Genome Resequencing Reveals Genetic Variation between the Parents of An Elite Hybrid Upland Cotton

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. DNA Isolation and Sequencing
2.3. Variant Detection and Annotation
2.4. Identification of Variations between 9053 and sGK9708
2.5. Co-localization with Previously Identified QTLs
2.6. Functional Annotation and Gene Expression Analyses
2.7. Validation of SNPs and InDels
3. Results
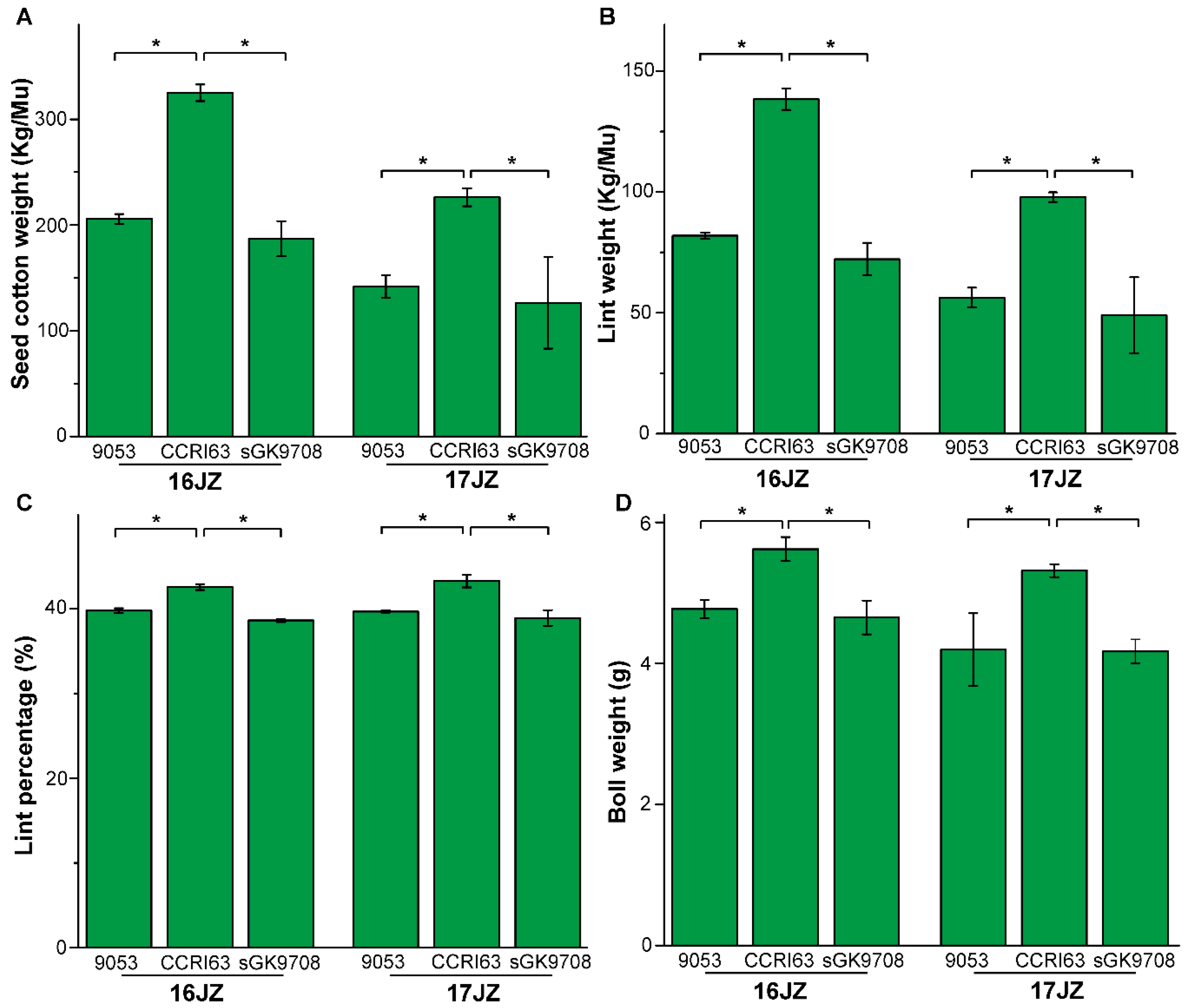
3.1. Agronomic Performance of Materials
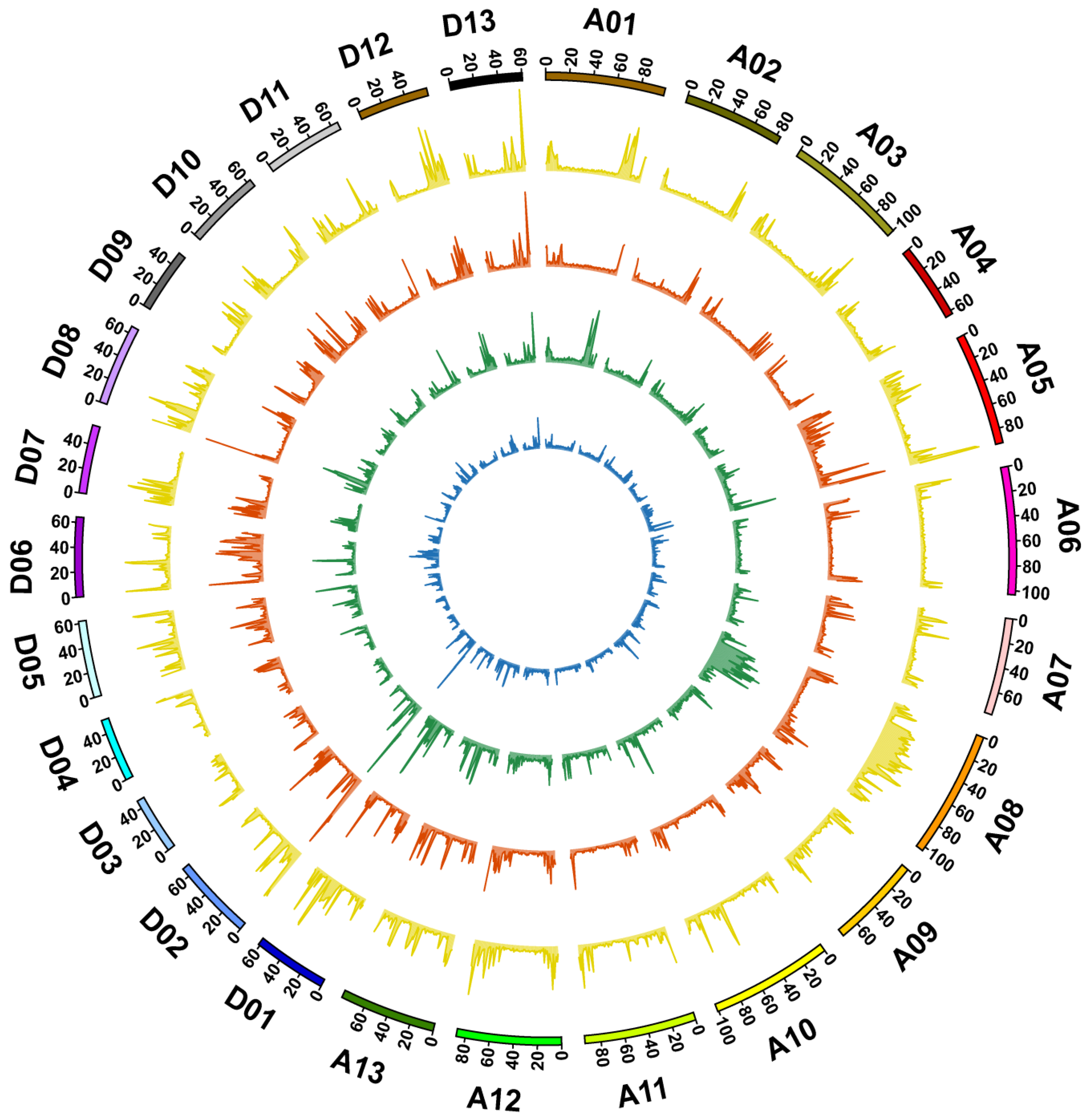
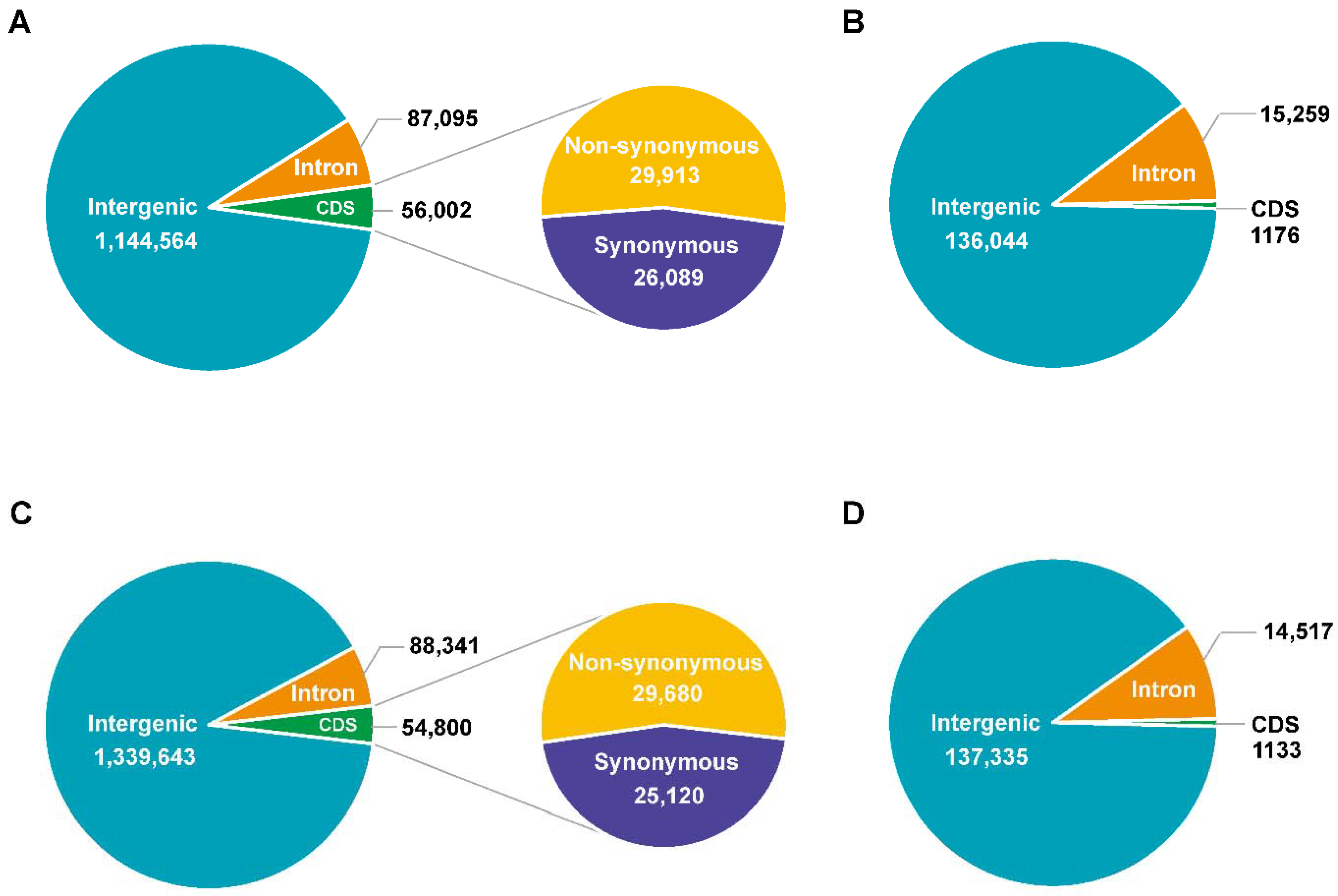
3.2. Genome Sequencing, Identification of Variation, and Annotation
3.3. Genetic Variations between 9053 and sGK9708
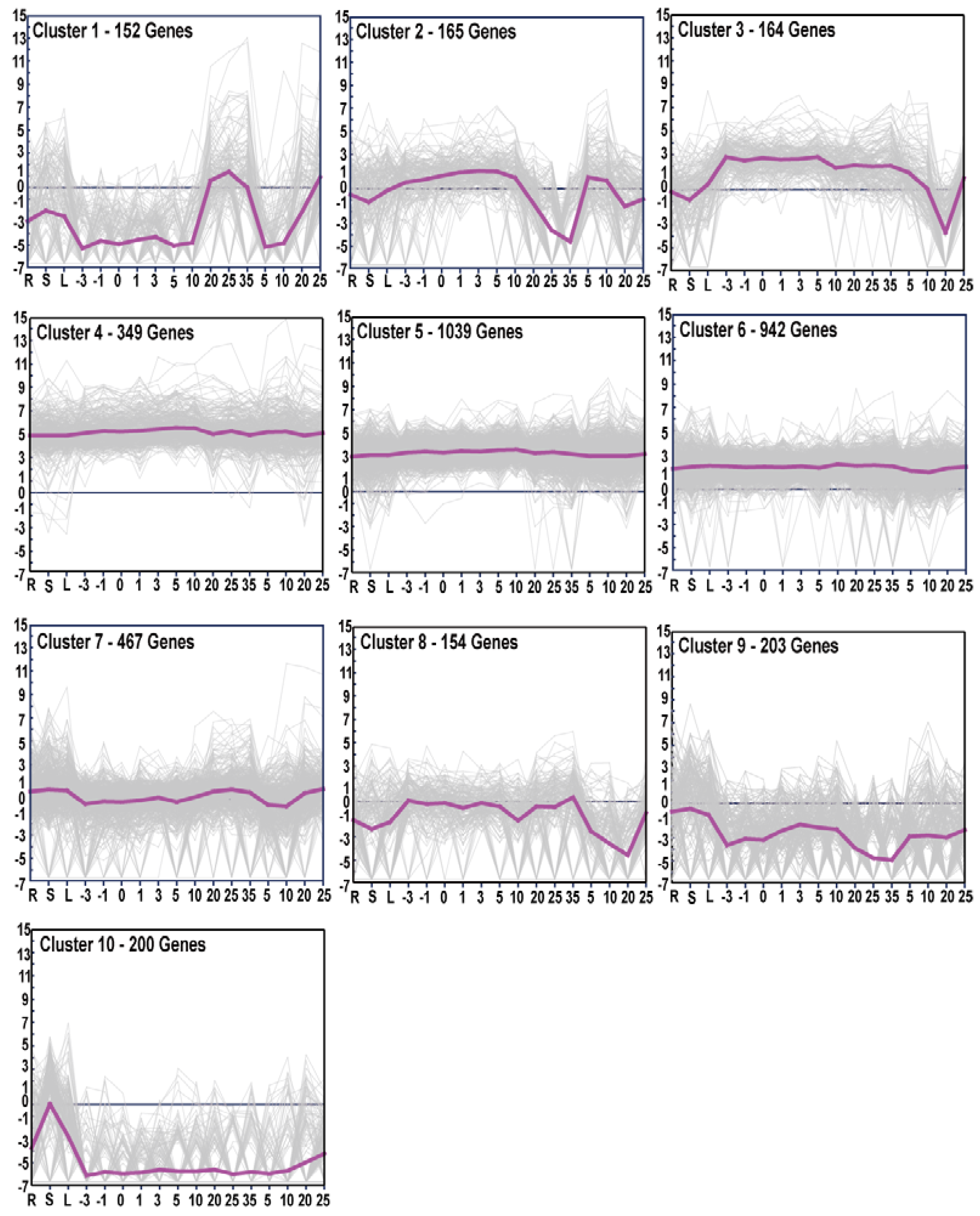
3.4. Gene Expression Patterns and GO Enrichment Analysis
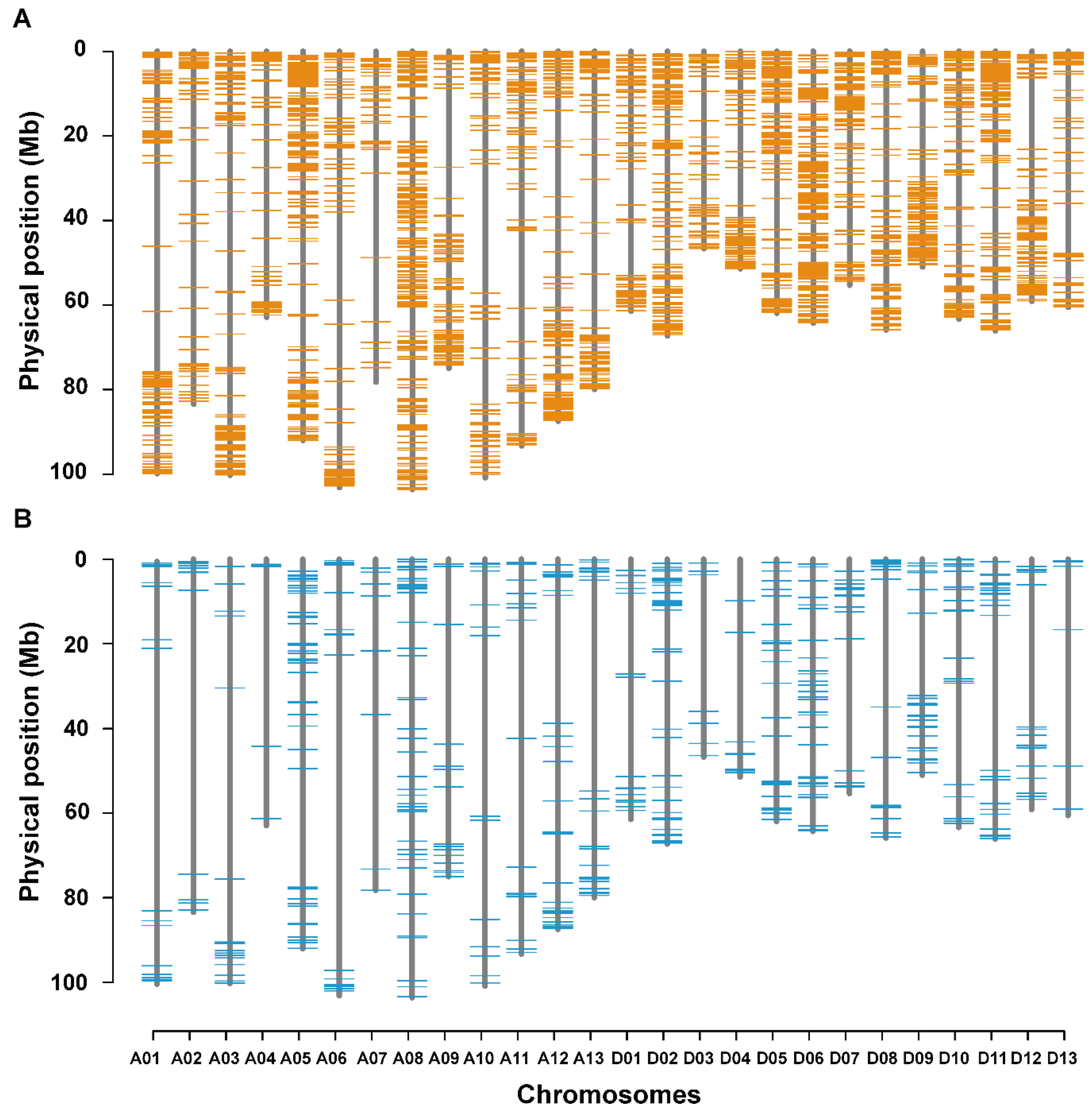
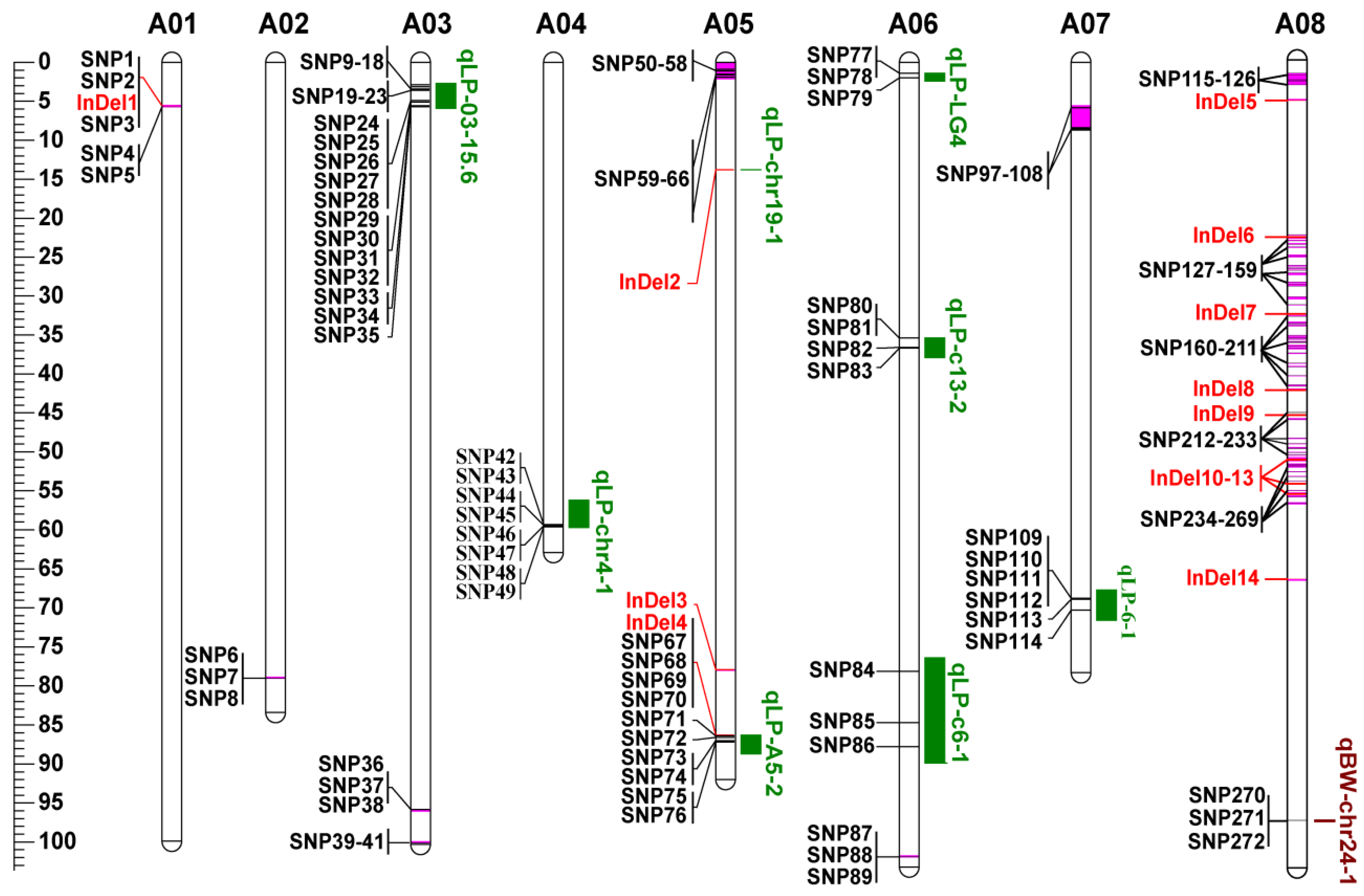
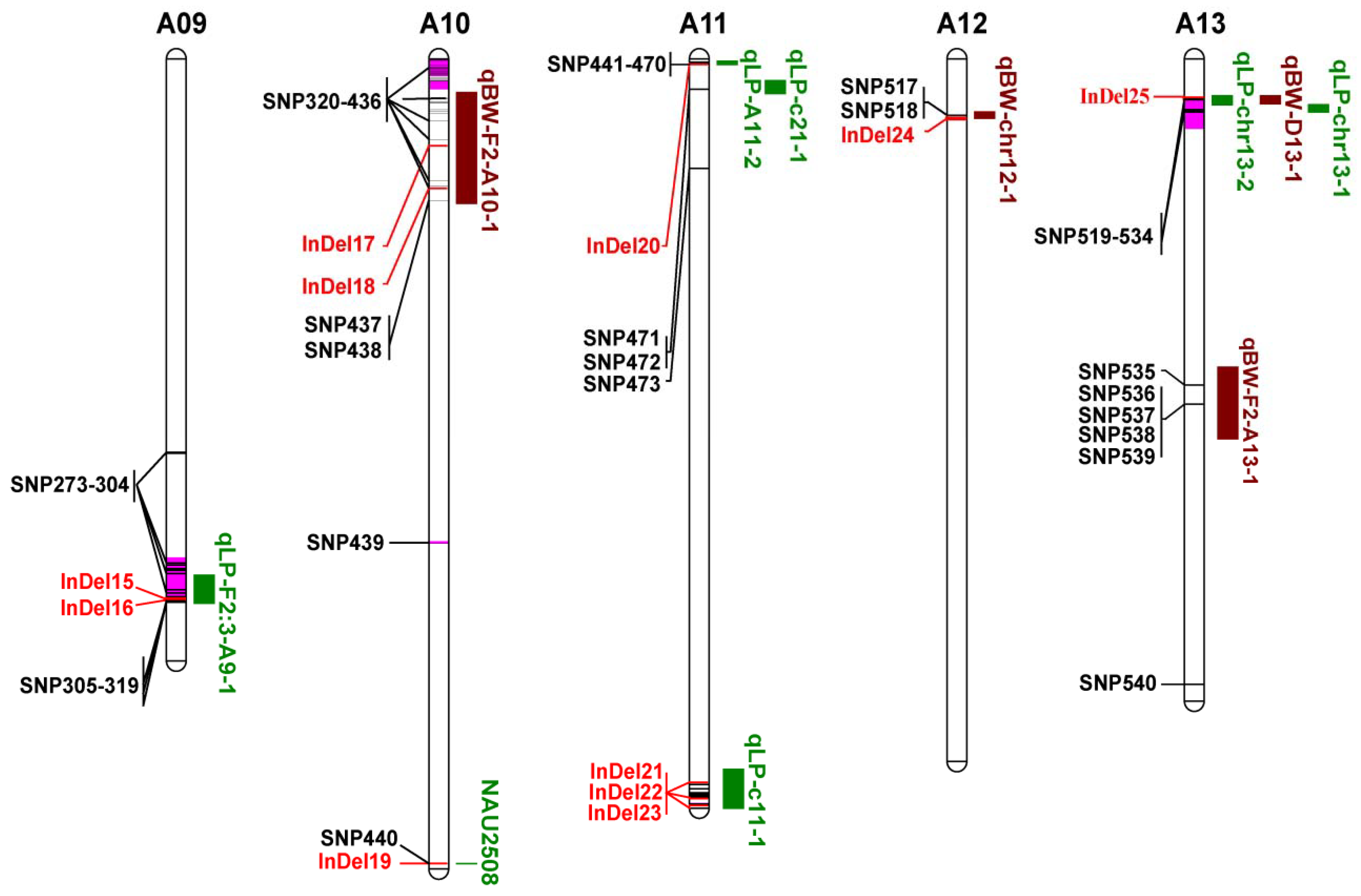
3.5. Co-localization of DNA Polymorphisms with QTLs for LP and BW
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chen, Z.J.; Scheffler, B.E.; Dennis, E.; Triplett, B.A.; Zhang, T.; Guo, W.; Chen, X.; Stelly, D.M.; Rabinowicz, P.D.; Town, C.D.; et al. Toward sequencing cotton (Gossypium) genomes. Plant Physiol. 2007, 145, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.B.; Li, Y.; Wang, B.; Chee, P.W. Recent advances in cotton genomics. Int. J. Plant Genom. 2008, 2008, 742304. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Wang, B.; Guo, W.; Qin, Y.; Wang, L.; Zhang, Y.; Zhang, T. Quantitative trait loci mapping for yield and its components by using two immortalized populations of a heterotic hybrid in Gossypium Hirsutum L. Mol. Breed. 2012, 29, 297–311. [Google Scholar] [CrossRef]
- Liang, Q.; Shang, L.; Wang, Y.; Hua, J. Partial dominance, overdominance and epistasis as the genetic basis of heterosis in upland cotton (Gossypium hirsutum L.). PLoS ONE 2015, 10, e0143548. [Google Scholar] [CrossRef] [PubMed]
- Shull, G.H. The Composition of a Field of Maize. J. Hered. 1908, os-4, 296–301. [Google Scholar] [CrossRef]
