
Maresin-1 Prevents Liver Fibrosis by Targeting Nrf2 and NF-κB, Reducing Oxidative Stress and Inflammation


 and
and
Abstract
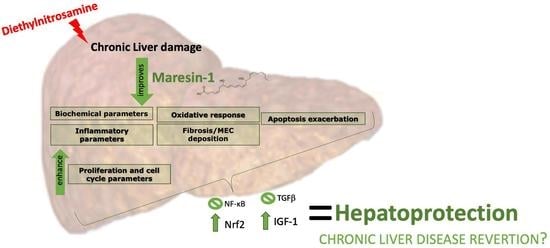
:
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Model of Fibrosis
2.3. Animals Preparation
2.4. Determination of Biochemical Parameters
2.5. Liver Histology
2.6. Immunohistochemistry Studies
2.7. Western Blot Analysis
2.8. Statistical Analysis
3. Results
3.1. MaR1 Administration Decrease Parameters of Chronic Liver Injury
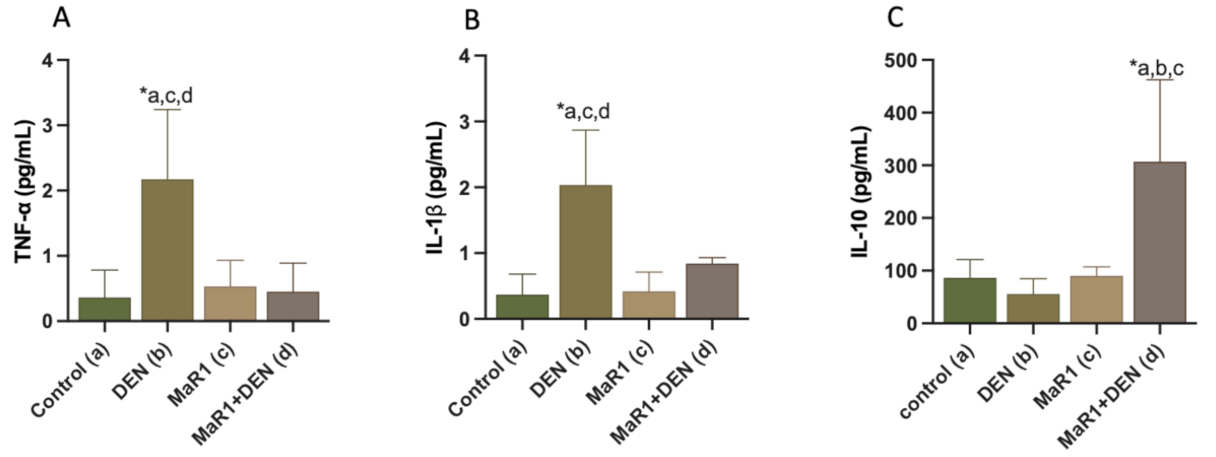
3.2. MaR1 Reduces Inflammatory Parameters in DEN-Induced Liver Injury
3.3. MaR1 Shows an Anti-Fibrogenic and Proliferative Response against DEN Injury
3.4. MaR1 Protects from Fibrosis Liver Injury through Nuclear Receptors and Growth Factors through Activation Mechanisms
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef]
- Zhou, W.C.; Zhang, Q.B.; Qiao, L. Pathogenesis of liver cirrhosis. World J. Gastroenterol. 2014, 20. [Google Scholar] [CrossRef] [PubMed]
- Parola, M.; Pinzani, M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol. Asp. Med. 2018, 65, 37–55. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.Q.; Ding, J.; Zhang, L.; Liu, C.M. Protective effects of ursolic acid in an experimental model of liver fibrosis through Nrf2/ARE pathway. Clin. Res. Hepatol. Gastroenterol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Luangmonkong, T.; Suriguga, S.; Mutsaers, H.A.M.; Groothuis, G.M.M.; Olinga, P.; Boersema, M. Targeting Oxidative Stress for the Treatment of Liver Fibrosis. Rev. Physiol. Biochem. Pharmacol. 2018, 175, 71–102. [Google Scholar] [CrossRef] [PubMed]
- Kolios, G.; Valatas, V.; Kouroumalis, E. Role of Kupffer cells in the pathogenesis of liver disease. World J. Gastroenterol. 2006, 12, 7413–7420. [Google Scholar] [CrossRef]
- Serhan, C.N.; Clish, C.B.; Brannon, J.; Colgan, S.P.; Chiang, N.; Gronert, K. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J. Exp. Med. 2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalli, J.; Zhu, M.; Vlasenko, N.A.; Deng, B.; Haeggström, J.Z.; Petasis, N.A.; Serhan, C.N. The novel 13S,14S-epoxy-maresinis converted by human macrophages to maresin 1 (MaR1), inhibits leukotriene A4 hydrolase (LTA4H), and shifts macrophage phenotype. FASEB J. 2013. [Google Scholar] [CrossRef] [Green Version]
- Dalli, J.; Ramon, S.; Norris, P.C.; Colas, R.A.; Serhan, C.N. Novel proresolving and tissue-regenerative resolvin and protectin sulfido-conjugated pathways. FASEB J. 2015. [Google Scholar] [CrossRef] [Green Version]
- Buckley, C.D.; Gilroy, D.; Serhan, C.N. Proresolving Lipid Mediators and Mechanisms in the Resolution of Acute Inflammation. Immunity 2014, 40, 315–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiurchiù, V.; Leuti, A.; Dalli, J.; Jacobsson, A.; Battistini, L.; MaCcarrone, M.; Serhan, C.N. Proresolving lipid mediators resolvin D1, resolvin D2, and maresin 1 are critical in modulating T cell responses. Sci. Transl. Med. 2016. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Chiang, N.; Dalli, J. New pro-resolving n-3 mediators bridge resolution of infectious inflammation to tissue regeneration. Mol. Aspects Med. 2018, 64, 1–17. [Google Scholar] [CrossRef]
