticles composite membrane for the fuel cell application. Int. J. Hydrogen Energy 2016, 41, 5793–5802. [Google Scholar] [CrossRef]
- Wang, F.; Hickner, M.; Ji, Q.; Harrison, W.; Mecham, J.; Zawodzinski, T.A. Synthesis of highly sulfonated poly(arylene ether sulfone) random (statistical) copolymers via direct polymerization. Macromol. Symp. 2001, 175, 387–396. [Google Scholar] [CrossRef]
- Xiao, L.; Chen, X.; Xu, J.J.; Chen, K.C.; Fang, J.H. Synthesis and properties of novel sidechain sulfonated poly(arylene ether sulfone)s for proton exchange membranes. J. Polym. Sci. Part A Polym. Chem. 2019, 57, 2304–2313. [Google Scholar] [CrossRef]
- Lade, H.; Kumar, V.; Arthanareeswaran, G.; Ismail, A.F. Sulfonated poly(arylene ether sulfone) nanocomposite electrolyte membrane for fuel cell applications: A review. Int. J. Hydrogen Energy 2017, 42, 1063–1074. [Google Scholar] [CrossRef]
- Yin, Y.; Du, Q.; Qin, Y.Z.; Zhou, Y.B.; Okamoto, K.I. Sulfonated polyimides with flexible aliphatic side chains for polymer electrolyte fuel cells. J. Membr. Sci. 2011, 367, 211–219. [Google Scholar] [CrossRef]
- Park, C.H.; Lee, S.Y.; Hwang, D.S.; Shin, D.W.; Cho, D.H.; Lee, K.H.; Kim, T.W.; Lee, M.; Kim, D.S. Nanocrack-regulated self-humidifying membranes. Nature 2016, 532, 480–483. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, X.; Wang, Z.Y.; Xie, X.F.; Qian, W. Preparation of graft poly(arylene ether sulfone)s-based copolymer with enhanced phase-separated morphology as proton exchange membranes via atom transfer radical polymerization. Polymers 2019, 11, 1297. [Google Scholar] [CrossRef] [Green Version]
- Haragirimana, A.; Ingabire, P.B.; Zhu, Y.X.; Lu, Y.; Li, N.; Hu, Z.X.; Chen, S.W. Four-polymer blend proton exchange membranes derived from sulfonated poly(aryl ether sulfone)s with various sulfonation degrees for application in fuel cells. J. Membr. Sci. 2019, 583, 209–219. [Google Scholar] [CrossRef]
- Haragirimana, A.; Li, N.; Ingabire, P.B.; Hu, Z.X.; Chen, S.W. Multi-component organic/inorganic blend proton exchange membranes based on sulfonated poly(arylene ether sulfone)s for fuel cells. Polymer 2020, 210, 123015. [Google Scholar] [CrossRef]
- Chen, R.M.; Li, G. New sulfonated poly(arylene ether sulfone) copolymers containing phenyl side chains as proton exchange membranes. New J. Chem. 2016, 40, 3755–3762. [Google Scholar] [CrossRef]
- Ding, J.; Chuy, C.; Holdcroft, S. Solid polymer electrolytes based on ionic graft polymers: Effect of graft chain length on nano-structured, ionic networks. Adv. Funct. Mater. 2002, 12, 389–394. [Google Scholar] [CrossRef]
- Peckham, T.J.; Holdcroft, S. Structure-morphology-property relationships of non-perfluorinated proton-conducting membranes. Adv. Mater. 2010, 22, 4667–4690. [Google Scholar] [CrossRef]
- Shin, D.W.; Guiver, M.D.; Lee, Y.M. Hydrocarbon-based polymer electrolyte membranes: Importance of morphology on ion transport and membrane stability. Chem. Rev. 2017, 117, 4759–4805. [Google Scholar] [CrossRef]
- Kim, K.; Heo, P.; Hwang, W.; Baik, J.; Sung, Y.; Lee, J. Cross-linked sulfonated poly(arylene ether sulfone) containing a flexible and hydrophobic bishydroxy perfluoropolyether crossLinker for high-performance proton exchange membrane. ACS Appl. Mater. Interfaces 2018, 10, 21788–21793. [Google Scholar] [CrossRef]
- Sharma, P.P.; Tinh, V.D.C.; Kim, D. Enhanced ion cluster size of sulfonated poly (arylene ether sulfone) for proton exchange membrane fuel cell application. Polymers 2021, 13, 1111. [Google Scholar] [CrossRef]