- Schnable, P.S.; Springer, N.M. Progress toward understanding heterosis in crop plants. Annu. Rev. Plant Boil. 2013, 64, 71–88. [Google Scholar] [CrossRef]
- Cheng, S.H.; Zhuang, J.Y.; Fan, Y.Y.; Du, J.H.; Cao, L.Y. Progress in research and development on hybrid rice: A super-domesticate in China. Ann. Bot. 2007, 100, 959–966. [Google Scholar] [CrossRef]
- Lai, J.; Li, R.; Xu, X.; Jin, W.; Xu, M.; Zhao, H.; Xiang, Z.; Song, W.; Ying, K.; Zhang, M.; et al. Genome-wide patterns of genetic variation among elite maize inbred lines. Nat. Genet. 2010, 42, 1027–1030. [Google Scholar] [CrossRef]
- Basunanda, P.; Radoev, M.; Ecke, W.; Friedt, W.; Becker, H.C.; Snowdon, R.J. Comparative mapping of quantitative trait loci involved in heterosis for seedling and yield traits in oilseed rape (Brassica napus L.). TAG. Theor. Appl. Genet. 2010, 120, 271–281. [Google Scholar] [CrossRef]
- Krieger, U.; Lippman, Z.B.; Zamir, D. The flowering gene SINGLE FLOWER TRUSS drives heterosis for yield in tomato. Nat. Genet. 2010, 42, 459–463. [Google Scholar] [CrossRef]
- He, S.; Sun, G.; Huang, L.; Yang, D.; Dai, P.; Zhou, D.; Wu, Y.; Ma, X.; Du, X.; Wei, S.; et al. Genomic divergence in cotton germplasm related to maturity and heterosis. J. Integr. Plant Boil. 2018. [CrossRef] [PubMed]
- Chen, R.; Davydov, E.V.; Sirota, M.; Butte, A.J. Non-synonymous and synonymous coding SNPs show similar likelihood and effect size of human disease association. PLoS ONE 2010, 5, e13574. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Xue, Y.; Li, J.; Deng, X.-W.; Zhang, Q. Rice functional genomics research in China. Philos. Trans. R. Soc. B Boil. Sci. 2007, 362, 1009–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Yan, W.; Xue, W.; Shao, D.; Xing, Y. Evolution and association analysis of Ghd7 in rice. PLoS ONE 2012, 7, e34021. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Zhao, H.; Ren, L.; Song, W.; Zeng, B.; Guo, J.; Wang, B.; Liu, Z.; Chen, J.; Li, W.; et al. Genome-wide genetic changes during modern breeding of maize. Nat. Genet. 2012, 44, 812–815. [Google Scholar] [CrossRef]