- Li, R.; Wang, Y.; Zhao, E.; Wu, K.; Li, W.; Shi, L.; Wang, D.; Xie, G.; Yin, Y.; Deng, M.; et al. Maresin 1, a Proresolving Lipid Mediator, Mitigates Carbon Tetrachloride-Induced Liver Injury in Mice. Oxid. Med. Cell. Longev. 2016, 2016. [Google Scholar] [CrossRef]
- Zhang, P.; Yin, Y.; Wang, T.; Li, W.; Li, C.; Zeng, X.; Yang, W.; Zhang, R.; Tang, Y.; Shi, L.; et al. Maresin 1 mitigates concanavalin A-induced acute liver injury in mice by inhibiting ROS-mediated activation of NF-κB signaling. Free Radic. Biol. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Soto, G.; Rodríguez, M.J.; Fuentealba, R.; Treuer, A.V.; Castillo, I.; González, D.R.; Zúñiga-Hernández, J. Maresin 1, a proresolving lipid mediator, ameliorates liver ischemia-reperfusion injury and stimulates hepatocyte proliferation in sprague-dawley rats. Int. J. Mol. Sci. 2020, 21, 540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, D.; Fu, G.; Li, W.; Sun, P.; Loughran, P.A.; Deng, M.; Scott, M.J.; Billiar, T.R. Maresin 1 protects the liver against ischemia/reperfusion injury via the ALXR/Akt signaling pathway. Mol. Med. 2021, 27. [Google Scholar] [CrossRef] [PubMed]
- Rius, B.; Duran-Güell, M.; Flores-Costa, R.; López-Vicario, C. The specialized proresolving lipid mediator maresin 1 protects hepatocytes from lipotoxic and hypoxia-induced endoplasmic reticulum stress. FASEB J. 2017, 31, 5384–5398. [Google Scholar] [CrossRef] [Green Version]
- Laiglesia, L.M.; Lorente-Cebrián, S.; Martínez-Fernández, L.; Sáinz, N.; Prieto-Hontoria, P.L.; Burrell, M.A.; Rodríguez-Ortigosa, C.M.; Martínez, J.A.; Moreno-Aliaga, M.J. Maresin 1 mitigates liver steatosis in ob/ob and diet-induced obese mice. Int. J. Obes. 2018. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, Z.-B.; Zheng, S.-S.; Hu, G.-Z.; Ding, C.-Y.; Zhang, Y.; Zhao, X.-H.; Ni, L.-M. Regression of liver fibrosis after biliary drainage in patients with choledocholith: A preliminary report. Hepatobiliary Pancreat. Dis. Int. 2005, 4, 104–107. [Google Scholar] [PubMed]
- Ellis, E.L.; Mann, D.A. Clinical evidence for the regression of liver fibrosis. J. Hepatol. 2012, 56, 1171–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Y.; Wang, M.; Chen, E.; Tang, H. Liver Regeneration: Analysis of the Main Relevant Signaling Molecules. Mediators Inflamm. 2017, 2017, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, N.H.; Heo, J.D.; Kim, T.B.; Rho, J.R.; Yang, M.H.; Jeong, E.J. Protective effects of ethyl acetate soluble fraction of Limonium tetragonum on diethylnitrosamine-induced liver fibrosis in rats. Biol. Pharm. Bull. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, M.J.; Herrera, F.; Donoso, W.; Castillo, I.; Orrego, R.; González, D.R.; Zúñiga-Hernández, J. Pro-resolving lipid mediator resolvin E1 mitigates the progress of diethylnitrosamine-induced liver fibrosis in sprague-dawley rats by attenuating fibrogenesis and restricting proliferation. Int. J. Mol. Sci. 2020, 21, 8827. [Google Scholar] [CrossRef] [PubMed]
- Korourian, S.; Hakkak, R.; Ronis, M.J.J.; Shelnutt, S.R.; Waldron, J.; Ingelman-Sundberg, M.; Badger, T.M. Diet and risk of ethanol-induced hepatotoxicity: Carbohydrate-fat relationships in rats. Toxicol. Sci. 1999, 47, 110–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishak, K.; Baptista, A.; Bianchi, L.; Callea, F.; De Groote, J.; Gudat, F.; Denk, H.; Desmet, V.; Korb, G.; MacSween, R.N.M.; et al. Histological grading and staging of chronic hepatitis. J. Hepatol. 1995. [Google Scholar] [CrossRef]