- Lee, K.H.; Chu, J.Y.; Mohanraj, V.; Kim, A.R.; Song, M.H.; Yoo, D.J. Enhanced ion conductivity of sulfonated poly(arylene ether sulfone) block copolymers linked by aliphatic chains constructing wide-range ion cluster for proton conducting electrolytes. Int. J. Hydrogen Energy 2020, 45, 29297–29307. [Google Scholar] [CrossRef]
- Yuan, D.; Qin, Y.J.; Li, S.S.; Du, S.H.; Xu, Y.Y.; Weng, Q.; Chen, P.; Chen, X.B.; An, Z.W. Enhanced performance of proton-conducting poly(arylene ether sulfone)s via multiple alkylsulfonated side-chains and block copolymer structures. J. Membr. Sci. 2021, 621, 118932. [Google Scholar] [CrossRef]
- Kreuer, K.D.; Paddison, S.J.; Spohr, E.; Schuster, M. Transport in proton ponductors for fuel-cell applications: Simulations, elementary reactions, and phenomenology. Chem. Rev. 2004, 104, 4637–4678. [Google Scholar] [CrossRef] [Green Version]
- Brandell, D.; Karo, J.; Liivat, A.; Thomas, J.O. Molecular dynamics studies of the Nafion®, Dow® and Aciplex® fuel-cell polymer membrane systems. J. Mol. Model 2007, 13, 1039–1046. [Google Scholar] [CrossRef]
- Sengupta, S.; Pant, R.; Komarov, P.; Venkatnathan, A.; Lyulin, A.V. Atomistic simulation study of the hydrated structure and transport dynamics of a novel multi acid side chain polyelectrolyte membrane. Int. J. Hydrogen Energy 2017, 42, 27254–27268. [Google Scholar] [CrossRef] [Green Version]
- Paddison, S.J.; Elliott, J.A. Molecular modeling of the short-side-chain perfluorosulfonic acid membrane. J. Phys. Chem. A 2005, 109, 7583–7593. [Google Scholar] [CrossRef]
- Paddison, S.J.; Elliott, J.A. On the consequences of side chain flexibility and backbone conformation on hydration and proton dissociation in perfluorosulfonic acid membranes. Phys. Chem. Chem. Phys. 2006, 8, 2193–2203. [Google Scholar] [CrossRef]
- Elliott, J.A.; Paddison, S.J. Modelling of morphology and proton transport in PFSA membranes. Phys. Chem. Chem. Phys. 2007, 9, 2602–2618. [Google Scholar] [CrossRef] [PubMed]
- Hristov, I.H.; Paddison, S.J.; Paul, R. Molecular modeling of proton transport in the short-side-chain perfluorosulfonic acid. J. Phys. Chem. B. 2008, 112, 2937–2949. [Google Scholar] [CrossRef] [PubMed]
- Clark II, J.F.; Paddison, S.J. The effect of side chain connectivity and local hydration on proton transfer in 3M perfluorosulfonic. Solid State Ion. 2012, 213, 83–91. [Google Scholar] [CrossRef]
- Savage, J.; Tse, Y.L.S.; Voth, G.A. Proton transport mechanism of perfluorosulfonic acid membranes. J. Phys. Chem. C 2014, 118, 17436–17445. [Google Scholar] [CrossRef]
- Feng, S.L.; Savage, J.; Voth, G.A. Effects of polymer morphology on proton solvation and transport in proton-exchange membranes. J. Phys. Chem. C 2012, 116, 19104–19116. [Google Scholar] [CrossRef]
- Savage, J.; Voth, G.A. Proton solvation and transport in realistic proton exchange membrane morphologies. J. Phys. Chem. C 2016, 120, 3176–3186. [Google Scholar] [CrossRef]
- Kim, D.J.; Park, C.H.; Nam, S.Y. Molecular dynamics simulations of modified PEEK polymeric membrane for fuel cell application. Int. J. Hydrogen Energy 2016, 41, 7641–7648. [Google Scholar] [CrossRef]
- Khodaparast-Kazeroonian, F.; Amjad-Iranagh, S.; Modarress, H. Molecular dynamics simulation study of carboxylated and sulfonated poly(arylene ether sulfone) membranes for fuel cell applications. Int. J. Hydrogen Energy 2015, 40, 15690–15703. [Google Scholar] [CrossRef]
- Li, X.; Wang, S.B.; Zhang, H.; Lin, C.; Xie, X.F.; Hu, C.X.; Tian, R. Sulfonated poly(arylene ether sulfone)s membranes with distinct microphase-separated morphology for PEMFCs. Int. J. Hydrogen Energy 2021, 46, 33978–33990. [Google Scholar] [CrossRef]