- Zheng, L.Y.; Guo, X.S.; He, B.; Sun, L.J.; Peng, Y.; Dong, S.S.; Liu, T.F.; Jiang, S.; Ramachandran, S.; Liu, C.M.; et al. Genome-wide patterns of genetic variation in sweet and grain sorghum (Sorghum bicolor). Genome Boil. 2011, 12, R114. [Google Scholar] [CrossRef]
- Paterson, A.H.; Wendel, J.F.; Gundlach, H.; Guo, H.; Jenkins, J.; Jin, D.; Llewellyn, D.; Showmaker, K.C.; Shu, S.; Udall, J.; et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 2012, 492, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wang, Z.; Li, F.; Ye, W.; Wang, J.; Song, G.; Yue, Z.; Cong, L.; Shang, H.; Zhu, S. The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet. 2012, 44, 1098–1103. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Fan, G.; Wang, K.; Sun, F.; Yuan, Y.; Song, G.; Li, Q.; Ma, Z.; Lu, C.; Zou, C.; et al. Genome sequence of the cultivated cotton Gossypium arboreum. Nat. Genet. 2014, 46, 567–572. [Google Scholar] [CrossRef]
- Li, F.; Fan, G.; Lu, C.; Xiao, G.; Zou, C.; Kohel, R.J.; Ma, Z.; Shang, H.; Ma, X.; Wu, J.; et al. Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. 2015, 33, 524–530. [Google Scholar] [CrossRef]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Hulse-Kemp, A.M.; Lemm, J.; Plieske, J.; Ashrafi, H.; Buyyarapu, R.; Fang, D.D.; Frelichowski, J.; Giband, M.; Hague, S.; Hinze, L.L.; et al. Development of a 63K SNP Array for Cotton and High-Density Mapping of Intraspecific and Interspecific Populations of Gossypium spp. G3 2015, 5, 1187–1209. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Wang, X.; Liu, Z.; Gu, Q.; Zhang, Y.; Li, Z.; Ke, H.; Yang, J.; Wu, J.; Wu, L.; et al. Genome-wide association study discovered genetic variation and candidate genes of fibre quality traits in Gossypium hirsutum L. Plant Biotechnol. J. 2017, 15, 982–996. [Google Scholar] [CrossRef] [PubMed]
- Page, J.T.; Liechty, Z.S.; Alexander, R.H.; Clemons, K.; Hulse-Kemp, A.M.; Ashrafi, H.; Van Deynze, A.; Stelly, D.M.; Udall, J.A. DNA sequence evolution and rare homoeologous conversion in tetraploid cotton. PLoS Genet. 2016, 12, e1006012. [Google Scholar] [CrossRef]