- Goodman, Z.D. Grading and staging systems for inflammation and fibrosis in chronic liver diseases. J. Hepatol. 2007, 47, 598–607. [Google Scholar] [CrossRef]
- DeRyckere, F.; Gannon, F. A one-hour minipreparation technique for extraction of DNA-binding proteins from animal tissues. BioTechniques 1994, 16, 405. [Google Scholar]
- Pinter, M.; Trauner, M.; Peck-Radosavljevic, M.; Sieghart, W. Cancer and liver cirrhosis: Implications on prognosis and management. ESMO Open 2016, 1, e000042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iredale, J.P.; Campana, L. Regression of Liver Fibrosis. Semin. Liver Dis. 2017, 37, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jalan, R.; Ginès, P.; Olson, J.C.; Mookerjee, R.; Moreau, R.; Garcia-Tsao, G.; Arroyo, V.; Kamath, P.S. Acute-on chronic liver failure. J. Hepatol. 2012, 57, 1336–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuberger, J. An update on liver transplantation: A critical review. J. Autoimmun. 2015, 66, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Pettinelli, P.; del Pozo, T.; Araya, J.; Rodrigo, R.; Araya, A.V.; Smok, G.; Csendes, A.; Gutierrez, L.; Rojas, J.; Korn, O.; et al. Enhancement in liver SREBP-1c/PPAR-α ratio and steatosis in obese patients: Correlations with insulin resistance and n-3 long-chain polyunsaturated fatty acid depletion. Biochim. Biophys. Acta-Mol. Basis Dis. 2009, 1792. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Fernández-Galilea, M.; Martínez-Fernández, L.; González-Muniesa, P.; Pérez-Chávez, A.; Martínez, J.A.; Moreno-Aliaga, M.J. Oxidative Stress and Non-Alcoholic Fatty Liver Disease: Effects of Omega-3 Fatty Acid Supplementation. Nutrients 2019, 11, 872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zúñiga, J.; Cancino, M.; Medina, F.; Varela, P.; Vargas, R.; Tapia, G.; Videla, L.A.; Fernández, V. N-3 PUFA supplementation triggers PPAR-α activation and PPAR-α/NF-κB interaction: Anti-inflammatory implications in liver ischemia-reperfusion injury. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [Green Version]
- Enguita, M.; Razquin, N.; Pamplona, R.; Quiroga, J.; Prieto, J.; Fortes, P. The cirrhotic liver is depleted of docosahexaenoic acid (DHA), a key modulator of NF-κB and TGFβ pathways in hepatic stellate cells. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Wang, Y.; Gao, H.; Li, D.; Jiang, R.; Ge, L.; Tong, C.; Xu, K. Associations among Dietary Omega-3 Polyunsaturated Fatty Acids, the Gut Microbiota, and Intestinal Immunity. Mediat. Inflamm. 2021, 2021, 1–11. [Google Scholar] [CrossRef]
- Costantini, L.; Molinari, R.; Farinon, B.; Merendino, N. Impact of Omega-3 Fatty Acids on the Gut Microbiota. Int. J. Mol. Sci. 2017, 18, 2645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depner, C.M.; Philbrick, K.A.; Jump, D.B. Docosahexaenoic acid attenuates hepatic inflammation, oxidative stress, and fibrosis without decreasing hepatosteatosis in a Ldlr-/- Mouse Model of Western Diet-Induced Nonalcoholic Steatohepatitis1-3. J. Nutr. 2013, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lytle, K.A.; Wong, C.P.; Jump, D.B. Docosahexaenoic acid blocks progression of western diet-induced nonalcoholic steatohepatitis in obese Ldlr-/- mice. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colomer, C.; Marruecos, L.; Vert, A.; Bigas, A.; Espinosa, L. NF-κB Members Left Home: NF-κB-Independent Roles in Cancer. Biomedicines 2017, 5, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenzuela, R.; Ortiz, M.; Hernández-Rodas, M.C.; Echeverría, F.; Videla, L.A. Targeting n-3 Polyunsaturated Fatty Acids in Non-Alcoholic Fatty Liver Disease. Curr. Med. Chem. 2020, 27. [Google Scholar] [CrossRef]
- Hernández-Rodas, M.C.; Valenzuela, R.; Echeverría, F.; Rincón-Cervera, M.Á.; Espinosa, A.; Illesca, P.; Muñoz, P.; Corbari, A.; Romero, N.; Gonzalez-Mañan, D.; et al. Supplementation with Docosahexaenoic Acid and Extra Virgin Olive Oil Prevents Liver Steatosis Induced by a High-Fat Diet in Mice through PPAR-α and Nrf2 Upregulation with Concomitant SREBP-1c and NF-kB Downregulation. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Zúñiga, J.; Venegas, F.; Villarreal, M.; Núñez, D.; Chandía, M.; Valenzuela, R.; Tapia, G.; Varela, P.; Videla, L.A.; Fernández, V. Protection against in vivo liver ischemia-reperfusion injury by n-3 long-chain polyunsaturated fatty acids in the rat. Free Radic. Res. 2010, 44. [Google Scholar] [CrossRef]
- Zitka, O.; Skalickova, S.; Gumulec, J.; Masarik, M.; Adam, V.; Hubalek, J.; Trnkova, L.; Kruseova, J.; Eckschlager, T.; Kizek, R. Redox status expressed as GSH:GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol. Lett. 2012, 4. [Google Scholar] [CrossRef] [Green Version]
- Vairetti, M.; Di Pasqua, L.G.; Cagna, M.; Richelmi, P.; Ferrigno, A.; Berardo, C. Changes in glutathione content in liver diseases: An update. Antioxidants 2021, 10, 364. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Glutathione synthesis. Biochim. et Biophys. Acta (BBA)-Gen. Subj. 2012, 1830, 3143–3153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miralles, C.; Busquets, X.; Santos, C.; Togores, B.; Hussain, S.; Rahman, I.; MacNee, W.; Agustí, A.G.N. Regulation of iNOS expression and glutathione levels in rat liver by oxygen tension. FEBS Lett. 2000, 476. [Google Scholar] [CrossRef] [Green Version]
- Iwakiri, Y. Nitric oxide in liver fibrosis: The role of inducible nitric oxide synthase. Clin. Mol. Hepatol. 2015, 21, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Mo, S.; Yi, Q.; Lai, J.; Liu, H.; Shi, Z. Role and mechanism of maresin-1 in acute lung injury induced by trauma-hemorrhagic shock. Med. Sci. Monit. 2020, 26. [Google Scholar] [CrossRef]
- Chatterjee, A.; Sharma, A.; Chen, M.; Toy, R.; Mottola, G.; Conte, M.S. The pro-resolving lipid mediator maresin 1 (MaR1) attenuates inflammatory signaling pathways in vascular smooth muscle and endothelial cells. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munir, F.; Jamshed, M.B.; Shahid, N.; Muhammad, S.A.; Bhandari, A.; Zhang, Q.Y. Protective effects of maresin 1 against inflammation in experimentally induced acute pancreatitis and related lung injury. Am. J. Physiol. - Gastrointest. Liver Physiol. 2019, 317. [Google Scholar] [CrossRef] [PubMed]