- Bahlakeh, G.; Nikazar, M. Molecular dynamics simulation analysis of hydration effects on microstructure and transport dynamics in sulfonated poly(2,6-dimethyl-1,4-phenylene oxide) fuel cell membranes. Int. J. Hydrogen Energy 2012, 37, 12714–12724. [Google Scholar] [CrossRef]
- Devanathan, R.; Venkatnathan, A.; Dupuis, M. Atomistic simulation of Nafion membrane. 2. Dynamics of water molecules and hydronium ions. J. Phys. Chem. B 2007, 111, 13006–13013. [Google Scholar] [CrossRef] [PubMed]
- Akbari, S.; Hamed Mosavian, M.T.; Moosavi, F.; Ahmadpour, A. Molecular dynamics simulation of Keggin HPA doped Nafion® 117 as a polymer electrolyte membrane. RSC Adv. 2017, 7, 44537–44546. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhao, Y.; Li, W.W.; Wang, S.B.; Liu, X.G.; Xie, X.F.; Chen, J.; Li, Q.F.; Jensen, J.O. Molecular dynamics simulation of radiation grafted FEP films as proton exchange membranes: Effects of the side chain length. Int. J. Hydrogen Energy 2017, 42, 29977–29987. [Google Scholar] [CrossRef]
- Bahlakeh, G.; Nikazar, M.; Hafezi, M.-J.; Dashtimoghadam, E.; Hasani-Sadrabadi, M.M. Molecular dynamics simulation study of proton diffusion in polymer electrolyte membranes based on sulfonated poly (ether ether ketone). Int. J. Hydrogen Energy 2012, 37, 10256–10264. [Google Scholar] [CrossRef]
- Pahari, S.; Choudhury, C.K.; Pandey, P.R.; More, M.; Venkatnathan, A.; Roy, S. Molecular dynamics simulation of phosphoric acid doped monomer of polybenzimidazole: A potential component polymer electrolyte membrane of fuel cell. J. Phys. Chem. B 2012, 116, 7357–7366. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer Cells | SPAES-2 | SPAES-6 | SPAES-10 | |
|---|---|---|---|---|
| Number of Chains | 10 | |||
| Number of SO3− | 30 | 90 | 150 | |
| Number of H3O+ | 30 | 90 | 150 | |
| Number of H2O | 60 | 180 | 300 | |
| Number of atoms | 6680 | 9380 | 12,080 | |
| Volume (Å3) | 76,726.60 | 105,689.08 | 134,630.27 | |
| Temperature (K) | Average | 300.01 | 300.01 | 299.99 |
| Std.Dev | 2.95 | 2.56 | 2.27 | |
| Density (g/cm3) | Average | 1.10 | 1.10 | 1.11 |
| Std.Dev | 0.02 | 0.02 | 0.02 | |
| SPAES-2 | SPAES-6 | SPAES-10 | |
|---|---|---|---|
| DH2O (×10−6 cm2/s) | 0.49 | 1.01 | 1.21 |
| DH3O+ (×10−7 cm2/s) | 1.15 | 1.16 | 0.61 |
| Membranes | SPAES-2 | SPAES-6 | SPAES-10 |
|---|---|---|---|
| O(H3O+)-S(SO3−) | 1.67 | 2.12 | 2.40 |
| O(H2O)-S(SO3−) | 2.45 | 3.51 | 5.66 |
| O(H2O)-O(H2O) | 1.81 | 3.42 | 7.69 |
| O(H2O)-O(H3O+) | 2.77 | 4.62 | 8.09 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Zhang, H.; Lin, C.; Tian, R.; Zheng, P.; Hu, C. Morphological Effect of Side Chain Length in Sulfonated Poly(arylene ether sulfone)s Polymer Electrolyte Membranes via Molecular Dynamics Simulation. Polymers 2022, 14, 5499. https://0-doi-org.brum.beds.ac.uk/10.3390/polym14245499
Li X, Zhang H, Lin C, Tian R, Zheng P, Hu C. Morphological Effect of Side Chain Length in Sulfonated Poly(arylene ether sulfone)s Polymer Electrolyte Membranes via Molecular Dynamics Simulation. Polymers. 2022; 14(24):5499. https://0-doi-org.brum.beds.ac.uk/10.3390/polym14245499
Chicago/Turabian StyleLi, Xue, Hong Zhang, Cheng Lin, Ran Tian, Penglun Zheng, and Chenxing Hu. 2022. "Morphological Effect of Side Chain Length in Sulfonated Poly(arylene ether sulfone)s Polymer Electrolyte Membranes via Molecular Dynamics Simulation" Polymers 14, no. 24: 5499. https://0-doi-org.brum.beds.ac.uk/10.3390/polym14245499