- Su, J.; Li, L.; Zhang, C.; Wang, C.; Gu, L.; Wang, H.; Wei, H.; Liu, Q.; Huang, L.; Yu, S. Genome-wide association study identified genetic variations and candidate genes for plant architecture component traits in Chinese upland cotton. TAG Theor. Appl. Genet. 2018, 131, 1299–1314. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Tu, L.; Lin, M.; Lin, Z.; Wang, P.; Yang, Q.; Ye, Z.; Shen, C.; Li, J.; Zhang, L.; et al. Asymmetric subgenome selection and cis-regulatory divergence during cotton domestication. Nat. Genet. 2017, 49, 579–587. [Google Scholar] [CrossRef] [Green Version]
- Fang, L.; Wang, Q.; Hu, Y.; Jia, Y.; Chen, J.; Liu, B.; Zhang, Z.; Guan, X.; Chen, S.; Zhou, B.; et al. Genomic analyses in cotton identify signatures of selection and loci associated with fiber quality and yield traits. Nat. Genet. 2017, 49, 1089–1098. [Google Scholar] [CrossRef]
- Team, R.D.C. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. Computing 2016, 14, 12–21. [Google Scholar]
- Yang, M.; Wang, X.; Ren, D.; Huang, H.; Xu, M.; He, G.; Deng, X.W. Genomic architecture of biomass heterosis in Arabidopsis. Proc. Natl. Acad. Sci. USA 2017, 114, 8101–8106. [Google Scholar] [CrossRef]
- Paterson, A.H.; Brubaker, C.L.; Wendel, J.F. A rapid method for extraction of cotton (Gossypium spp.) genomic DNA suitable for RFLP or PCR analysis. Plant Mol. Boil. Report. 1993, 11, 122–127. [Google Scholar] [CrossRef]
- Yu, J.; Jung, S.; Cheng, C.H.; Ficklin, S.P.; Lee, T.; Zheng, P.; Jones, D.; Percy, R.G.; Main, D. CottonGen: A genomics, genetics and breeding database for cotton research. Nucleic Acids Res. 2014, 42, D1229–D1236. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Said, J.I.; Knapka, J.A.; Song, M.; Zhang, J. Cotton QTLdb: A cotton QTL database for QTL analysis, visualization, and comparison between Gossypium hirsutum and G. hirsutum × G. barbadense populations. Mol. Genet. Genom. 2015, 290, 1615–1625. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Liang, C.; Meng, Z.; Sun, G.; Meng, Z.; Guo, S.; Zhang, R. CottonFGD: An integrated functional genomics database for cotton. BMC Plant Boil. 2017, 17, 101. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Gong, Q.; Wang, L.; Jin, Y.; Xi, J.; Li, Z.; Qin, W.; Yang, Z.; Lu, L.; Chen, Q.; et al. Genome-wide study of YABBY genes in upland cotton and their expression patterns under different stresses. Front. Genet. 2018, 9, 33. [Google Scholar] [CrossRef]
- Sturn, A.; Quackenbush, J.; Trajanoski, Z. Genesis: Cluster analysis of microarray data. Bioinformatics 2002, 18, 207–208. [Google Scholar] [CrossRef]
- You, F.M.; Huo, N.; Yong, Q.G.; Luo, M.C.; Ma, Y.; Hane, D.; Lazo, G.R.; Dvorak, J.; Anderson, O.D. BATCHPRIMER3: A high throughput web application for PCR and sequencing primer design. BMC Bioinform. 2008, 9, 253. [Google Scholar] [CrossRef]
- Jeanmougin, F.; Thompson, J.D.; Gouy, M.; Higgins, D.G.; Gibson, T.J. Multiple sequence alignment with Clustal X. Trends Biochem. Sci. 1998, 23, 403–405. [Google Scholar] [CrossRef]
- Jain, M.; Moharana, K.C.; Shankar, R.; Kumari, R.; Garg, R. Genomewide discovery of DNA polymorphisms in rice cultivars with contrasting drought and salinity stress response and their functional relevance. Plant Biotechnol. J. 2014, 12, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yang, S.; Gong, J.; Zhao, Y.; Feng, Q.; Gong, H.; Li, W.; Zhan, Q.; Cheng, B.; Xia, J.; et al. Genomic analysis of hybrid rice varieties reveals numerous superior alleles that contribute to heterosis. Nat. Commun. 2015, 6, 6258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Zhao, W.; Jiang, C.; Wang, X.; Xiong, H.; Todorovska, E.G.; Yin, Z.; Chen, Y.; Wang, X.; Xie, J.; et al. Genetic architecture and candidate genes for deep-sowing tolerance in rice revealed by non-syn GWAS. Front. Plant Sci. 2018, 9, 332. [Google Scholar] [CrossRef] [PubMed]