- Cezar, T.L.C.; Martinez, R.M.; da Rocha, C.; Melo, C.P.B.; Vale, D.L.; Borghi, S.M.; Fattori, V.; Vignoli, J.A.; Camilios-Neto, D.; Baracat, M.M.; et al. Treatment with maresin 1, a docosahexaenoic acid-derived pro-resolution lipid, protects skin from inflammation and oxidative stress caused by UVB irradiation. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Nordgren, T.M.; Heires, A.J.; Wyatt, T.A.; Poole, J.A.; LeVan, T.D.; Cerutis, D.R.; Romberger, D.J. Maresin-1 reduces the pro-inflammatory response of bronchial epithelial cells to organic dust. Respir. Res. 2013, 14. [Google Scholar] [CrossRef] [Green Version]
- Spite, M.; Claria, J.; Serhan, C.N. Resolvins, Specialized Proresolving Lipid Mediators, and Their Potential Roles in Metabolic Diseases. Cell Metab. 2013, 19, 21–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Investig. 2018, 128, 2657–2669. [Google Scholar] [CrossRef]
- Mariqueo, T.; Zúñiga-Hernández, J. Omega-3 derivatives, specialized pro-resolving mediators: Promising therapeutic tools for the treatment of pain in chronic liver disease. Prostaglandins, Leukot. Essent. Fat. Acids 2020, 158, 102095. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.H.; Shin, K.O.; Kim, J.Y.; Khadka, D.B.; Kim, H.J.; Lee, Y.M.; Cho, W.J.; Cha, J.Y.; Lee, B.J.; Lee, M.O. A maresin 1/RORα/12-lipoxygenase autoregulatory circuit prevents inflammation and progression of nonalcoholic steatohepatitis. J. Clin. Investig. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baiocchini, A.; Montaldo, C.; Conigliaro, A.; Grimaldi, A.; Correani, V.; Mura, F.; Ciccosanti, F.; Rotiroti, N.; Brenna, A.; Montalbano, M.; et al. Extracellular matrix molecular remodeling in human liver fibrosis evolution. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy-Ullrich, J.E.; Suto, M.J. Thrombospondin-1 regulation of latent TGF-β activation: A therapeutic target for fibrotic disease. Matrix Biol. 2018, 68–69, 28–43. [Google Scholar] [CrossRef] [PubMed]
- McQuitty, C.E.; Williams, R.; Chokshi, S.; Urbani, L. Immunomodulatory Role of the Extracellular Matrix Within the Liver Disease Microenvironment. Front. Immunol. 2020, 11, 2903. [Google Scholar] [CrossRef]
- Sun, Q.; Wu, Y.; Zhao, F.; Wang, J. Maresin 1 inhibits transforming growth factor-β1-induced proliferation, migration and differentiation in human lung fibroblasts. Mol. Med. Rep. 2017, 16, 1523–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, S.; Gao, C.; Long, Y.; Huang, W.; Chen, J.; Fan, F.; Jiang, C.; Xu, Y. Maresin 1 Mitigates High Glucose-Induced Mouse Glomerular Mesangial Cell Injury by Inhibiting Inflammation and Fibrosis. Mediators Inflamm. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Guicciardi, M.E. Apoptosis: A mechanism of acute and chronic liver injury. Gut 2005, 54, 1024–1033. [Google Scholar] [CrossRef] [Green Version]
- Malhi, H.; Gores, G.J. Cellular and Molecular Mechanisms of Liver Injury. Gastroenterology 2008, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell Death and Cell Death Responses in Liver Disease: Mechanisms and Clinical Relevance. Gastroenterology 2014, 147, 765–783.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Dixit, V.M. Death Receptors: Signaling and Modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Lin, B. Pathophysiological Significance of Hepatic Apoptosis. ISRN Hepatol. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Canbay, A.; Feldstein, A.E.; Higuchi, H.; Werneburg, N.; Grambihler, A.; Bronk, S.F.; Gores, G.J. Kupffer Cell Engulfment of Apoptotic Bodies Stimulates Death Ligand and Cytokine Expression. Hepatology 2003, 38. [Google Scholar] [CrossRef]
- Serhan, C.N.; Dalli, J.; Karamnov, S.; Choi, A.; Park, C.-K.; Xu, Z.-Z.; Ji, R.-R.; Zhu, M.; Petasis, N.A. Macrophage proresolving mediator maresin 1 stimulates tissue regeneration and controls pain. FASEB J. 2012, 26, 1755–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.W.; Yu, S.H.; Fretwurst, T.; Larsson, L.; Sugai, J.V.; Oh, J.; Lehner, K.; Jin, Q.; Giannobile, W.V. Maresin 1 Promotes Wound Healing and Socket Bone Regeneration for Alveolar Ridge Preservation. J. Dent. Res. 2020, 99. [Google Scholar] [CrossRef] [PubMed]
- Wahyuni, T.; Kobayashi, A.; Tanaka, S.; Miyake, Y.; Yamamoto, A.; Bahtiar, A.; Mori, S.; Kametani, Y.; Tomimatsu, M.; Matsumoto, K.; et al. Maresin-1 induces cardiomyocyte hypertrophy through IGF-1 paracrine pathway. Am. J. Physiol.-Cell Physiol. 2021, 321. [Google Scholar] [CrossRef] [PubMed]
- Allard, J.B.; Duan, C. IGF-binding proteins: Why do they exist and why are there so many? Front. Endocrinol. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osher, E.; Macaulay, V.M. Therapeutic Targeting of the IGF Axis. Cells 2019, 8, 895. [Google Scholar] [CrossRef] [Green Version]
- Yakar, S.; Rosen, C.J.; Beamer, W.G.; Ackert-Bicknell, C.L.; Wu, Y.; Liu, J.L.; Ooi, G.T.; Setser, J.; Frystyk, J.; Boisclair, Y.R.; et al. Circulating levels of IGF-1 directly regulate bone growth and density. J. Clin. Investig. 2002, 110. [Google Scholar] [CrossRef]
- Takahashi, Y. Essential roles of growth hormone (GH) and insulin-like growth factor-I (IGF-I) in the liver [Review]. Endocr. J. 2012, 59, 955–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, Y. The Role of Growth Hormone and Insulin-Like Growth Factor-I in the Liver. Int. J. Mol. Sci. 2017, 18, 1447. [Google Scholar] [CrossRef] [PubMed]
- Puche, J.E.; Castilla, I. Human conditions of insulin-like growth factor-I (IGF-I) deficiency. J. Transl. Med. 2012, 10, 1–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Miao, X.; Zhu, D.; Li, D.; Zhang, Y.; Song, C.; Liu, K. Insulin-like growth factor-1 and non-alcoholic fatty liver disease: A systemic review and meta-analysis. Endocrine 2019, 65, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Fiore, E.; Domínguez, L.M.; Bayo, J.; Malvicini, M.; Atorrasagasti, C.; Rodriguez, M.; Cantero, M.J.; García, M.; Yannarelli, G.; Mazzolini, G. Human umbilical cord perivascular cells-derived extracellular vesicles mediate the transfer of IGF-I to the liver and ameliorate hepatic fibrogenesis in mice. Gene Ther. 2020, 27. [Google Scholar] [CrossRef] [PubMed]
- Fiore, E.J.; Bayo, J.M.; Garcia, M.G.; Malvicini, M.; Lloyd, R.; Piccioni, F.; Rizzo, M.; Peixoto, E.; Sola, M.B.; Atorrasagasti, C.; et al. Mesenchymal stromal cells engineered to produce IGF-I by recombinant adenovirus ameliorate liver fibrosis in mice. Stem Cells Dev. 2015, 24. [Google Scholar] [CrossRef] [PubMed]