- Said, J.I.; Song, M.; Wang, H.; Lin, Z.; Zhang, X.; Fang, D.D.; Zhang, J. A comparative meta-analysis of QTL between intraspecific Gossypium hirsutum and interspecific G. hirsutum × G. barbadense populations. Mol. Genet. Genom. 2015, 290, 1003–1025. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Nie, X.; Shen, C.; You, C.; Li, W.; Zhao, W.; Zhang, X.; Lin, Z. Population structure and genetic basis of the agronomic traits of upland cotton in China revealed by a genome-wide association study using high-density SNPs. Plant Biotechnol. J. 2017, 15, 1374–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Cao, L.; Wang, S.; Li, Y.; Shi, X.; Liu, H.; Li, L.; Zhang, Z.; Fowke, L.C.; Wang, H.; et al. Downregulation of multiple CDK inhibitor ICK/KRP genes upregulates the E2F pathway and increases cell proliferation, and organ and seed sizes in Arabidopsis. Plant J. Cell Mol. Boil. 2013, 75, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Meijon, M.; Satbhai, S.B.; Tsuchimatsu, T.; Busch, W. Genome-wide association study using cellular traits identifies a new regulator of root development in Arabidopsis. Nat. Genet. 2014, 46, 77–81. [Google Scholar] [CrossRef]
- Wang, W.; Mauleon, R.; Hu, Z.; Chebotarov, D.; Tai, S.; Wu, Z.; Li, M.; Zheng, T.; Fuentes, R.R.; Zhang, F.; et al. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature 2018, 557, 43–49. [Google Scholar] [CrossRef]
- Ma, Z.; He, S.; Wang, X.; Sun, J.; Zhang, Y.; Zhang, G.; Wu, L.; Li, Z.; Liu, Z.; Sun, G.; et al. Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield. Nat. Genet. 2018, 50, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Jin, X.; Zhu, D.; Lin, Z. Uncovering SNP and indel variations of tetraploid cottons by SLAF-seq. BMC Genom. 2017, 18, 247. [Google Scholar] [CrossRef]
- Subbaiyan, G.K.; Waters, D.L.; Katiyar, S.K.; Sadananda, A.R.; Vaddadi, S.; Henry, R.J. Genome-wide DNA polymorphisms in elite indica rice inbreds discovered by whole-genome sequencing. Plant Biotechnol. J. 2012, 10, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Batley, J.; Barker, G.; O’Sullivan, H.; Edwards, K.J.; Edwards, D. Mining for single nucleotide polymorphisms and insertions/deletions in maize expressed sequence tag data. Plant Physiol. 