- Riis, S.; Murray, J.B.; O’Connor, R. IGF-1 Signalling Regulates Mitochondria Dynamics and Turnover through a Conserved GSK-3β-Nrf2-BNIP3 Pathway. Cells 2020, 9, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey-Downs, L.C.; Mitschelen, M.; Sosnowska, D.; Toth, P.; Pinto, J.T.; Ballabh, P.; Valcarcel-Ares, M.N.; Farley, J.A.; Koller, A.; Henthorn, J.C.; et al. Liver-Specific Knockdown of IGF-1 Decreases Vascular Oxidative Stress Resistance by Impairing the Nrf2-Dependent Antioxidant Response: A Novel Model of Vascular Aging. J. Gerontol. Ser. A Boil. Sci. Med. Sci. 2011, 67, 313–329. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Mo, Z.C.; Yin, K.; Zhao, G.J.; Lv, Y.C.; Ouyang, X.P.; Jiang, Z.S.; Fu, Y.; Tang, C.K. Epigallocatechin-3-gallate prevents TNF-α-induced NF-κB activation thereby upregulating ABCA1 via the Nrf2/Keap1 pathway in macrophage foam cells. Int. J. Mol. Med. 2012. [Google Scholar] [CrossRef] [Green Version]
- Ryoo, I.G.; Ha, H.; Kwak, M.K. Inhibitory role of the KEAP1-NRF2 pathway in TGFbβ1-stimulated renal epithelial transition to fibroblastic cells: A modulatory effect on SMAD signaling. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Yang, Y. Activation of Nrf2/AREs-mediated antioxidant signalling, and suppression of profibrotic TGF-β1/Smad3 pathway: A promising therapeutic strategy for hepatic fibrosis—A review. Life Sci. 2020, 256, 117909. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Li, H.; Liu, Q.; Liu, F.; Tang, L.; Li, C.; Yuan, Y.; Zhan, Y.; Xu, W.; Li, W.; et al. Nuclear factor p65 interacts with Keap1 to repress the Nrf2-ARE pathway. Cell. Signal. 2011. [Google Scholar] [CrossRef]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-κB and underlies its chemo-resistance. Blood 2012. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Grupos | |||||
|---|---|---|---|---|---|---|
| Control | DEN | MaR1 | MaR1 + DEN | |||
| Albumin g/dL | 2.39 ± 0.57 | 1.61 ± 0.18 * | (a,c,d) | 2.53 ± 0.59 | 2.07 ± 0.42 | |
| AST UI/L | 90.87 ± 30.68 | 199.7 ± 54.94 * | (a,c,d) | 85.49 ± 29.54 | 107.0 ± 28.32 * | (a,b,c) |
| ALT UI/L | 46.7 ± 23.57 | 89.58 ± 40.63 * | (a,c,d) | 55.47 ± 33.63 | 50.35 ± 23.46 | |
| γGT UI/L | 1.63 ± 0.52 | 2.2 ± 0.45 | 1.67 ± 0.52 | 1.43 ± 0.53 | ||
| AP UI/L | 591.3 ± 138.6 | 917.8 ± 285 * | (a,c) | 568.5 ± 130.5 | 747.0 ± 168.4 | |
| LDH UI/L | 332.7 ± 202.8 | 620.6 ± 239.3 | 531.6 ± 441.3 | 599.4 ± 420.6 | ||
| Hepatic index | 4.610 ± 0.63 | 5.480 ± 0.21 * | (a,b,c) | 4.515 ± 0.27 | 4.84 ± 0.152 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez, M.J.; Sabaj, M.; Tolosa, G.; Herrera Vielma, F.; Zúñiga, M.J.; González, D.R.; Zúñiga-Hernández, J. Maresin-1 Prevents Liver Fibrosis by Targeting Nrf2 and NF-κB, Reducing Oxidative Stress and Inflammation. Cells 2021, 10, 3406. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10123406
Rodríguez MJ, Sabaj M, Tolosa G, Herrera Vielma F, Zúñiga MJ, González DR, Zúñiga-Hernández J. Maresin-1 Prevents Liver Fibrosis by Targeting Nrf2 and NF-κB, Reducing Oxidative Stress and Inflammation. Cells. 2021; 10(12):3406. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10123406
Chicago/Turabian StyleRodríguez, María José, Matías Sabaj, Gerardo Tolosa, Francisca Herrera Vielma, María José Zúñiga, Daniel R. González, and Jessica Zúñiga-Hernández. 2021. "Maresin-1 Prevents Liver Fibrosis by Targeting Nrf2 and NF-κB, Reducing Oxidative Stress and Inflammation" Cells 10, no. 12: 3406. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10123406