2003, 132, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zhu, Z.; Zhou, X.; Yan, Y.; Dong, Z.; Cui, D. High-throughput sequencing reveals single nucleotide variants in longer-kernel bread wheat. Front. Plant Sci. 2016, 7, 1193. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jin, X.; Zhang, B.; Shen, C.; Lin, Z. Enrichment of an intraspecific genetic map of upland cotton by developing markers using parental RAD sequencing. DNA Res. Int. J. Rapid Publ. Rep. Genes Genomes 2015, 22, 147–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, C.B.; Bhareti, P.; Muthamilarasan, M.; Mukherjee, M.; Khan, Y.; Rathi, P.; Prasad, M. Genome-wide SNP identification and characterization in two soybean cultivars with contrasting Mungbean Yellow Mosaic India Virus disease resistance traits. PLoS ONE 2015, 10, e0123897. [Google Scholar] [CrossRef] [PubMed]
- Landry, C.R.; Lemos, B.; Rifkin, S.A.; Dickinson, W.J.; Hartl, D.L. Genetic properties influencing the evolvability of gene expression. Science 2007, 317, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Wray, G.A. The evolutionary significance of cis-regulatory mutations. Nat. Rev. Genet. 2007, 8, 206–216. [Google Scholar] [CrossRef]
- Keurentjes, J.J.; Fu, J.; Terpstra, I.R.; Garcia, J.M.; van den Ackerveken, G.; Snoek, L.B.; Peeters, A.J.; Vreugdenhil, D.; Koornneef, M.; Jansen, R.C. Regulatory network construction in Arabidopsis by using genome-wide gene expression quantitative trait loci. Proc. Natl. Acad. Sci. USA 2007, 104, 1708–1713. [Google Scholar] [CrossRef]
- Jiang, Y.; Schmidt, R.H.; Zhao, Y.; Reif, J.C. A quantitative genetic framework highlights the role of epistatic effects for grain-yield heterosis in bread wheat. Nat. Genet. 2017, 49, 1741–1746. [Google Scholar] [CrossRef]
- Wan, Q.; Guan, X.; Yang, N.; Wu, H.; Pan, M.; Liu, B.; Fang, L.; Yang, S.; Hu, Y.; Ye, W.; et al. Small interfering RNAs from bidirectional transcripts of GhMML3_A12 regulate cotton fiber development. New Phytol. 2016, 210, 1298–1310. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Tian, Y.; Wan, Q.; Fang, L.; Guan, X.; Chen, J.; Hu, Y.; Ye, W.; Zhang, H.; Guo, W.; et al. Genetics and evolution of MIXTA genes regulating cotton lint fiber development. New Phytol. 2018, 217, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhang, C.; Yang, X.; Liu, K.; Wu, Z.; Zhang, X.; Zheng, W.; Xun, Q.; Liu, C.; Lu, L.; et al. PAG1, a cotton brassinosteroid catabolism gene, modulates fiber elongation. New Phytol. 2014, 203, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.X.; Chen, Y.; Zeng, W.; Li, Y.; Gao, L.; Li, D.D.; Bacic, A.; Xu, W.L.; Li, X.B. The cotton beta-galactosyltransferase 1 (GalT1) that galactosylates arabinogalactan proteins participates in controlling fiber development. Plant J. Cell Mol. Biol. 2017, 89, 957–971. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total Reads | Mapped Reads | Average Depth | Q30 | Coverage (%) |
|---|---|---|---|---|---|
| 9053 | 1,197,221,488 | 1,191,826,523 | 59.9 | 88.89 | 98.34 |
| sGK9708 | 1,098,196,338 | 1,092,627,188 | 64.37 | 90.84 | 97.73 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, C.; Li, W.; Wang, Z.; Pei, X.; Liu, Y.; Ren, Z.; He, K.; Zhang, F.; Sun, K.; Zhou, X.; et al. Genome Resequencing Reveals Genetic Variation between the Parents of An Elite Hybrid Upland Cotton. Agronomy 2018, 8, 305. https://0-doi-org.brum.beds.ac.uk/10.3390/agronomy8120305
Song C, Li W, Wang Z, Pei X, Liu Y, Ren Z, He K, Zhang F, Sun K, Zhou X, et al. Genome Resequencing Reveals Genetic Variation between the Parents of An Elite Hybrid Upland Cotton. Agronomy. 2018; 8(12):305. https://0-doi-org.brum.beds.ac.uk/10.3390/agronomy8120305
Chicago/Turabian StyleSong, Chengxiang, Wei Li, Zhenyu Wang, Xiaoyu Pei, Yangai Liu, Zhongying Ren, Kunlun He, Fei Zhang, Kuan Sun, Xiaojian Zhou, and et al. 2018. "Genome Resequencing Reveals Genetic Variation between the Parents of An Elite Hybrid Upland Cotton" Agronomy 8, no. 12: 305. https://0-doi-org.brum.beds.ac.uk/10.3390/agronomy8120305